


Abstract
Raloxifene and 4-hydroxytamoxifen (4-OHT) are important estrogen-related drugs used in the treatment of osteoporosis and breast cancer. Sulfation is involved in the metabolism and inactivation of both compounds in human tissues, although the sulfotransferase (SULT) isoforms involved in their conjugation have not been well described. The ability of seven expressed SULT isoforms to sulfate raloxifene and 4-OHT was investigated. Raloxifene was conjugated by all seven SULT isoforms tested, whereas 4-OHT was conjugated only by SULTs 1A1, 1E1, and 2A1. Characterization of raloxifene and 4-OHT sulfation demonstrates that sulfation can occur at therapeutic concentrations. SULT1E1 displayed the lowest Km (0.2 μM) for 4-OHT sulfation and SULT2A1 the lowest (0.3 μM) for raloxifene sulfation. SULT1E1 was the only isoform exhibiting detectable levels of raloxifene disulfation activity. Modeling of the interactions of raloxifene in the active site of SULT1E1 indicates that both hydroxyl groups of raloxifene can be readily positioned in proximity to the sulfonyl group of 3′-phosphoadenosine 5′-phosphosulfate and the catalytically important His107 residue. Both raloxifene and 4-OHT sulfation activities were detectable in all human liver cytosols tested. 4-OHT sulfation was detected in cytosol prepared from endometrial biopsies of normal women obtained during the proliferative and secretory phases of the same menstrual cycle. In contrast, raloxifene sulfation was detectable only in secretory phase cytosols in association with SULT1E1 activity. In summary, several human SULT isoforms are capable of sulfating raloxifene and 4-OHT. Tissue-specific expression of the individual SULT isoforms may have important roles in the regulation of the activity of these compounds.
Raloxifene (Evista) is a selective estrogen receptor modulator used in the treatment of osteoporosis, while tamoxifen is used in the treatment and prevention of breast cancer. Although both agents antagonize the effects of estrogen in breast tissue and mimic the effects of estrogen in bone, tamoxifen induces significant stimulation of uterine tissue, whereas raloxifene does not (Jordan and Morrow, 1999). The lack of uterine stimulation should potentially give raloxifene a therapeutic advantage as a more selective agent. Since both compounds are estrogen antagonists, the ongoing clinical STAR (study of tamoxifen and raloxifene) trial is being conducted to determine whether raloxifene is more or less effective than tamoxifen in reducing the incidence of breast cancer in women who are at an increased risk of developing this disease (Bentrem and Jordan, 2002; Kelminski, 2002; Wickerham, 2003). However, to understand the specific mechanisms of action of these compounds, it is important to analyze the pathways involved in their inactivation and metabolism.
Conjugation of estrogens, hormone replacement therapy (HRT) agents, and therapeutic estrogenic compounds with a sulfonate group is an important mechanism in the regulation of the activity and disposition of these compounds. Addition of the sulfonate group results in the inability to bind and activate steroid hormone receptors as well as an increase in water solubility and excretion (Falany, 1997). Therefore, sulfation of raloxifene and 4-hydroxytamoxifen (4-OHT), a major active metabolite of tamoxifen, may be critical in regulating their physiological functions, especially in hormone-responsive target tissues, because these compounds are structurally and functionally steroid analogs (Labrie et al., 2001). In human tissues, a family of cytosolic sulfotransferases (SULTs) is responsible for the conjugation of small xenobiotic compounds, many therapeutic drugs, and endogenous compounds including steroids, thyroid hormones, and monoamine neurotransmitters (Falany, 1997; Glatt et al., 2001). The role of sulfation in the metabolism and regulation of the activity of raloxifene and 4-OHT has not been well studied.
Several members of the SULT family catalyze the sulfation of estrogens, therapeutic estrogenic compounds, and HRT agents. SULT1E1 is responsible for the high affinity sulfation of β-estradiol (E2), estrone, and 17α-ethinylestradiol (Falany et al., 1995; Schrag et al., 2004). Several members of the phenol SULT1 family also conjugate the physiological estrogens, although with substantially lower affinities (Falany et al., 1994). Agents used in therapy as antiestrogens or as HRT agents are also substrates for sulfation by several members of the SULT2 family. 4-OHT is reportedly sulfated by SULT1A1 (Chen et al., 2002), whereas tibolone and its active metabolites are sulfated by several SULTs including SULT1E1, SULT2A1, and SULT2B1b (Meloche and Falany, 2001). Raloxifene sulfation has been reported in human Caco-2 cells, although the SULT isoforms involved were not identified (Jeong et al., 2004).
In this study, the ability of seven expressed human SULT isoforms to conjugate raloxifene and 4-OHT was investigated. Since sulfation is essentially a steroid inactivation reaction, the tissue-specific effects of tamoxifen and raloxifene may depend upon their sulfation patterns and affinities as well as the SULT isoforms present in target tissues. The activity and kinetic properties of the expressed SULTs were investigated as well as sulfation activity in liver and endometrial cytosol. The results indicate that several of the cytosolic SULTs are involved in the sulfation of 4-OHT and raloxifene, although SULT1E1 seems to be predominantly involved in the sulfation of raloxifene and SULTs 1A1 and 2A1 in the conjugation of 4-OHT.
Materials and Methods
Materials. Raloxifene hydrochloride and 4-OHT were purchased from Sigma Chemical Co. (St. Louis, MO). 3′-Phosphoadenosine 5′-phosphosulfate (PAPS) was obtained from Dr. Sanford Singer (University of Dayton, Dayton, OH). [35S]PAPS (2.2 Ci/mmol) was purchased from New England Nuclear (Boston, MA). LK6DF 60 Å silica gel thin-layer chromatography (TLC) plates with a layer thickness of 250 μm were obtained from Whatman Inc. (Clifton, NJ). All other chemicals used were of reagent grade from Fisher Scientific (Norcross, GA).
Sulfation Assays. Sulfation activity was determined using raloxifene or 4-OHT as substrate with each of the seven different bacterially expressed human SULT isoforms. With the exception of ST2B1b, the SULTs were expressed in Escherichia coli using the pKK233-2 vector to generate the native form of the enzyme and then purified by DEAE-Sepharose chromatography to obtain a preparation suitable for enzymatic characterization (Falany et al., 1989, 1995; Wilborn et al., 1993; Wang et al., 1998). Because of the instability of the native ST2B1b enzyme expressed in E. coli, SULT2B1b assays were performed with a His-tagged construct that confers stability to this particular SULT (Meloche and Falany, 2001). Sulfation assays using raloxifene or 4-OHT as the substrate were performed with each of the expressed human SULTs (SULT2A1, SULT1E1, SULT2B1b, SULT1A1, SULT1A3, SULT1B1, SULT1C2) with the reaction and isolation procedure that is used with nonradiolabeled substrates by using [35S]PAPS as the sulfate donor with the subsequent resolution of 35S-sulfates by TLC (Falany et al., 1994, 2004; Meloche et al., 2002). SULT1A1 reactions contained the appropriate substrate dissolved in ethanol, 50 mM Tris-HCl, pH 7.4 and 25 μM PAPS in a final volume of 62.5 μl; all other SULT reactions were supplemented with the addition of 10 mM MgCl2. The final ethanol concentration in the reactions was <0.1%. Control reactions were run with no substrate but contained the appropriate volume of the ethanol vehicle. Reactions were incubated for the appropriate time at 37°C and then terminated by spotting a 50-μl aliquot of each reaction on a silica gel TLC plate. The plate was developed in methylene chloride/MeOH/ammonium hydroxide (85:15:5 by volume) and the radiolabeled sulfated products were localized by autoradiography. The sulfated products were scraped into scintillation fluid, and radioactivity was determined by scintillation spectroscopy (Falany et al., 1995; Meloche et al., 2001, 2002). Initially, both substrates were screened for activity with each of the SULTs; then, for the appropriate SULTs, substrate concentration curves were performed. For determination of apparent Km values, reactions were monitored for linearity with respect to both time and protein concentration. Since cytosolic SULTs frequently display substrate inhibition with high affinity substrates, experiments to establish kinetic parameters were run at low substrate concentrations in the linear range to minimize the effects of substrate inhibition (Zhang et al., 1998). Km values were calculated using the Enzyme Kinetics program (Trinity Software, Inc., Plymouth, NH).
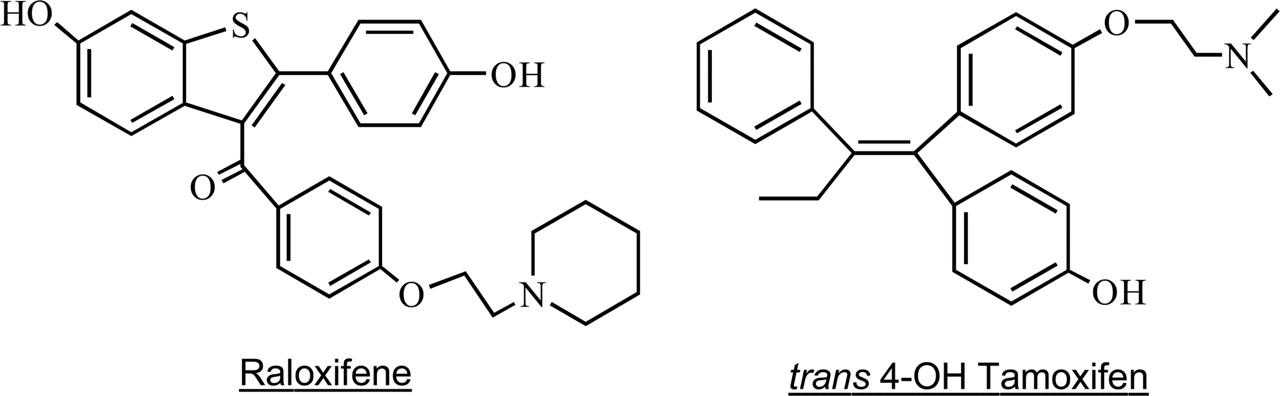
Structures of raloxifene and trans-4-hydroxytamoxifen.
Mass Spectroscopy. To identify 4-OHT-sulfate and the multiple sulfated raloxifene reaction products, nonradioactive reactions were run in parallel with reactions containing [35S]PAPS that were monitored by TLC. Identification of 4-OHT-sulfate and raloxifene mono- and disulfates was carried out by HPLC-mass spectroscopy using a Sciex 3000 mass spectrometer with two PerkinElmer series 200 micropumps (PerkinElmer Life and Analytical Sciences, Boston, MA) and a Keystone Scientific Aquasil (100 × 2 mm ID) analytical column with a C-18 guard column (Thermo Electron Corporation, Waltham, MA). The mobile phases were: A, 5 mM ammonium acetate; and B, acetonitrile. The gradient profile was 0 to 1 min, 80% A; 1 to 5 min, 10% A (linear); 5 to 7.5 min 10% A; and 7.5 to 10 min 80% A (step). Mass spectroscopy results were analyzed with the Analyst 1.4 software (Applied Biosystems, Foster City, CA).
Modeling of Raloxifene Binding with SULT1E1. The models of raloxifene-bound human SULT1E1 used in the docking studies were derived from the crystal structure of human SULT1E1 bound to 3′,5′-diphosphoadenosine (PAP) and a polychlorinated biphenyl (PCB) inhibitor (4,4′-hydroxy-3,5,3′,5′-tetrachlorobiphenyl) (Kester et al., 2000; Shevtsov et al., 2003). The orientation of this PCB within the active site is very similar to that of E2 in mouse SULT1E1, which shows high homology with human SULT1E1 (Kakuta et al., 1997). To date, mouse SULT1E1 is the only SULT1E1 isoform cocrystallized with E2 and PAP.
Raloxifene possesses one double ring and one single ring, each of which contains a phenolic hydroxyl group capable of accepting the sulfuryl group from PAPS (Fig. 1). The two human SULT1E1 · raloxifene · PAPS models used as input for the Affinity (Accelrys, Burlington, MA) docking program were constructed by elaborating PAP into the PAPS binding site and positioning the nucleophilic hydroxyl of raloxifene (on either ring) in the position that the 4-OH of the PCB occupies in the SULT1E1 · PAP · PCB structure (Shetsov et al., 2003). Protons were added to the structure by the program (pH 7), and electrostatic potentials and charges were assigned using the Insight Consistent Force Field (CFF) (Hagler and Ewig, 1994), which includes terms for sulfur and phosphorus. An energetically minimized configuration was calculated for each of the two raloxifene models using the Discovery 3 minimization package [0.001 convergence, Polak-Ribiere Conjugate Gradient algorithm (Press et al., 1986), no charge cutoffs] before Affinity docking.
At least 40 randomly oriented docked structures with energies no greater than 30 kcal above the starting structure were generated using the Affinity module of Insight. The energy of each orientation was calculated using the CFF package and modeled within a solvent grid by the cell multipole method (0.5 grid spacing, 3.00 grid buffer size, and 0.5 RESTR buffer size 7).
Sulfation of Raloxifene and 4-OHT by Human Liver and Endometrial Cytosols. Liver specimens were provided by the Tissue Procurement Service of the Comprehensive Cancer Center at University of Alabama at Birmingham and frozen at –80°C. Cytosols were prepared in 5 mM phosphate buffer, pH 7.4, containing 10% glycerol, as described previously (Falany et al., 1995; Wang et al., 1998) and stored frozen at –80°C until assayed. Similarly, endometrial cytosols were prepared in the same buffer from endometrial pipelle biopsies collected from normal, healthy patients during the proliferative and secretory phases of the same menstrual cycle (Falany et al., 1998). Cytosolic protein levels were determined using the Bradford method (Bradford, 1976) with gamma globulin as a standard. Sulfation assays were carried out using the TLC method described above for raloxifene and 4-OHT with each liver and endometrial cytosol. Reactions were run in triplicate at a 5 μM substrate concentration for 20 min using liver or endometrial cytosol as the enzyme source, and then analyzed by the TLC method and quantified by scintillation spectroscopy.
Results
Sulfation of Raloxifene and 4-OHT by Expressed Human SULTs. The structures of raloxifene and 4-OHT (Fig. 1) suggest that one or more of the human SULTs involved in phenol or estrogen conjugation may readily sulfate these compounds. Therefore, the ability of seven expressed isoforms of human cytosolic SULT to conjugate raloxifene or 4-OHT was investigated. As summarized in Table 1, all seven SULT isoforms tested, SULT1A1, 1A3, 1B1, 1C2, 1E1, 2A1, and 2B1b, were capable of conjugating raloxifene, whereas 4-OHT was sulfated only by SULT1A1, SULT1E1, and SULT2A1. In comparison, E2 sulfation at a concentration of 20 nM was detected using SULT1A1, SULT1A3, and SULT1E1 (Falany et al., 1994, 1995), although only SULT1E1 conjugates E2 with an affinity in the low nanomolar range (Falany et al., 1995). At micromolar E2 concentrations, SULT1C2 and SULT2A1 have also been reported to conjugate E2 (Falany et al., 1995). Thus, although both of these HRT agents can be sulfated, each has a different profile for sulfation by human SULTs that also is different from that for E2 sulfation. In addition, the ability of the major liver SULTs 1A1 and 2A1 to conjugate both raloxifene and 4-OHT suggests that there may be significant first-pass metabolism of the orally administered drugs.
Sulfation of raloxifene, 4-hydroxytamoxifen, and E2 by expressed human SULTs
A positive sign (+) indicates that the compound was sulfated by the SULT isoform indicated. N.D. indicates that no sulfated product was detected.
Table 2 shows the apparent Km values for raloxifene and 4-OHT sulfation determined with each SULT isoform that showed sulfation activity. Substrate inhibition is a common feature of the SULTs and occurs more readily with high affinity substrates (Zhang et al., 1998). Concentrations of raloxifene and 4-OHT were used in the assays to limit the effects of substrate inhibition if it occurred. SULT2A1 had the lowest Km (0.3 μM) for raloxifene sulfation. SULTs 1A1 and 1B1 had Km values for raloxifene sulfation in the 1 to 2 μM range, whereas SULT1C2 had the highest Km (10.3 μM). For all the SULTs, the range of Km values with raloxifene (0.3–10.3 μM) was in the low micromolar range and, thus, all have a potential role in the sulfation of therapeutic concentrations of raloxifene. The Km value for raloxifene sulfation by SULT1E1 could not be reliably estimated because SULT1E1 forms both the mono- and disulfates of raloxifene (described below). For 4-OHT sulfation, SULT1E1 has the lowest Km (0.2 μM). SULT1A1 and SULT2A1 both have slightly higher Km values for 4-OHT sulfation of 0.8 and 1.6 μM, respectively. The Km values are also in the low micromolar concentration range, suggesting the potential sulfation of therapeutic 4-OHT concentrations.
Km values for sulfation of raloxifene and 4-OHT by human cytosolic SULTs
The SULTs were expressed in E. coli XL1-blue cells using the pKK233-2 vector and purified by DEAE-Sepharose chromatography. Km values were determined using substrate concentrations between 0.025 and 1 μM to avoid substrate inhibition. The PAPS concentration was 20 μM. Apparent Km values were calculated using the Enzyme Kinetics program (Trinity Software). The values represent means ± standard deviation of three separate determinations.
Figure 2A shows the formation of raloxifene monosulfate using a range of raloxifene concentrations by the three human SULTs that also sulfate 4-OHT. Raloxifene is most rapidly sulfated by SULT2A1, whereas SULT1E1 and SULT1A1 conjugate raloxifene at lower rates. SULT2A1 also had the lowest Km for raloxifene of the seven SULT isoforms (Table 2). Although SULT2A1 had the highest affinity for raloxifene, substrate inhibition was not observed at these concentrations. In comparison, substrate inhibition was observed with SULT1A1 and SULT1E1 at concentrations above 10 μM (Fig. 2A).
Concentration curves for the sulfation of 4-OHT by SULT1A1, SULT1E1, and SULT2A1 are shown in Fig. 2B. In contrast to raloxifene, 4-OHT is more rapidly sulfated by SULT1E1 as compared with SULT2A1 and SULT1A1. SULT1E1 also has the lowest Km value for 4-OHT sulfation of the three isoforms (Table 2). At 4-OHT concentrations up to 30 μM, substrate inhibition was not observed with any of these three isoforms.
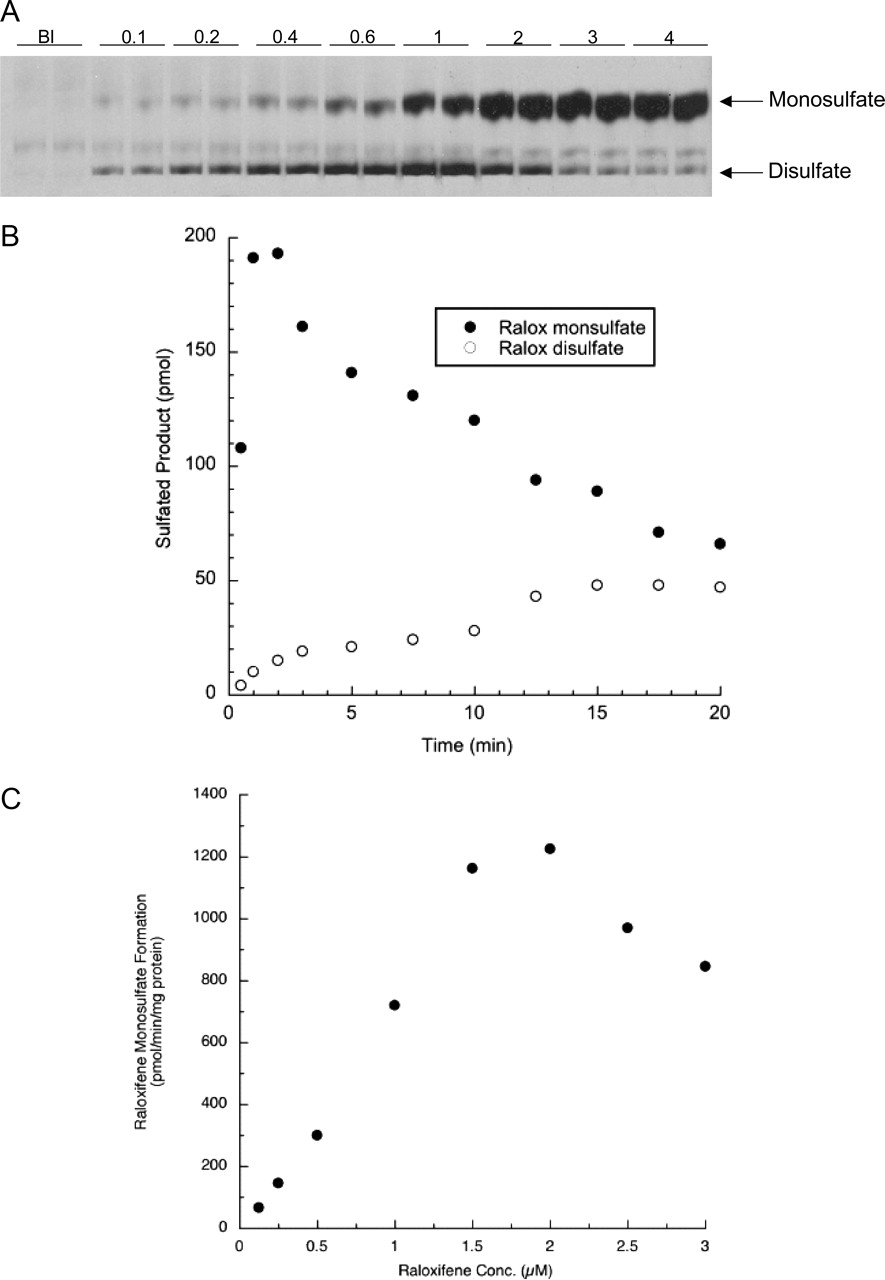
Sulfation of Raloxifene at Two Sites by SULT1E1. Raloxifene has two phenolic hydroxyl groups (Fig. 1), and, thus, the potential exists for the formation of two different monosulfates as well as a disulfate. In contrast, 4-OHT has only a single hydroxyl that can be sulfated. Results from TLC assays demonstrated that SULT1E1 was capable of forming raloxifene disulfate, whereas the other SULT isoforms tested form detectable levels of only raloxifene monosulfates. Figure 3A is an autoradiograph of a TLC plate with SULT1E1 reactions at raloxifene concentrations from 0.1 to 4 μM, showing the formation of raloxifene monosulfates and disulfate. The two potential raloxifene monosulfates travel at the same Rf in the TLC system and, thus, are indistinguishable. Figure 3B shows a time course of raloxifene monosulfate and disulfate formation. Under the reaction conditions used, raloxifene was rapidly monosulfated and then converted to the disulfate. The time course indicates a reaction mechanism in which raloxifene is monosulfated, released from the enzyme, and then rebound in an orientation allowing conjugation of the free hydroxyl. Figure 3C shows raloxifene monosulfate formation at concentrations from 0.1 to 3 μM, under conditions where disulfate formation was not detectable. At low raloxifene concentrations, slight sigmoid kinetics were observed, and at concentrations above 2 μM, substrate inhibition was detected. The kinetics of E2 high affinity sulfation by SULT1E1 indicates that SULT1E1 is inhibited by E2 binding to a noncatalytic allosteric binding site (Zhang et al., 1998); possibly one or both of the monosulfates are also capable of binding to an allosteric regulatory site.

Sulfation of increasing concentrations of raloxifene and 4-OHT by the human SULT1A1, 1E1, and 2A1 isoforms. The SULT isoforms were expressed and purified as described under Materials and Methods. Reactions were carried out using increasing concentrations of raloxifene or 4-OHT and 20 μM [35S]PAPS. Reaction products were resolved by TLC. A shows the formation of raloxifene monosulfate with raloxifene as substrate, and B displays the results with 4-OHT as substrate. Each point is the average of two reactions.
Identification of Raloxifene Sulfates by MS/MS. Since raloxifene can be monosulfated and then sulfated at the second hydroxyl group to form the disulfate, the selectivity for formation of the initial monosulfate was investigated. Figure 4 shows the LC-MS analysis of the raloxifene sulfation by SULT1E1 at a raloxifene concentration of 2.5 μM. Based upon the reaction kinetics presented in Fig. 3B, this substrate concentration should result in the formation of both raloxifene monosulfates and disulfate. The identity of raloxifene disulfate was confirmed, as well as the presence of two monosulfates by LC-MS/MS. The two monosulfates migrated with slightly different retention times during chromatography on the C-18 column before mass spectroscopic analysis. The levels of these two monosulfates in the reaction mixture differ by a factor of approximately 7.5, suggesting that disulfate formation may be derived primarily from resulfation of one of the monosulfated forms.
The identity of the raloxifene monosulfates formed by three of the SULT isoforms that did not form detectable levels of raloxifene disulfate in the TLC assay was examined. Reactions were carried out at 1 μM raloxifene, and the sulfated products were analyzed by LC-MS. SULT2A1 formed only the faster eluting raloxifene monosulfate observed during the LC procedure (data not shown). SULTs 1A1 and 1B1 also formed primarily the faster eluting monosulfate, although trace amounts of the second, slower eluting monosulfate were occasionally observed. The identities of the hydroxyl groups used to form the raloxifene monosulfates could not be confirmed by LC-MS/MS because of similar fragmentation patterns. The sulfate moieties were sensitive to cleavage during LC-MS/MS and were removed before fragmentation of raloxifene.
Modeling of Raloxifene Binding to SULT1E1. As demonstrated in Fig. 3, SULT1E1 is capable of forming raloxifene disulfate as well as both monosulfates. Analysis of the reaction products by LC-MS indicates that one monosulfate is present in greater amounts (Fig. 4). This suggests that raloxifene may be binding in the active site of SULT1E1 in an orientation favoring formation of one of the monosulfates. To better understand how each of the hydroxyl groups of raloxifene can be positioned in the active site of SULT1E1 to react with the sulfuryl group of PAPS, raloxifene was docked into the active site of human SULT1E1 using the Affinity docking algorithm. The Affinity algorithm randomly rotates and translates the initial inserted position of raloxifene about its center of mass to generate a new docking orientation, energy-minimizes the new position by adjusting individual bond-lengths and bond-angles in both raloxifene and the surrounding ligands, and computes the energy (ΔH) of the final minimized structures (long-distance nonbonding forces are included in the minimization calculations; see Materials and Methods for further details). The algorithm found raloxifene orientations in the active site that seem to be capable of allowing interaction with PAPS such that either the single- or double-aromatic ring hydroxyls of raloxifene are within ∼3 Å of the sulfuryl group. In these structures, the nucleophilic hydroxyls of either ring are also placed in proximity to lysine 47 and histidine 107, which are critical for catalysis (Kakutu et al., 1997). The lowest predicted energy raloxifene single- and double-ring reactive structures are shown in Fig. 5. These structures support the ability of SULT1E1 to bind raloxifene in catalytically active complexes such that either hydroxyl group can be sulfonated and does not significantly favor the formation of one monosulfate.
Sulfation of Raloxifene and 4-OHT by Human Tissue Cytosols. Sulfation is recognized as an important reaction in the metabolism of 4-OHT (Chen et al., 2002; Nowell et al., 2002). However, little is known concerning the sulfation of raloxifene and 4-OHT in human tissues. Since all SULT isoforms tested were capable of sulfating raloxifene, but only three conjugated 4-OHT, this suggested that there are probably tissue-specific differences in the sulfation of these compounds. Therefore, the ability of human liver and endometrial tissues to conjugate raloxifene and 4-OHT was examined. Table 3 shows the sulfation of raloxifene and 4-OHT by several representative human liver cytosols. All the human liver cytosols assayed sulfated both raloxifene and 4-OHT at generally similar rates. Raloxifene sulfation occurred at rates from 0.47 to 1.87 pmol/min/mg protein, whereas 4-OHT was sulfated at rates from 0.16 to 2.02 pmol/min/mg protein. The range of rates for each substrate is indicative of varying levels of the major SULT isoforms in human liver cytosol. Comparison of the raloxifene and 4-OHT sulfation rates indicated that there was not a significant correlation in the levels of the two activities in the different liver samples.

Raloxifene sulfation catalyzed by SULT1E1. A is an autoradiograph of a TLC plate showing the formation of raloxifene monosulfates and disulfate by SULT1E1 at increasing raloxifene concentrations. The reactions contained 10 μM [35S]PAPS and were run to completion to increase the formation of the disulfate. Each point is the average of two reactions. Bl refers to a blank reaction that did not contain raloxifene. B shows the time course of the formation of raloxifene monosulfates and disulfate. The reaction contained 2.5 μM raloxifene and 10 μM [35S]PAPS. Under these conditions, raloxifene was rapidly monosulfated and then more slowly converted to the disulfate. C shows the sulfation of increasing concentrations of raloxifene by SULT1E1 under conditions where disulfate formation is not detectable. Reactions contained 1.0 μM raloxifene, 10 μM [35S]PAPS, and 25% of the SULT1E1 enzyme used in Fig. 2 (2 μg) and the reactions were carried out for 2 min to avoid raloxifene disulfate formation.
Sulfation of raloxifene and 4-hydroxytamoxifen by human liver cytosols
Cytosols were prepared from several human livers and assayed for sulfation activity with 5 μM raloxifene or 4-OHT as substrate, as described under Materials and Methods. The values represent means ± standard deviation of three reactions.
Human endometrium is an important estrogen-responsive tissue and has been reported to express several SULT isoforms. SULT1E1 expression in endometrium is only detectable during the secretory phase of the menstrual cycle, whereas SULT1A1 is expressed in both the proliferative and secretory phases (Falany et al., 1998). Cytosol prepared from secretory and proliferative endometrium obtained from normal healthy women during the same menstrual cycle was assayed for sulfation activity with raloxifene and 4-OHT as substrates. As shown in Table 4, raloxifene sulfation is not detected in proliferative endometrial cytosol but occurs in secretory endometrial cytosol at rates of approximately 4 to 22 pmol/min/mg protein. This cyclical pattern of raloxifene sulfation correlates to SULT1E1 expression in these tissues phases (Falany et al., 1998). In contrast, 4-OHT sulfation was detected in both proliferative and secretory endometrial cytosols in all four samples. In two samples, 4-OHT sulfation is higher in secretory endometrium, and in two samples it is higher in the proliferative endometrial cytosol. This pattern is consistent with the expression of SULT1A1 activity in these samples. These results indicate that raloxifene and 4-OHT are differentially sulfated in normal cycling endometrial tissue.
Sulfation of raloxifene and 4-hydroxytamoxifen by human proliferative and secretory endometrial cytosols
Cytosols were prepared from endometrial Pipelle biopsies of four patients collected during the proliferative (Pro) and secretory (Sec) phases of the menstrual cycle (Zhang et al., 1998). Cytosols were assayed in triplicate for sulfation activity with 5 μM raloxifene or 4-OHT as substrate, as described under Materials and Methods. The values represent means ± standard deviation of four reactions.
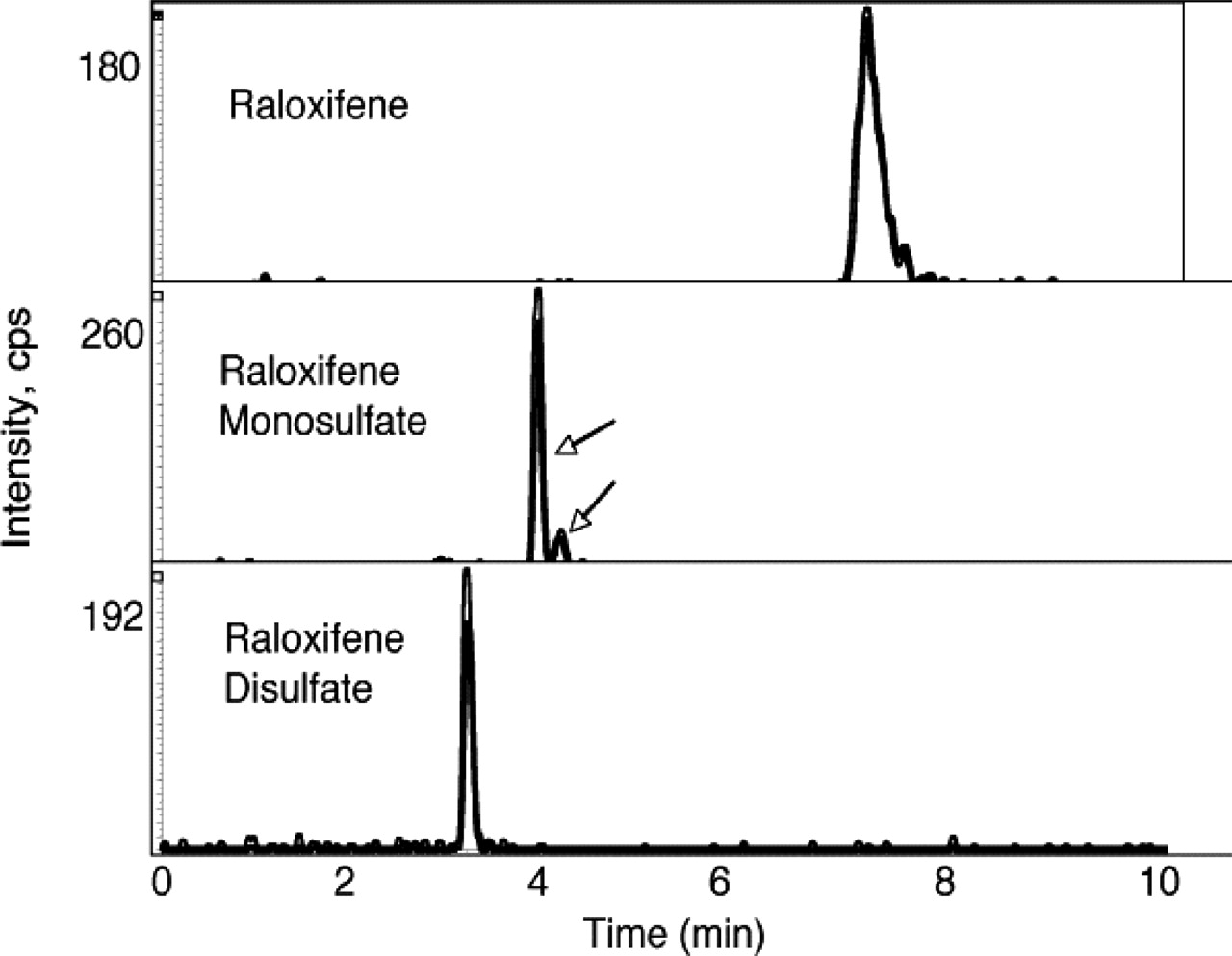
LC-MS analysis of raloxifene monosulfates and disulfate formation by SULT1E1. A reaction was carried out with 2.5 μM raloxifene substrate to generate both raloxifene monosulfate and disulfate. The reaction products were analyzed by LC-MS as described under Materials and Methods. Shown are the selected ion chromatographs for raloxifene (A), the two raloxifene monosulfates (B), and raloxifene disulfate (C) detected in the same LC-MS procedure.

Predicted lowest energy reactive structures of raloxifene at the active site of SULT1E1. A, Single-ring hydroxyl in a reactive position. B, double-ring hydroxyl in a reactive position. The amine moiety of lysine 47 and the imadozolium moiety of histidine 107 are positioned to assist sulfuryl transfer to raloxifene. For clarity, only the hydrogen atoms on potentially reactive moieties are indicated. Color scheme: green, carbon; red, oxygen; white, hydrogen; blue, nitrogen; yellow, sulfur; purple, phosphorus. The protein backbone is indicated with a pink ribbon.
Discussion
The sulfation of estrogens, HRT agents, and therapeutic estrogenic compounds is an important mechanism in their metabolism and disposition and plays a critical role in modulating the physiological effects of these compounds (Raftogianis et al., 2000). Because sulfation is critical in the metabolism of E2 at physiological concentrations that are involved in interaction with the estrogen receptors (Zhang et al., 1998; Kotov et al., 1999), it may play a similar role in modulating the effects of HRT agents such as tamoxifen and raloxifene. The sulfation of raloxifene and 4-OHT by multiple expressed SULT isoforms and by human liver and endometrial cytosols indicates that these reactions are probably occurring in human tissues.
The SULT isoforms abundant in the GI tract and liver, such as SULT1A1, 1A3, 1B1 and 2A1, have important roles in drug and xenobiotic sulfation after absorption of compounds from the GI tract. The liver and GI tract SULT activity is important in generating the plasma levels of sulfated compounds. In contrast, several of the SULT isoforms function in the tissue-specific inactivation or synthesis of steroid and thyroid hormones that may be important in defining the tissues affected by a drug or hormone (Visser, 1996; Darras et al., 1999; Kotov et al., 1999). Expression of the individual SULT isoforms in different tissues has been proposed to regulate the tissue-specific activity of E2 as well as HRT agents such as tibolone (Kotov et al., 1999; Parker, 1999; Vos et al., 2002; Falany et al., 2004). In this study, the ability of raloxifene to be sulfated by all seven SULT isoforms investigated and 4-OHT by three of the major hepatic SULT isoforms suggests that sulfation may have an important role in regulating the activity and distribution of these drugs at both a systemic level (liver) and a tissue-specific level (endometrium). The two most abundant human liver SULTs are SULT1A1 and SULT2A1 (Falany et al., 1989, 1990). There are approximately 5- to 6-fold variations in the levels of immunoreactive protein for these SULT isoforms in human liver. DHEA sulfation activity in human liver cytosol is a good indicator of SULT2A1 expression, and p-nitrophenol sulfation has been used as an indicator of SULT1A1, although concerns have been raised as to its specificity (Tabrett and Coughtrie, 2003). No significant correlation was observed among DHEA, PNP, raloxifene, or 4-OHT sulfation activities in the human livers in this study (data not shown).
4-OHT is an active metabolite of tamoxifen and is capable of binding and activating the estrogen receptors (Levenson et al., 1998; Nikov et al., 2001). Sulfation of 4-OHT prevents receptor activation. Chen et al. (2002), using a PNP-linked assay, reported that SULT1A1, 1A3, and 1E1, but not 2A1, were capable of sulfating 4-OHT. The PNP-linked assay depends on the SULT isoforms to also use PNP-sulfate, to sulfate the PAP formed by the same enzymes during the sulfation of 4-OHT. Activity is determined by monitoring the appearance of PNP absorbance. The linked assay is generally used to assay the activity of SULT isoforms with phenol SULT activity and those that use PNP as a substrate. The lack of detectable activity with SULT2A1 may be due to its inability to use PNP as a substrate (Falany et al., 1989; Comer et al., 1993). Km values for 4-OHT sulfation with the linked assay were 59 μM, 28 μM, and 24 μM for SULT1A1, 1A3, and 1E1, respectively (Chen et al., 2002). Using a TLC assay to resolve sulfated products as well as LC-MS to identify 4-OHT-sulfate formation, expressed SULT1A1, 1E1, and 2A1 were capable of sulfating 4-OHT, whereas 1A3, 1B1, 1C2, and 2B1b did not generate detectable 4-OHT-sulfate. The Km values determined with the TLC assay for SULT1A1, 1E1, and 2A1 were 0.8 μM, 0.2 μM, and 1.6 μM, respectively. The Km values determined with the TLC assay are similar to the values obtained for prototypical substrates for these enzymes and are in a more physiological concentration range (Falany et al., 1989; Comer et al., 1993).
Raloxifene is rapidly absorbed after oral administration and is considered to undergo first-pass metabolism via glucuronidation, since only raloxifene glucuronides are found in serum (Hochner-Celnikier, 1999; Kemp et al., 2002). Although only raloxifene glucuronides are reported in human serum, only 6% of administered raloxifene, including the glucuronide conjugates, is recovered in urine (Hochner-Celnikier, 1999). Little information has been reported concerning raloxifene sulfation, although significant amounts of raloxifene sulfate were found in rat liver after raloxifene administration (Dodge et al., 1997). In human intestinal Caco-2 cells, raloxifene sulfate was the major metabolite formed (Jeong et al., 2004), whereas the formation of raloxifene glucuronide was not observed. The observation that raloxifene was sulfated by all seven of the human SULTs examined in this study as well as in liver and endometrial cytosols suggests the reaction is occurring in human tissues.
Raloxifene and 4-OHT are conjugated by different SULT isoforms (Table 1), which may explain, in part, how they can have similar effects on breast and bone but different effects in the uterus. Based upon the SULT isoforms present in a tissue, regulation of the intracellular activity of raloxifene and 4-OHT may occur, as is the case with E2 and tibolone (Kotov et al., 1999; Vos et al., 2002). E2 activity in human endometrium is regulated in part by changes in SULT1E1 expression during the menstrual cycle (Falany et al., 1998). Similar to the expression of SULT1E1, raloxifene sulfation was detectable only in endometrial cytosol obtained during the secretory phase of the menstrual cycle. In contrast, 4-OHT sulfation activity was present in both proliferative and secretory endometrial samples. Detectable levels of SULT1E1 are present only in the secretory phase, whereas SULT1A1 expression is not related to the phase of the menstrual cycle (Falany et al., 1998). Thus, in endometrial tissue, 4-OHT sulfation apparently does not correlate with the cyclical expression of SULT1E1, whereas raloxifene sulfation does. This suggests that in endometrial cytosol, 4-OHT is conjugated by SULT1A1. The activity of 4-OHT would be inhibited by SULT1A1 activity throughout the menstrual cycle, whereas raloxifene would be more active during the proliferative phase and inhibited by SULT1E1 activity during the secretory phase. Although raloxifene glucuronides have been reported in human plasma, little has been reported concerning the expression of UGTs in human endometrium; thus, it is difficult to evaluate the role of glucuronidation in the conjugation of raloxifene and 4-OHT in this tissue. Recently, Lepine et al. (2004) reported the expression of UGT2B7 in epithelial cells of endometrium obtained from postmenopausal women. UGT2B7 is involved in the conjugation of estrogens including E2 and E1, although its activity toward raloxifene and 4-OHT is not known.
Analysis of the kinetics of raloxifene sulfation by SULT1E1 is complicated by the formation of both raloxifene monosulfates and disulfate at low raloxifene concentrations. At low concentrations, raloxifene monosulfation by SULT1E1 showed sigmoid kinetics. This pattern may result from the different rates of formation of the two monosulfates; however, the rates of formation at these concentrations were too low to allow identification of the individual monosulfates. LC-MS analysis of raloxifene sulfation at higher concentrations indicates that the formation of one monosulfate is favored, although it is not established which monosulfate was used in the formation of the disulfate. A modeling approach was used to investigate whether one hydroxyl group was more efficiently bound in the active site of SULT1E1. The orientation of raloxifene in the active site of SULT1E1 so that the hydroxyl groups were in position to interact with the sulfuryl group of PAPS was accomplished using the Affinity docking algorithm. The algorithm found raloxifene structures that seem to be capable of reacting with PAPS, structures in which either the single- or double-aromatic ring hydroxyl of raloxifene are located within ∼3 Å of the sulfuryl group. In these structures, the nucleophilic hydroxyls of either ring are also placed in proximity of lysine 47 and histidine 107, which are critical for catalysis (Kakuta et al., 1997). The minimal energy differences for optimal binding were almost identical, suggesting that both hydroxyls of raloxifene could be efficiently bound in reactive orientations.
What is not shown in Fig. 5 is the considerable diversity of orientations among similar-energy structures. The superimposed active site raloxifene structures form a volume that essentially fills the crevice-like binding pocket available in SULT1E1 (the PDB files of the six lowest-energy single- and double-ring reactive structures are available in supplemental material). SULT1E1 accepts a relatively large class of structurally diverse compounds as substrates, so that it is not surprising that the docking program predicts a large number of possible orientations. The fact that many different reactive structures are found by the program implies that the active site is flexible and therefore capable of searching to achieve optimal positioning of the reactive groups. This plasticity then allows for the enzyme to bind and sulfate raloxifene at two different positions. Minor structural differences or an inability to accommodate the charged sulfonate group in the active site must limit the ability of the other human SULT isoforms to bind the raloxifene monosulfates and form the disulfate.
Sulfation is important in both the systemic metabolism and distribution of drugs and xenobiotics, as well as in regulating the activity of steroid-like compounds, at the tissue and cellular levels. The ability of multiple expressed human SULTs to conjugate both raloxifene and 4-OHT in vitro indicates that a better understanding of the role of sulfation in their metabolism is warranted. Of particular interest is the differential sulfation of raloxifene and 4-OHT by the proliferative and secretory endometrial cytosols. This difference may be related to the endometrial stimulation associated with tamoxifen therapy.
Footnotes
-
This research was supported in part by U.S. Public Health Service Grants GM38953 to C.N.F. and GM54469 to T.S.L.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.006551.
-
ABBREVIATIONS: HRT, hormone replacement therapy; SULT, sulfotransferase; 4-OHT, 4-hydroxytamoxifen; PAPS, 3′-phosphoadenosine 5′-phosphosulfate; PCB, polychlorinated biphenyl; PNP, 4-nitrophenol; UGT, UDP-glucuronosyltransferase; LC-MS, liquid chromatographymass spectroscopy; E2, β-estradiol; TLC, thin-layer chromatography; PAP, 3′,5′-diphosphoadenosine; MS/MS, tandem mass spectrometry; GI, gastrointestinal.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received July 14, 2005.
- Accepted December 12, 2005.

- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}