


Abstract
Gemfibrozil more potently inhibits CYP2C9 than CYP2C8 in vitro, and yet the opposite inhibitory potency is observed in the clinic. To investigate this apparent paradox, we evaluated both gemfibrozil and its major metabolite, an acyl-glucuronide (gemfibrozil 1-O-β-glucuronide) as direct-acting and metabolism-dependent inhibitors of the major drug-metabolizing cytochrome P450 enzymes (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) in human liver microsomes. Gemfibrozil most potently inhibited CYP2C9 (IC50 of 30 μM), whereas gemfibrozil glucuronide most potently inhibited CYP2C8 (IC50 of 24 μM). Unexpectedly, gemfibrozil glucuronide, but not gemfibrozil, was found to be a metabolism-dependent inhibitor of CYP2C8 only. The IC50 for inhibition of CYP2C8 by gemfibrozil glucuronide decreased from 24 μM to 1.8 μM after a 30-min incubation with human liver microsomes and NADPH. Inactivation of CYP2C8 by gemfibrozil glucuronide required NADPH, and proceeded with a KI (inhibitor concentration that supports half the maximal rate of enzyme inactivation) of 20 to 52 μM and a kinact (maximal rate of inactivation) of 0.21 min–1. Potent inhibition of CYP2C8 was also achieved by first incubating gemfibrozil with alamethicin-activated human liver microsomes and UDP-glucuronic acid (to form gemfibrozil glucuronide), followed by a second incubation with NADPH. Liquid chromatography-tandem mass spectrometry analysis established that human liver microsomes and recombinant CYP2C8 both convert gemfibrozil glucuronide to a hydroxylated metabolite, with oxidative metabolism occurring on the dimethylphenoxy moiety (the group furthest from the glucuronide moiety). The results described have important implications for the mechanism of the clinical interaction reported between gemfibrozil and CYP2C8 substrates such as cerivastatin, repaglinide, rosiglitazone, and pioglitazone.
There have been several reports of clinical interactions between gemfibrozil (e.g., Lopid, Parke-Davis) and CYP2C8 substrates such as cerivastatin, repaglinide, rosiglitazone, and pioglitazone (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005). Reports on the in vitro inhibitory potential of gemfibrozil demonstrated that this lipid-lowering drug is a more potent inhibitor of CYP2C9 than of CYP2C8 (Wen et al., 2001; Wang et al., 2002; Fujino et al., 2003). However, in the clinic, gemfibrozil is a more potent inhibitor of CYP2C8 than of CYP2C9. Coadministration of gemfibrozil with the CYP2C9 substrate warfarin does not increase the plasma concentrations of either R- or S-warfarin (in fact, it actually decreases them) (Lilja et al., 2005). An important step in providing a potential explanation for why gemfibrozil is a more potent inhibitor of CYP2C9 than CYP2C8 in vitro but is a more potent inhibitor of CYP2C8 than CYP2C9 in vivo was provided by Shitara et al. (2004), who demonstrated that gemfibrozil 1-O-β-glucuronide is a more potent inhibitor than gemfibrozil of CYP2C8. These same authors demonstrated that gemfibrozil 1-O-β-glucuronide inhibits in vitro the CYP2C8-mediated metabolism of cerivastatin, as well as the OATP2-mediated uptake of cerivastatin (Shitara et al., 2004). Other recent reports of glucuronide metabolites interacting directly with CYP2C8 have been published, including the CYP2C8-mediated hydroxylation of the acyl glucuronide of diclofenac, and the 17β-O-glucuronide of estradiol (Kumar et al., 2002; Delaforge et al., 2005).
This study was designed to compare the in vitro inhibitory potential of gemfibrozil and its 1-O-β-glucuronide toward the major human drug-metabolizing P450 enzymes, namely CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4. In addition to confirming that gemfibrozil glucuronide is a more potent inhibitor of CYP2C8 than is gemfibrozil, we discovered that gemfibrozil glucuronide is a metabolism-dependent inhibitor of CYP2C8, suggesting that the clinical interactions between gemfibrozil and CYP2C8 substrates involve, at least in part, the conversion of gemfibrozil to an acyl glucuronide followed by the NADPH-dependent inactivation of CYP2C8 (Fig. 1).
The conversion of gemfibrozil to its acyl glucuronide, and the subsequent metabolism-dependent inhibition of CYP2C8 by gemfibrozil glucuronide, could also be demonstrated in vitro by incubating gemfibrozil with alamethicin-activated human liver microsomes and UDP-glucuronic acid (to form the acyl glucuronide), followed by incubation with NADPH. This two-step procedure may serve as a generic method to screen drugs for their ability to be converted to a glucuronide that inhibits P450 enzymes either directly or in a metabolism-dependent manner.

Metabolism of gemfibrozil.
Materials and Methods
Chemicals and Reagents. Gemfibrozil was purchased from Sigma-Aldrich (St. Louis, MO). Gemfibrozil 1-O-β-glucuronide was prepared as described below. Human liver microsomes (pooled from 16 donors) were prepared and characterized at XenoTech LLC (Lenexa, KS), and recombinant CYP2C8 (Bactosomes) was obtained from Cypex (Dundee, Scotland). All other reagents were obtained from commercial sources, most of which have been described elsewhere (Robertson et al., 2000; Madan et al., 2002).
Synthesis of Gemfibrozil Glucuronide. Gemfibrozil (4 mM) was incubated at 37°C for 5 h with liver 9000g supernatant fraction from Aroclor 1254-induced rats (7.2 mg/ml) in a 50-ml reaction mixture containing 100 mM glycine buffer (pH 8.6), 10% glycerol (v/v), 2.2% acetonitrile (v/v), and 8 mM UDP-glucuronic acid. The reaction was quenched with 2 ml of 2 N HCl. Gemfibrozil glucuronide was purified by semipreparative HPLC on a YMC ODSA30 × 100 mm, S5 column (YMC, Inc., Wilmington, NC) at a flow rate of 10 ml/min. The step-wise linear gradient was 10% to 20% solvent B over 5 min, and from 20% to 90% solvent B over 34 min, where solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile. Fractions containing gemfibrozil glucuronide were pooled and lyophilized. Gemfibrozil glucuronide was obtained as a white solid (44% yield, HPLC purity: >99% at 275 nm). Liquid chromatography/mass spectrometry (electrospray ionization [M – H]–): m/z 425; MS2: m/z 425→249, 175; MS3: m/z 425→249→121. High resolution MS: C21H30O9, calculated m/z for [M + Na+]: 449.17875, found 449.1777 (error, –2.4 ppm). 1H NMR (400 MHz, DMSO-d6 + D2O) δ ppm 1.15 (s, 3 H), 1.15 (s, 3H), 1.64 (s, 2 H), 2.06 (s, 3H), 2.22 (s, 3H), 3.17 (t, J = 8.56 Hz, 1 H), 3.27 (t, J = 8.80 Hz, 1 H), 3.33 (t, J = 9.05 Hz, 1 H), 3.70 (d, J = 9.29 Hz, 1 H), 3.83–3.91 (m, J = 4.89 Hz, 1 H), 5.34 (d, J = 8.31 Hz, 1 H), 6.60 (d, J = 7.34 Hz, 1 H), 6.68 (s, 1H), 6.96 (d, J = 7.34 Hz, 1 H).
P450 Enzyme Inhibition.IC50 Determination. Unless indicated otherwise, P450 activities in human liver microsomes were determined according to previously published procedures (Pearce et al., 1996; Robertson et al., 2000; Madan et al., 2002; Toren et al., 2004). In general, incubations were conducted in 400-μl incubation mixtures (pH 7.4) containing high purity water, potassium phosphate buffer (50 mM), MgCl2 (3 mM), EDTA (1 mM), NADP (1 mM), glucose 6-phosphate (5 mM), glucose-6-phosphate dehydrogenase (1 unit/ml), marker substrate, and human liver microsomes (50–100 μg protein/ml). The concentration of each marker substrate was based on previously determined Km values as follows: 20 μM phenacetin (for CYP1A2), 50 μM bupropion (for CYP2B6), 15 μM paclitaxel (for CYP2C8), 4 μM diclofenac (for CYP2C9), 35 μM S-mephenytoin (for CYP2C19), 5 μM dextromethorphan (for CYP2D6), 100 μM testosterone (for CYP3A4), and 3 μM midazolam (second substrate for CYP3A4). All incubations were conducted at 37°C for 5 min before addition of the stop reagent with internal standard, which in most cases was a deuterated analog of the metabolite measured. Positive controls for both direct and metabolism-dependent inhibition were included at a single concentration.
Aliquots of the stock and/or working solutions of either gemfibrozil or its 1-O-β-glucuronide were manually added to buffer mixtures containing the components described above, but prior to addition of the NADPH-generating system. Fresh solutions were prepared on the day of the experiment.
To evaluate the potential for metabolism-dependent inhibition, gemfibrozil or its 1-O-β-glucuronide (at the same concentrations used to evaluate direct inhibition) were incubated at 37°C with human liver microsomes and an NADPH-generating system for approximately 30 min. Reactions were initiated by the addition of the NADPH-generating system. After the 30-min preincubation, the marker substrate (at a concentration approximately equal to its Km) was added, and the incubation was continued for 5 min to measure residual P450 activity.
KI and kinact Determination. To determine the KI and kinact values for the inactivation of CYP2C8, various concentrations of gemfibrozil glucuronide (0.25–64 μM) were incubated for 2 to 40 min with pooled human liver microsomes (0.1 and 1.0 mg/ml) at 37°C. After the preincubation, an aliquot (40 μl) was transferred to another incubation tube (final volume 400 μl) containing 10 μM paclitaxel and an NADPH-generating system to measure residual CYP2C8 activity. This procedure diluted the microsomes to 10 or 100 μg/ml and diluted gemfibrozil glucuronide to one tenth its original concentration to minimize any direct inhibitory effects on CYP2C8. The incubations were then continued for 2 min to allow for conversion of paclitaxel to 6α-hydroxypaclitaxel. A short incubation with paclitaxel was used to minimize additional inactivation of CYP2C8 when measuring residual CYP2C8 activity. Reactions were carried out in triplicate.
Metabolism of Gemfibrozil Glucuronide. Gemfibrozil glucuronide (64 μM) was incubated for 0 or 30 min with pooled human liver microsomes (1 mg/ml) or recombinant CYP2C8 (∼40 pmol/ml) in potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), NADP (1 mM), glucose 6-phosphate (5 mM), and glucose-6-phosphate dehydrogenase (1 unit/ml). The samples were stopped with an equal volume of acetonitrile and 4% formic acid, and precipitated protein was removed by centrifugation. Aliquots of these samples were analyzed on an API-4000 Q-trap (see Analytical Methods, below) using a TurboIonSpray source in the negative ionization mode. Data acquisition was conducted using Analyst software (version 1.4) with information-dependent-acquisition scan function. Constant neutral loss survey scans at 176 Da were used to trigger product ion data acquisition on the gemfibrozil glucuronide. Separations were performed using a Develosil RP 150 × 2.0 mm analytical column (Phenomenex, Torrance, CA) with a linear gradient over 45 min.
Two-Step Activation of Gemfibrozil to a CYP2C8 Inhibitor. Having obtained evidence that gemfibrozil glucuronide is a metabolism-dependent inhibitor of CYP2C8, we evaluated whether such inhibition could be achieved starting with gemfibrozil in a two-step process. Accordingly, gemfibrozil was incubated with human liver microsomes in the presence of UDP-glucuronic acid (to support the formation of gemfibrozil glucuronide) and then in the presence of NADPH (to support the metabolism-dependent inactivation of CYP2C8 by gemfibrozil glucuronide). Pooled human liver microsomes (1.1 mg) were preincubated with 27.5 μg of alamethicin (a pore-forming peptide that increases microsomal UDP-glucuronosyltransferase activity without compromising P450 activity (Fisher et al., 2000)) on ice in a total volume of 2 ml. Aliquots (250 μl) of the alamethicin-activated microsomes were transferred to a second set of tubes containing 2.0 ml of potassium phosphate buffer (55 mM, pH 7.4), MgCl2 (3.3 mM), EDTA (1.1 mM), saccharic acid 1,4-lactone (100 μM), and gemfibrozil (30 or 100 μM) for a final microsomal protein concentration of 50 μg/ml. Glucuronidation reactions were started by the addition of 250 μl of a 40 mM solution of UDP-glucuronic acid (final concentration = 4 mM). After a 0- or 60-min incubation at 37°C, the samples were placed on ice. The samples were then divided into 0- and 30-min NADPH-preincubation samples. After a 0- or 30-min preincubation with NADPH, an aliquot (40 μl) was transferred to another incubation tube (final volume 400 μl) containing paclitaxel (10 μM) and an NADPH-generating system to measure residual CYP2C8 activity. This procedure diluted the microsomal protein to 5 μg/ml and diluted gemfibrozil and its glucuronide to one tenth their original concentration to minimize any direct inhibitory effects on CYP2C8. The incubations were then continued for 5 min to allow for conversion of paclitaxel to 6α-hydroxypaclitaxel.
Analytical Methods. All analyses were performed with an Applied Biosystems/Sciex API2000, API3000, or API-4000 Q-trap HPLC-MS/MS system equipped with an electrospray (TurboIonSpray) ionization source (Applied Biosystems, Foster City, CA), two LC-10ADvp pumps with an SCL-10Advp controller, SIL-HTA autosampler, and DGU-14 solvent degasser (Shimadzu, Columbia, MD). The HPLC column used was a Develosil RPAqueous, 5 μm, 50 × 2.0 mm column (Phenomenex), which was preceded by a direct connection guard column with a C8, 4.0 mm × 2.0 mm cartridge (Phenomenex).
Statistical Analyses. Data for the IC50 determinations were processed with a validated customized add-in (DI IC50 LCMS Template version 1.1) for the computer program Microsoft Excel (Office 2000; Microsoft Inc., Redmond, WA). When inhibition of P450 enzymes was observed, the data were processed to determine IC50 values by nonlinear regression with XLFit (Version 3.0; IDBS, Limited, Surrey, UK).
Data for the KI and kinact investigation were analyzed to determine the rate of enzyme inactivation at each inhibitor concentration tested (Kitz and Wilson, 1962). Based on this method, the natural log of the ratio of the residual activity (Ei) to the control activity (Et) (where the residual activity is the rate after a defined incubation period with the inhibitor) was plotted against incubation time for each concentration of inhibitor. Linear regression (GraFit, Version 4.021; Erithacus Software, Ltd., Horley, Surrey, UK) was then performed such that the negative slope represents the observed initial rate of enzyme inactivation at each concentration of inhibitor. The rates of inactivation were then plotted against the inhibitor concentration and the data fitted by nonlinear regression (GraFit, Version 4.021; Erithacus Software, Ltd.) according to the following equation (Jones et al., 1999):  This equation is analogous to the Michaelis-Menten equation, where λ represents the rate constant for inactivation at each inhibitor concentration, [I]isthe inhibitor concentration, KI is the inhibitor concentration that produces half the maximal rate of inactivation (analogous to Km), and kinact represents the maximal rate of inactivation (analogous to Vmax). This equation assumes that there is negligible change in inhibitor concentration during incubation and that the loss of enzyme activity is due to inactivation by a first-order process.
This equation is analogous to the Michaelis-Menten equation, where λ represents the rate constant for inactivation at each inhibitor concentration, [I]isthe inhibitor concentration, KI is the inhibitor concentration that produces half the maximal rate of inactivation (analogous to Km), and kinact represents the maximal rate of inactivation (analogous to Vmax). This equation assumes that there is negligible change in inhibitor concentration during incubation and that the loss of enzyme activity is due to inactivation by a first-order process.
Results
Effect of Gemfibrozil and Gemfibrozil 1-O-β-glucuronide on Selected P450 Enzymes.IC50 Determinations. Gemfibrozil and gemfibrozil glucuronide were evaluated as direct-acting and metabolism-dependent inhibitors of the major P450 enzymes in human liver microsomes, namely, CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4, the activities of which were assessed with marker substrates at concentrations equal to Km. The results are summarized in Table 1. Gemfibrozil was a more potent inhibitor of CYP2C9 than of CYP2C8 (IC50 value of 30 μM versus 120 μM, respectively), whereas the converse was true of gemfibrozil glucuronide (the IC50 value was 24 μM for CYP2C8 and >300 μM for CYP2C9).
Inhibition of the major P450 enzymes in human liver microsomes by gemfibrozil and its 1-O-β-glucuronide
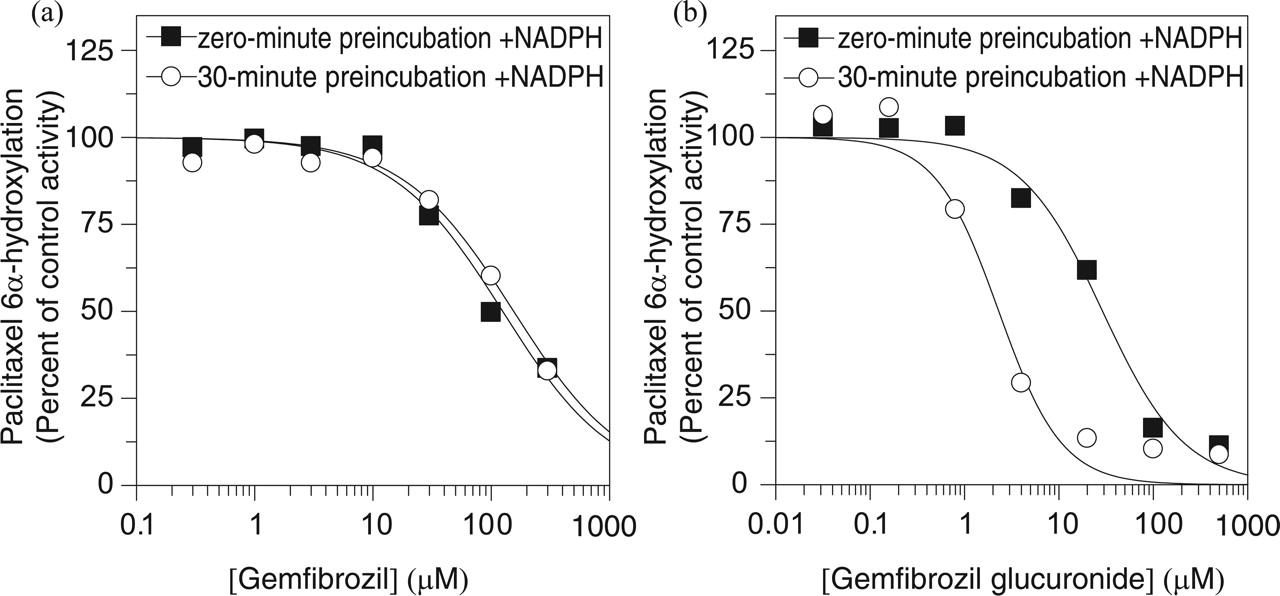
Gemfibrozil inhibited CYP1A2 (IC50 99 μM) and CYP2C19 (IC50 100 μM) with slightly greater potency than it inhibited CYP2C8 (IC50 120 μM), but it did not inhibit CYP2B6 or CYP2D6 (IC50 > 300 μM), nor did it inhibit CYP3A4, regardless of whether testosterone or midazolam was used as marker substrate. As summarized in Table 1, preincubation of gemfibrozil with NADPH-fortified human liver microsomes for 30 min did not increase its inhibitory effect, suggesting that gemfibrozil is not a metabolism-dependent inhibitor of the P450 enzymes examined. This is further illustrated in Fig. 2a, which shows that preincubation had no effect on the potency with which gemfibrozil inhibited CYP2C8 activity toward paclitaxel.
With the notable exception of CYP2C8, gemfibrozil glucuronide was less potent than gemfibrozil at inhibiting all of the P450 enzymes examined. Consequently, gemfibrozil glucuronide was a more selective inhibitor than gemfibrozil in that gemfibrozil glucuronide was at least an order of magnitude more potent as an inhibitor of CYP2C8 than the other P450 enzymes examined. Furthermore, the potency with which gemfibrozil glucuronide inhibited CYP2C8 increased by an additional order of magnitude after a 30-min preincubation with NADPH-fortified human liver microsomes, as shown in Fig. 2b. No such increase in inhibitory potency was apparent with any of the other P450 enzymes examined.
The marked increase in CYP2C8 inhibition caused by preincubating gemfibrozil glucuronide with human liver microsomes for 30 min was dependent on the presence of NADPH. In the presence of NADPH, the 30-min preincubation resulted in more than a 10-fold shift in IC50 (from 24 to 1.8 μM), whereas in its absence, the preincubation actually resulted in a slight decrease in inhibitory potency [the IC50 value was 48 μM (results not shown)].
Inactivation of CYP2C8 by Gemfibrozil 1-O-β-glucuronide.KI and kinact Determination. The results in Fig. 2b suggest that gemfibrozil glucuronide is a metabolism-dependent inhibitor of CYP2C8. Accordingly, experiments were performed to determine KI (the concentration of gemfibrozil glucuronide supporting the half-maximal rate of CYP2C8 inactivation) and kinact (the first order rate constant for CYP2C8 inactivation). The results are summarized in Fig. 3. The inactivation of CYP2C8 was dependent on the concentration of gemfibrozil glucuronide (over the range examined, which was 1.25–64 μM), and the time course conformed to a first order process (as indicated by the linearity of plots of the log of the residual enzyme activity against time). The KI values differed slightly depending on whether gemfibrozil glucuronide was preincubated with human liver microsomes at 0.1 mg/ml (and subsequently diluted 10-fold to determine CYP2C8 activity) or 1.0 mg/ml (and subsequently diluted 10-fold to determine CYP2C8 activity). The KI value was 20 μM in the former case and 52 μM in the latter. In both cases, the kinact value was 0.21 min–1, meaning that, in the presence of saturating concentrations of gemfibrozil glucuronide, 21% of CYP2C8 was inactivated every minute. As a metabolism-dependent inhibitor, gemfibrozil glucuronide inhibited CYP2C8 with roughly the same potency regardless of whether the concentration of human liver microsomes was 0.1 or 1.0 mg/ml. If the mechanism-based inhibition of CYP2C8 involved product inhibition (i.e., reversible inhibition of CYP2C8 by a metabolite of gemfibrozil glucuronide), then gemfibrozil glucuronide would have been expected to cause more inhibition when incubated with a 10-fold higher concentration of human liver microsomes, which was not the case. This, in turn, suggests but does not prove that gemfibrozil glucuronide is an irreversible or quasi-irreversible inactivator of CYP2C8.
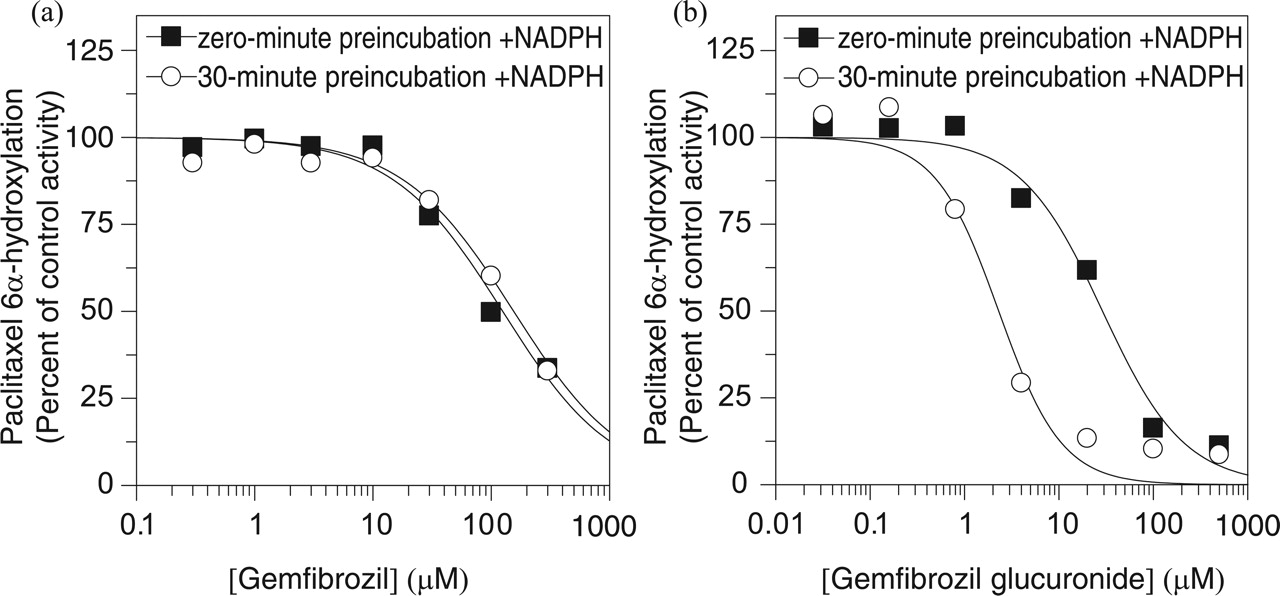
Evaluation of gemfibrozil and gemfibrozil glucuronide as direct-acting and metabolism-dependent inhibitors of CYP2C8. a, gemfibrozil inhibited CYP2C8 in pooled human liver microsomes (0.05 mg/ml) with IC50 values of 120 μM (without preincubation) and 150 μM (with a 30-min preincubation with NADPH); b, gemfibrozil glucuronide inhibited CYP2C8 in pooled human liver microsomes (0.05 mg/ml) with an IC50 values of 24 μM (without preincubation) and 1.8 μM with a 30-min preincubation with NADPH. Each symbol represents the average of duplicate determinations.
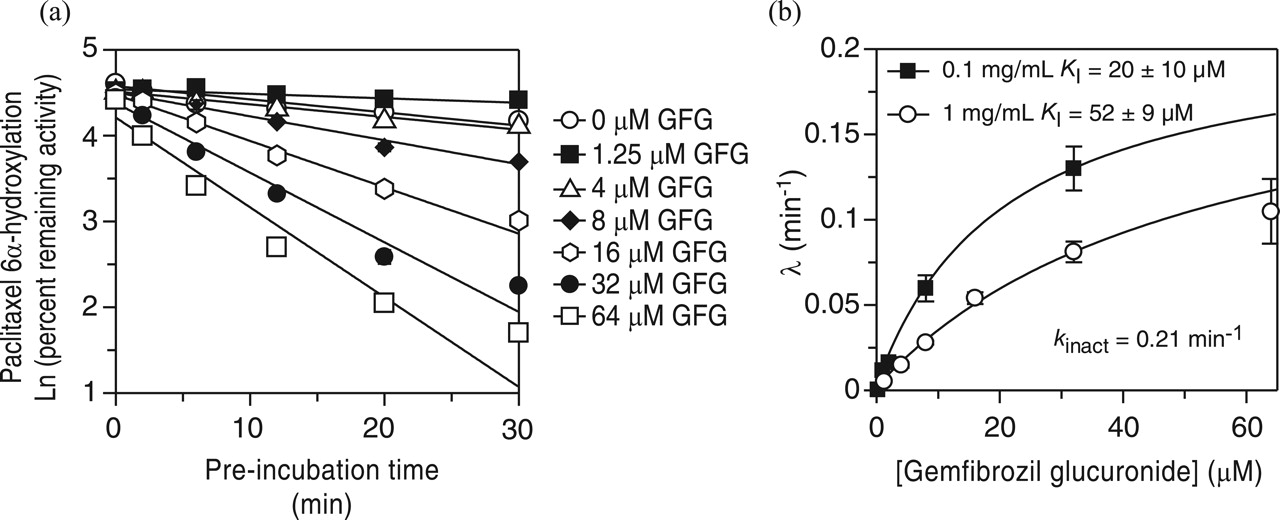
Determination of KI and kinact for the metabolism-dependent inhibition of CYP2C8 by gemfibrozil glucuronide. a, gemfibrozil glucuronide (GFG) was preincubated (at concentrations indicated) with NADPH and pooled human liver microsomes (1 mg/ml) for the time points indicated at 37°C. After the preincubation, aliquots (40 μl) were removed and residual CYP2C8 activity was determined as described under Materials and Methods. Individual points represent the average of triplicate determinations ± standard deviation. The graph in b represents the direct plot of the initial rates of inactivation of CYP2C8 from two experiments, the first with 0.1 mg/ml pooled human liver microsomes, and the second with 1 mg/ml (the latter is shown in graph a). Values are the slopes of the initial rates of inactivation (λ) at each concentration of gemfibrozil glucuronide, shown ± standard error.
Metabolism of Gemfibrozil Glucuronide. In the presence of NADPH, human liver microsomes and recombinant CYP2C8 both converted gemfibrozil glucuronide to a hydroxylated metabolite, as determined by LC-MS/MS. The mass fragmentation pattern of this metabolite is shown in Fig. 4. The presence of the ion at m/z 265 is due to the loss of a 176-Da fragment from the hydroxy-gemfibrozil glucuronide molecular ion of 441 Da, which is characteristic of collision-induced dissociation of the glucuronic acid moiety. The presence of the ion at m/z 137 indicates that hydroxylation of gemfibrozil glucuronide occurred at the dimethylphenoxy moiety.
Preincubation of Gemfibrozil with UDP-Glucuronic Acid and Alamethicin. As shown in Fig. 5, incubating gemfibrozil with alamethicin-activated human liver microsomes and UDP-glucuronic acid increased its ability to inhibit CYP2C8, presumably because of the formation of gemfibrozil glucuronide. Incubating these same samples with NADPH caused a further loss of CYP2C8 activity, presumably because of the metabolism-dependent inhibition of CYP2C8 by gemfibrozil glucuronide (Fig. 5).

LC-MS/MS analysis of the metabolite formed from gemfibrozil glucuronide by human liver microsomes or recombinant CYP2C8. The full scan of MS spectra of putative hydroxylated gemfibrozil glucuronide is shown, along with the proposed fragmentation of the molecule. The samples were incubated with human liver microsomes or recombinant CYP2C8, NADPH, and gemfibrozil glucuronide (64 μM) for 60 min.

Two-stage activation of gemfibrozil by human liver microsomes in the presence of UDP-glucuronic acid and alamethicin (the glucuronidation step) and then NADPH (the cytochrome P450 step). Gemfibrozil (30 or 100 μM) was preincubated for 0 or 60 min with pooled human liver microsomes, UDP-glucuronic acid, and alamethicin, and an additional 0 or 30 min with NADPH before measuring residual CYP2C8 activity, as described under Materials and Methods. It should be noted that the preincubated samples were diluted 10-fold before measuring CYP2C8 activity, hence, the final concentration of gemfibrozil was 10 μM or less. Samples without preincubation served as controls. Values represent the average of duplicate determinations.
Discussion
For the purpose of prioritizing clinical drug interaction studies, the U.S. Food and Drug Administration frequently evaluates in vitro P450 inhibition data on the basis of rank order, which tacitly assumes that the P450 enzyme inhibited most potently in vitro will be the same enzyme most potently inhibited in vivo (Bjornsson et al., 2003; Huang, 2004). This approach has proven reliable on numerous occasions, especially when a drug candidate has been evaluated as both a direct-acting and a metabolism-dependent inhibitor of the major drug-metabolizing P450 enzymes in human liver microsomes. Gemfibrozil represents an exception to the rule, because it inhibits CYP2C9 more potently than it inhibits CYP2C8 in vitro, and yet the opposite is observed in vivo, where gemfibrozil does not inhibit the CYP2C9-dependent metabolism of warfarin and has only minimal effect on the metabolism of the CYP2C9-substrate nateglinide (Lilja et al., 2005; Niemi et al., 2005), but does inhibit the metabolism of several CYP2C8 substrates, including cerivastatin, repaglinide, rosiglitazone, and pioglitazone (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005). In the present study, we have confirmed the previous finding of Shitara et al. (2004) that gemfibrozil 1-O-β-glucuronide is a direct inhibitor of CYP2C8 and, moreover, that the potency with which gemfibrozil inhibits P450 enzymes is shifted from CYP2C9 to CYP2C8 by glucuronidation (Table 1). Furthermore, we have shown that gemfibrozil glucuronide is an NADPH-dependent, mechanism-based inhibitor of CYP2C8 (Figs. 2b and Fig. 3). We propose that this mechanism-based inhibition of CYP2C8 by gemfibrozil glucuronide explains, at least in part, the observed clinical interactions between multiple-dose gemfibrozil and the aforementioned CYP2C8 substrates (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005).
The mechanism by which gemfibrozil glucuronide inactivates CYP2C8 remains to be determined, but some mechanistic information is apparent from the results of the present study. Gemfibrozil glucuronide is an acyl glucuronide, and some acyl glucuronides can rapidly rearrange to form reactive aldehydes that form Schiff's bases (covalent adducts) with lysine residues on proteins (Li and Benet, 2003). Accordingly, we evaluated gemfibrozil glucuronide as both a time-dependent and metabolism-dependent inhibitor of CYP2C8 (by incubating human liver microsomes with gemfibrozil glucuronide in the absence and presence of NADPH, respectively). This experiment established that gemfibrozil glucuronide is a metabolism-dependent inhibitor, not simply a time-dependent inhibitor, of CYP2C8. In addition, normal Michaelis-Menten-type kinetics were observed in the experiment to determine kinact (Fig. 3b), making the spontaneous formation of reactive metabolites a less likely explanation for the increase in the inhibition of CYP2C8. Furthermore, gemfibrozil glucuronide has an apparent half-life of 44 h in aqueous buffer (pH 7.4, 37°C), making it one of the least reactive acyl glucuronides (Ebner et al., 1999). Finally, incubating gemfibrozil glucuronide with 10 times the concentration of human liver microsomes (1.0 versus 0.1 mg/ml) did not increase its inhibitory effect, suggesting that the mechanism-based inactivation of CYP2C8 is not due to product inhibition but is due to irreversible or quasi-irreversible inactivation of CYP2C8.
The results of the present study suggest that the clinically significant interactions observed between gemfibrozil and CYP2C8 substrates are due, at least in part, to the NADPH-dependent inactivation of CYP2C8 by gemfibrozil 1-O-β-glucuronide. A method to predict in vivo inhibition from quasi-irreversible or irreversible inhibition was reported by Lu and colleagues (eq. 1 in Lu et al., 2003) as follows:  where [I] is the inactivator concentration, and kinact and KI are parameters estimable by nonlinear regression, (fm · fm,P450) represents the fraction of a hypothetical concomitantly administered drug metabolized by a given P450 enzyme, and ke is the rate constant for enzyme degradation [which has not been determined experimentally, but which was assumed to be 0.0008 min–1, which in turn assumes a half-life of 21 h (Mayhew et al., 2000)]. This relationship relies on certain assumptions, including 1) that the conditions of the well stirred pharmacokinetic model are met, 2) that the substrate exhibits linear pharmacokinetics and is metabolized only in the liver, and 3) that complete absorption from the gastrointestinal tract occurs (Mayhew et al., 2000; Lu et al., 2003). As illustrated in Fig. 6, the in vitro kinact (0.21 min–1) and KI (51 μM) data were used to simulate the effects of gemfibrozil glucuronide on the pharmacokinetics of concomitantly administered CYP2C8 substrates (AUCpo(i)-to-AUCpo(c) ratio). For these simulations, we used various published values of the in vivo concentrations of gemfibrozil glucuronide: 1) 89 μM, which is the average unbound (free) Cmax in liver; 2) 20 μM, which is the total (free + bound) in plasma; and 3) 2.3 μM, which is the unbound (free) Cmax in plasma (Shitara et al., 2004). We did not base these simulations on an estimate of the concentration of free gemfibrozil glucuronide in the intrahepatic blood, as has been proposed by others (Kanamitsu et al., 2000; Ito et al., 2004), because this approach was developed for orally administered drugs, whereas gemfibrozil glucuronide is presumably formed in the liver. On the basis of these simulations, for a drug whose clearance was 95% dependent on metabolism by CYP2C8 (i.e., fm · fm,CYP2C8 = 0.95), gemfibrozil glucuronide would be predicted to cause an 8-fold to a 19-fold increase in AUC, depending on which concentration of gemfibrozil glucuronide is used. The -fold increase in AUC of a victim drug would be less for drugs whose clearance was less dependent on metabolism by CYP2C8 as shown in Fig. 6. In agreement, the AUC ratio for repaglinide, cerivastatin, pioglitazone, and rosiglitazone is ∼8.0 (range 5.5–15), 4.4 (range 1.1–8.0), 3.2 (range 2.3–6.5), and 2.3 (range 1.5–2.8), respectively (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005). Conversely, the -fold increase in AUC of a victim drug would be greater if the half-life of CYP2C8 were longer than 21 h (i.e., if ke were smaller), or if the lower estimate of KI (20 μM instead of 51 μM) were used in the simulations. This predicted effect when using either the average unbound (free) Cmax concentration in liver or the total (free + bound) plasma concentration of gemfibrozil glucuronide is larger than that observed clinically, whereas the predicted effect using the unbound (free) Cmax in plasma is similar to the observed clinical effect. The results of the present study also provide a likely explanation for the discordant results reported by Prueksaritanont et al. (2005), who recently evaluated gemfibrozil as a P450 enzyme inducer in primary cultures of human hepatocytes. These investigators observed that treatment of human hepatocytes with 250 μM gemfibrozil caused an increase in the levels of mRNA encoding CYP3A4 (3.8-fold) and CYP2C8 (4.9-fold). Gemfibrozil induced CYP3A4 activity (testosterone 6β-hydroxylation) to the same extent that it induced CYP3A4 mRNA. In contrast, rather than inducing CYP2C8 activity (amodiaquine N-dealkylation) to the same extent that it induced CYP2C8 mRNA, gemfibrozil caused a marked decrease (>60%) in CYP2C8 activity. The discrepancy between the gemfibrozil-induced increase in CYP2C8 mRNA and the decrease in CYP2C8 activity can be rationalized on the basis that gemfibrozil up-regulated the transcription of CYP2C8 (and CYP3A4) but was converted by human hepatocytes to its acyl glucuronide, which subsequently inactivated CYP2C8 (but not CYP3A4).
where [I] is the inactivator concentration, and kinact and KI are parameters estimable by nonlinear regression, (fm · fm,P450) represents the fraction of a hypothetical concomitantly administered drug metabolized by a given P450 enzyme, and ke is the rate constant for enzyme degradation [which has not been determined experimentally, but which was assumed to be 0.0008 min–1, which in turn assumes a half-life of 21 h (Mayhew et al., 2000)]. This relationship relies on certain assumptions, including 1) that the conditions of the well stirred pharmacokinetic model are met, 2) that the substrate exhibits linear pharmacokinetics and is metabolized only in the liver, and 3) that complete absorption from the gastrointestinal tract occurs (Mayhew et al., 2000; Lu et al., 2003). As illustrated in Fig. 6, the in vitro kinact (0.21 min–1) and KI (51 μM) data were used to simulate the effects of gemfibrozil glucuronide on the pharmacokinetics of concomitantly administered CYP2C8 substrates (AUCpo(i)-to-AUCpo(c) ratio). For these simulations, we used various published values of the in vivo concentrations of gemfibrozil glucuronide: 1) 89 μM, which is the average unbound (free) Cmax in liver; 2) 20 μM, which is the total (free + bound) in plasma; and 3) 2.3 μM, which is the unbound (free) Cmax in plasma (Shitara et al., 2004). We did not base these simulations on an estimate of the concentration of free gemfibrozil glucuronide in the intrahepatic blood, as has been proposed by others (Kanamitsu et al., 2000; Ito et al., 2004), because this approach was developed for orally administered drugs, whereas gemfibrozil glucuronide is presumably formed in the liver. On the basis of these simulations, for a drug whose clearance was 95% dependent on metabolism by CYP2C8 (i.e., fm · fm,CYP2C8 = 0.95), gemfibrozil glucuronide would be predicted to cause an 8-fold to a 19-fold increase in AUC, depending on which concentration of gemfibrozil glucuronide is used. The -fold increase in AUC of a victim drug would be less for drugs whose clearance was less dependent on metabolism by CYP2C8 as shown in Fig. 6. In agreement, the AUC ratio for repaglinide, cerivastatin, pioglitazone, and rosiglitazone is ∼8.0 (range 5.5–15), 4.4 (range 1.1–8.0), 3.2 (range 2.3–6.5), and 2.3 (range 1.5–2.8), respectively (Backman et al., 2002; Niemi et al., 2003a,b; Jaakkola et al., 2005). Conversely, the -fold increase in AUC of a victim drug would be greater if the half-life of CYP2C8 were longer than 21 h (i.e., if ke were smaller), or if the lower estimate of KI (20 μM instead of 51 μM) were used in the simulations. This predicted effect when using either the average unbound (free) Cmax concentration in liver or the total (free + bound) plasma concentration of gemfibrozil glucuronide is larger than that observed clinically, whereas the predicted effect using the unbound (free) Cmax in plasma is similar to the observed clinical effect. The results of the present study also provide a likely explanation for the discordant results reported by Prueksaritanont et al. (2005), who recently evaluated gemfibrozil as a P450 enzyme inducer in primary cultures of human hepatocytes. These investigators observed that treatment of human hepatocytes with 250 μM gemfibrozil caused an increase in the levels of mRNA encoding CYP3A4 (3.8-fold) and CYP2C8 (4.9-fold). Gemfibrozil induced CYP3A4 activity (testosterone 6β-hydroxylation) to the same extent that it induced CYP3A4 mRNA. In contrast, rather than inducing CYP2C8 activity (amodiaquine N-dealkylation) to the same extent that it induced CYP2C8 mRNA, gemfibrozil caused a marked decrease (>60%) in CYP2C8 activity. The discrepancy between the gemfibrozil-induced increase in CYP2C8 mRNA and the decrease in CYP2C8 activity can be rationalized on the basis that gemfibrozil up-regulated the transcription of CYP2C8 (and CYP3A4) but was converted by human hepatocytes to its acyl glucuronide, which subsequently inactivated CYP2C8 (but not CYP3A4).

Simulation of the effect of gemfibrozil 1-O-β-glucuronide on the pharmacokinetics of CYP2C8 substrates. The effect of gemfibrozil glucuronide on CYP2C8 substrates (AUCpo(i)-to-AUCpo(c) ratio) was simulated based on eq. 1 from Lu et al. (2003), as described in the text. The y-axis represents the -fold increase in AUC of the victim drug (i.e., the drug whose clearance is inhibited by gemfibrozil glucuronide). The x-axis represents the fraction of clearance determined by CYP2C8 (e.g., 0.9 means 90% of the victim drug's clearance is determined by CYP2C8). The three simulations (A, B, C) are based on three estimates of the in vivo concentration of gemfibrozil glucuronide as described in the text.
The observation that gemfibrozil is an enzyme inducer may help explain the effects of gemfibrozil on warfarin metabolism. Gemfibrozil (600 mg b.i.d. for 8 days) was evaluated for its ability to inhibit the CYP2C9-dependent metabolism of warfarin (because CYP2C9 is the enzyme most potently inhibited by gemfibrozil), but, unexpectedly, gemfibrozil was found to increase, not decrease, the metabolic clearance of warfarin (Lilja et al., 2005). Although Prueksaritanont et al. (2005) did not assess the effects of gemfibrozil on CYP2C9, this enzyme is typically up-regulated together with CYP3A4 and CYP2C8, and it seems reasonable to speculate that gemfibrozil increases the metabolic clearance of warfarin by inducing CYP2C9.
The biotransformation of drugs and other xenobiotics frequently occurs in two sequential phases. During phase I metabolism, enzymes that catalyze oxidative, reductive, or hydrolytic reactions introduce or expose a functional group that is subsequently conjugated, often with a highly water-soluble moiety such as glucuronic or sulfonic acid, during phase II metabolism (Parkinson, 2001). Although phase I metabolism generally precedes phase II, there are reports of phase II metabolites undergoing oxidation by P450 enzymes. Examples include the oxidation of sulfate conjugates of testosterone, dehydroepiandrosterone, and estrogens (Ingelman-Sundberg et al., 1975; Kitada et al., 1987; Ohmori et al., 1998). In one case, cytochrome P450 (namely, CYP2C12, which is expressed in female but not male rats) catalyzes the oxidation of a steroid di-sulfate (namely 5α-androstane-3α,17β-diol-3,17-disulfate) (Ryan et al., 1984). More recently, CYP2C8 has been shown to catalyze the oxidation of two glucuronides, namely, the acyl glucuronide of diclofenac and the 17β-glucuronide of estradiol (Kumar et al., 2002; Delaforge et al., 2005). In both cases, hydroxylation occurs well away from the glucuronide moiety (4′-hydroxylation in the case of diclofenac glucuronide and 2-hydroxylation in the case of estradiol-17β-glucuronide). In the case of the aglycones (i.e., prior to glucuronidation), these same reactions are catalyzed by CYP2C9 (Leemann et al., 1993; Lee et al., 2003) and even CYP2C8, albeit at a much slower rate (Lee et al., 2003; Melet et al., 2004). It remains to be determined whether, as a general rule, substrates for CYP2C9 become substrates for CYP2C8 once they have been glucuronidated. The active site of CYP2C8 has been mapped by X-ray crystallography and determined to be considerably larger than that of CYP2C9 (based on the rigid crystal structures of the enzymes, which does not take into account the potential flexibility of their active sites). As such, the former is capable of accommodating large substrates, including glucuronides, in its active site, with metabolism predicted to occur distal to the glucuronide moiety (Melet et al., 2004; Schoch et al., 2004).
In the presence of NADPH, human liver microsomes and recombinant CYP2C8 both converted gemfibrozil glucuronide to a hydroxylated metabolite. Hydroxylation occurred well away from the glucuronide moiety on the dimethylphenoxy ring of gemfibrozil. This distal site of hydroxylation is analogous to that reported for the glucuronides of diclofenac and estradiol, and is consistent with the proposed orientation of large acidic substrates in the active site of CYP2C8 (Melet et al., 2004). From the mass fragmentation pattern, we cannot discern whether CYP2C8 oxidizes one of the methyl groups (aliphatic hydroxylation) or the phenol ring (aromatic hydroxylation). The latter reaction could produce a substituted hydroquinone, which is a candidate for the putative reactive metabolite that ostensibly inactivates CYP2C8. However, methyl-hydroxylation and subsequent oxidation to the carboxylic acid has also been reported as a major route of gemfibrozil metabolism (Shitara et al., 2004). Further experiments will be required to ascertain whether formation of the hydroxylated metabolite or its subsequent metabolism leads to the inactivation of CYP2C8. It also remains to be determined whether inactivation of CYP2C8 by gemfibrozil glucuronide involves covalent modification of the protein and, if so, which amino acid residue or residues are modified.
Finally, because it was considered that formation of glucuronide metabolites that can cause clinically significant inhibition of P450 enzymes might be a more widespread phenomenon than was heretofore appreciated, we investigated an experimental design that might be applied generally during preclinical P450 inhibition testing. We have shown that, in a manner analogous to preincubating test drugs with human liver microsomes and NADPH (a method commonly used to identify metabolism-dependent inhibitors), a test drug (gemfibrozil, in this case) can be preincubated with human liver microsomes first with UDP-glucuronic acid (in the presence of alamethicin to stimulate UDP-glucuronosyltransferase activity) and then with NADPH before measuring P450 activity (CYP2C8 activity, in this case). Along similar lines, Kajosaari et al. (2005) used the first step of this approach to show that glucuronidation of gemfibrozil by alamethicin-activated human liver microsomes increased the degree of inhibition of CYP2C8. Our results show that the ability of gemfibrozil to inhibit CYP2C8 increased after the “glucuronidation step” and increased even further after the subsequent “cytochrome P450 step” (Fig. 5). This two-step procedure might prove useful in screening drug candidates for their potential to be converted to glucuronides that can function as direct-acting or metabolism-dependent inhibitors of CYP2C8 (or any of the other drug-metabolizing P450 enzymes in human liver microsomes).
Acknowledgments
We thank Kyle Tholen, Rob Grbac, Jason Barricklow, Mark Horrigan, and Greg Loewen for assistance with the experimental work, and Kammie Settle for indispensable assistance with the graphics.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.007633.
-
ABBREVIATIONS: P450, cytochrome P450; IC50, inhibitor concentration that causes 50% inhibition; HPLC, high-performance liquid chromatography; LC-MS/MS, liquid chromatography-tandem mass spectrometry; KI, inhibitor concentration that supports half the maximal rate of inactivation; kinact, maximal rate of enzyme inactivation.
- Received October 11, 2005.
- Accepted November 15, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}