


Abstract
CYP2C19 is an important enzyme for human drug metabolism, and it also participates in the metabolism of endogenous substrates, whereas the CYP2C18 enzyme is not expressed in human liver despite high mRNA expression. Mice transgenic for the human CYP2C18 and CYP2C19 genes were generated. Quantitative mRNA analysis showed CYP2C18 and CYP2C19 transcripts in liver, kidneys, and heart to be expressed in a sexually dimorphic manner, with male mice having 2- to 100-fold higher levels. Transcript levels in the small intestine were somewhat higher than liver but were similar in both sexes. Transgene mRNA expression was much lower in lung and brain and least in the heart. Immunoblotting using an antipeptide antiserum, reactive with human CYP2Cs and mouse CYP2C70, revealed increased immunoreactive protein in liver microsomes from heterozygous transgenic male mice and a concomitant increase in 5′-hydroxylation of R-omeprazole and S-mephenytoin intrinsic clearance, consistent with CYP2C19 overexpression. A CYP2C18-specific antiserum showed that this enzyme was not expressed in livers or kidneys from heterozygous transgenic mice, but the antiserum had high affinity for recombinant CYP2C18 expressed in COS-7 cells. It is concluded that 1) both the CYP2C18 and CYP2C19 genes are subject to sexually dimorphic regulation in murine liver, kidney, and heart; 2) the CYP2C18 protein is not expressed in murine liver or kidney despite high levels of the corresponding mRNA; and 3) this transgenic model may be suitable for studying sex-dependent regulation of the human CYP2C genes and possibly serve as an in vivo model for CYP2C19-dependent drug metabolism.
Cytochrome P450 (P450) 2C (CYP2C) enzymes are heme-containing monooxygenases responsible for the metabolism of many endogenous and xenobiotic compounds. In humans, the CYP2C subfamily contains four members: CYP2C8, CYP2C9, CYP2C18, and CYP2C19, which together are involved in the metabolism of approximately 20% of clinically used drugs (Goldstein, 2001). In addition to exogenous substrates, CYP2C enzymes metabolize arachidonic acid and some steroids (Nebert and Russell, 2002), and the action of CYP2C9 has been implicated in the regulation of vascular tone (Hillig et al., 2003).
Each member of the human CYP2C subfamily has been shown to be genetically polymorphic. Polymorphisms within the CYP2C19 gene have been shown to affect the metabolism of many commonly used drugs such as the anticonvulsant mephenytoin, proton pump inhibitors such as omeprazole, the anxiolytic agent diazepam, certain antidepressants, and the antimalarial drug proguanil (Ingelman-Sundberg et al., 2007). The success rate of treatment with omeprazole has been highly correlated with CYP2C19 genotype (Furuta et al., 1998; Klotz, 2006).
Natural and synthetic glucocorticoids (agonists and antagonists), as well as other clinically important drugs, induce the hepatic CYP2B, CYP2C, and CYP3A subfamilies in humans. Induction can lead to clinically important drug-drug interactions (Pascussi et al., 2003). The constitutive androgen receptor and pregnane X receptor are known to be involved in the regulation of CYP2C19, suggesting a constitutive regulation by endogenous hormones and by drugs such as rifampicin and dexamethasone (Gerbal-Chaloin et al., 2001; Chen et al., 2003). Drug-drug interactions, together with genetic polymorphisms in CYP2C enzymes, greatly contribute to the variation in oral bioavailability and systemic clearance of CYP2C substrates. This variation is particularly undesirable for drugs with narrow therapeutic indices.
The 4′-hydroxylation of S-mephenytoin is a well established in vitro and in vivo metric of CYP2C19 enzymatic activity (Meier et al., 1985; Walsky and Obach, 2004). S-Mephenytoin 4′-hydroxylation has been shown to take place at a higher rate in human liver microsomes than in microsomes from mice (Bogaards et al., 2000). S-Mephenytoin metabolism, measured as S-mephenytoin disappearance, has been shown to be similar between male and female mice of five commonly used laboratory strains (Löfgren et al., 2004). Furthermore, the 5′-hydroxylation of omeprazole is a similarly well established marker for CYP2C19 activity (Niioka et al., 2007).
To obtain a better in vivo model system to study the role of human CYP2C enzymes in drug pharmacology and toxicology and to investigate the transcriptional regulation of these genes, we have generated a gene-addition transgenic mouse containing the human CYP2C18 and CYP2C19 genes. We find that both human CYP2C genes are subject to tissue-specific sexually dimorphic transcriptional regulation. Furthermore, the CYP2C19 gene, but not the CYP2C18 gene, is expressed in the mouse liver to yield a catalytically active enzyme as indicated by S-mephenytoin and R-omeprazole metabolism.
Materials and Methods
Chemicals and Antibodies. Oligonucleotide polymerase chain reaction (PCR) primers were purchased from Invitrogen (Paisley, Scotland). Bovine serum albumin and β-NADPH were obtained from Sigma Chemicals Co. (St. Louis, MO). S-Mephenytoin and the reference (±)-4′-hydroxymephenytoin were purchased from Toronto Research Chemicals (North York, ON, Canada). R-Omeprazole and internal standard (H 259/36; AstraZeneca, Mölndal, Sweden) were gifts from Dr. Leif Bertilsson (Karolinska Institutet, Stockholm, Sweden). 5′-Hydroxyomeprazole was supplied by AstraZeneca directly. An antiserum specific against CYP2C18 was donated by Dr. Eric Johnson (The Scripps Research Institute, La Jolla, CA) (Richardson et al., 1997). An antipeptide antiserum (α-hCYP2C), targeted against the four C-terminal amino acids of the four human CYP2Cs and coincidentally mouse CYP2C70, was donated by Dr. Robert Edwards (Section of Experimental Medicine and Toxicology, Imperial College, London, UK) (Edwards et al., 1998). In liver microsomes from transgenic mice this antiserum is expected to react only with CYP2C70, CYP2C18 (if present), and CYP2C19, whereas in human liver microsomes the serum will react also with CYP2C8 and CYP2C9. Recombinant CYP2C18 standard (supersomes) was purchased from BD Biosciences (San Jose, CA). All the other chemicals and reagents were of analytical grade from commercial sources.
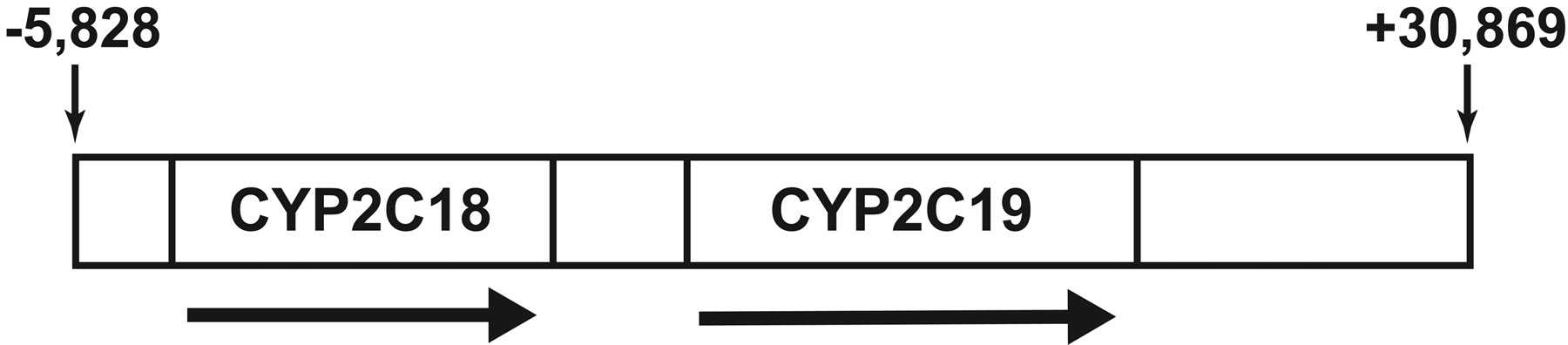
Animals and Generation of Transgenic CYP2C18/19 Mice. Transgenic mice containing the human CYP2C18 and CYP2C19 genes were generated from human bacterial artificial chromosome (BAC) clone RP11-466J14, which was reported to contain both complete CYP2C18 and CYP2C19 genes. The BAC clone was confirmed by both restriction digestion and sequencing of the 5′ and 3′ ends, revealing the 5′ end to begin 5828 bases upstream from the start of CYP2C18 exon 1 (RefSeq: NM_000772) and the 3′ end to contain an additional 30,869 bases downstream from the end of CYP2C19 exon 9 (RefSeq: NM_000769) (Fig. 1). BAC DNA was isolated (Midi Prep, Qiagen, Valencia, CA) and digested with the restriction enzyme NotI to release the insert, which was then isolated on a Sepharose gel column. Purified BAC DNA was injected into C57BL/6 eggs, and founders were identified by genotyping of genomic DNA (gDNA) extracted from tail biopsies (described below). The subsequent transgenic CYP2C18/19 mice were reared and studied at several locations: AstraZeneca Transgenic Centre (Mölndal, Sweden), AstraZeneca Bioscience (Södertälje, Sweden), and Karolinska Institutet (Stockholm, Sweden). All the transgenic CYP2C18/19 mice, with the exception of the 9-week old mice used as one group in the S-mephenytoin microsomal metabolism studies, were offspring from a transgenic CYP2C18/19 mouse and wild-type (wt) breeding and thus were heterozygous for the transgene. A small number of the transgenic 9-week-old mice used in the S-mephenytoin studies were heterozygous offspring from crossing two transgenic mice.

Schematic representation of BAC RP11-466J14, used to generate the CYP2C18/19 transgenic mice. The 5′ end of the BAC is located 5828 bp upstream of exon 1 of CYP2C18. The 3′ end is located 30,869 bp downstream of exon 9 of CYP2C19. The arrows indicate the gene orientations.
PCR Genotyping. To identify mice possessing the CYP2C18/19 transgene, gDNA was extracted from tail or ear tissue using either established protocols (Sambrook and Russell, 2001) or a commercial kit (DNeasy Tissue, Qiagen). Amplification of gDNA was performed in a 20-μl reaction volume containing 10 μl of HiFi PCR MasterMix (ABgene House, Surrey, UK), 250 nM each primer, and 1 μl of gDNA. The four different gene specific primer pairs are listed in Table 1. The PCR thermoprofile consisted of initial denaturation for 2 min at 94°C followed by 30 cycles of 10 s at 94°C, 20 s at 60°C, and 45 s at 68°C followed by a 3-min extension at 70°C. Amplification products were visualized with ethidium bromide/agarose gel electrophoresis.
Primer sequences
Total RNA Isolation and cDNA Synthesis for Real-Time PCR. Transgenic and wt control mice were killed via carbon dioxide inhalation. Whole livers, kidneys, heart, lung, brain, and 2- to 3-cm sections of proximal small intestine (∼duodenum) from 26- to 31-week-old male and female mice were placed in RNAlater (Qiagen) according to the manufacturer's recommendations. At the same time, sections of either ear or tail were removed for gDNA isolation and genotyping (described above). Total RNA was extracted from approximately 10 mg of tissue using a commercially available kit (RNeasy, Qiagen). DNA was removed with DNase digestion (Qiagen). The quality of extracted RNA was assessed by UV spectrophotometry and direct visualization via formaldehyde-agarose gel electrophoresis. RNA was reverse-transcribed into first-strand cDNA using 0.5 μg of total RNA, 4 μl of 5× reaction buffer, 5 μM oligo(dT)18, 0.5 mM dNTPs, 10 mM DTT, 1 μl of RNaseOut (Invitrogen, Carlsbad, CA), and 200 U of Superscript II RNase H-reverse transcriptase (Invitrogen) in a 20-μl reaction volume. Reactions were incubated at 42°C for 60 min followed by inactivation at 70°C for 10 min. To address the possibility of gDNA contamination, reverse-transcription reactions were also run in the absence of reverse transcriptase.
Primers Design for Real-Time PCR. The primer pairs for the amplification of CYP2C19 and CYP2C18 transcripts were targeted to sequences showing relatively low homology with the endogenous murine CYP2C isoforms using a multiple sequence alignment (ClustalW). Multiple potential primers for real-time PCR were evaluated with the following criteria: observation of a single melting curve peak, visualization of a single amplicon of the appropriate length following agarose gel electrophoresis, direct sequencing of amplicons, amplification efficiencies >95%, and the absence of amplification products using either liver cDNA derived from wt mice or reverse transcriptase minus controls. Ultimately, the best CYP2C19 primer pair was obtained by minor modification of those used previously by Klose et al. (1999). Murine hypoxanthine guanine phosphoribosyltransferase (HPRT) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as normalization factors. Primer sequences are shown in Table 1.
Real-Time Quantitative PCR. To quantitate the expression of CYP2C18 and CYP2C19 transcripts, reaction mixtures for real-time PCR were 25 μl and contained the following: 12.5 μl 2× SYBR Green Master Mix (Applied Biosystems, Foster City, CA), 0.25 μl of cDNA, and the appropriate primer pair (400 nM). The PCR thermoprofile consisted of initial activation of the polymerase at 95°C for 10 min followed by 40 PCR cycles of 95°C for 20 s, 58°C for 30 s, and 72°C for 30 s. Assays were performed in triplicate on an Applied Biosystems 7500 Fast Real-Time PCR system. Relative CYP2C transgene expression levels were determined using qBase version 1.3.5 (Hellemans et al., 2007). CYP2C18 and CYP2C19 expression levels were normalized using the geometric mean of HPRT and GAPDH according to established methodologies (Vandesompele et al., 2002).
Fluorescent in Situ Hybridization Analysis. Two heterozygous mice were crossed, and primary mouse embryonic fibroblasts were obtained and cultured according to established methods. Four cell lines were established, two for heterozygous animals and two for homozygous animals. After 5 days of cell culture (Dulbecco's modified Eagle's medium, 10% fetal bovine serum), chromosome preparations were made according to standard cytogenetic techniques. Approximately 30 μl of the chromosome preparation was dropped onto a slide, air-dried, and baked at 60°C for 1 h followed by 4′,6-diamidino-2-phenylindole (DAPI) staining for ∼10 min.
Approximately 2 μg of DNA from two different preparations of linearized BAC clone insert was labeled with either 20 mM biotin-dUTP or 20 mM digoxigenin-dUTP by a standard nick translation procedure (90 min, 15°C). Probe length was analyzed on a 1% agarose gel. The probes showed the optimal average length of ∼300 base pair (bp) after nick translation. Approximately 50 ng of DNA in 10 μl of hybridization buffer (50% formamide, 2× SSC, 10% dextran sulfate) was applied to chromosomes fixed on the slide, mounted with a coverslip, and sealed with rubber cement. Probe DNA and chromosomes were denatured together at 72°C for 3 min. Hybridization was overnight at 37°C in a wet chamber. After hybridization, the coverslip was removed, and the slide was washed in 2× SSC for 8 min. Slides were then incubated at 72°C in 0.4× SSC/0.1% Tween 20 for 1 min. For probe detection, both an antidigoxigenin antibody coupled with rhodamine and avidin coupled with fluorescein isothiocyanate were incubated for 30 min at 37°C in 4× SSC/0.1% Tween/5% bovine serum albumin followed by two 10-min washes in 4× SSC/0.1% Tween. Slides were then stained in DAPI for 10 min. DAPI-stained chromosomes and in situ hybridization signals were analyzed on a Zeiss Axioplan II microscope (Carl Zeiss GmbH, Jena, Germany). For an optimal identification of chromosome banding, DAPI-stained metaphases were imaged before probe hybridization. Each image field (blue, green, and orange) was recorded separately with a black/white charge-coupled device camera (SenSys, Photometrics, Tucson, AZ). Chromosomes and fluorescent in situ hybridization (FISH) signals were then displayed in false colors, and images were merged with SmartCapture X software (Digital Scientific, Cambridge, UK).
Gene Copy Number Determination. Transgene copy number was approximated using a real-time PCR approach based on existing methodologies (De Preter et al., 2002; Hoebeeck et al., 2007). Briefly, the efficiency and specificity of primer pairs for CYP2C19, human IL2 (hIL2), and mouse IL2 (mIL2) genomic sequences were evaluated using serial 2-fold dilutions of mouse and human gDNA ranging from 2 to 0.25 ng (primer sequences shown in Table 1). gDNA was quantified fluorometrically using PicoGreen (Invitrogen). Reaction mixtures were 25 μl and contained the following: 12.5 μl2× SYBR Green Master Mix (Applied Biosystems), human or mouse gDNA, and the appropriate primer pair (400 nM). The PCR thermoprofile consisted of initial activation of the polymerase at 95°C for 10 min followed by 40 PCR cycles of 95°C for 15 s, 60°C for 1 min followed by a melting curve to verify the presence of a single amplification product. Assays were performed in triplicate on an Applied Biosystems 7500 Fast Real-Time PCR system. Subsequently, 1 ng of gDNA from four heterozygous mice and four humans was amplified using the same PCR conditions, and the relative amounts of CYP2C19 and hIL2 were determined for each human gDNA sample along with CYP2C19 and mIL2 in each mouse gDNA sample. To calculate the mouse CYP2C19 copy number relative to the human gDNA samples, the following equation was used: relative copy number difference = (1+E)-ΔCt 2C19 /(1+E)-ΔCt IL2 (E = efficiency of the PCR reaction, ΔCt 2C19 = difference in mean CYP2C19 threshold cycle values between mouse and human gDNA samples, and ΔCt IL2 = difference in mean IL2 threshold cycle values between mouse and human gDNA samples).
Microsomal Preparation. Microsomes were prepared from individual male and female mouse livers and kidneys according to standard methodologies (Maines, 1999). Briefly, tissues were homogenized in 15 to 20 ml of buffer (10 mM sodium/potassium phosphate/1.14% KCl, pH 7.4) at 0 to 4°C followed by centrifugation (10,000g, 10 min) and subsequent centrifugation of the supernatant (105,000g for 90 min). Microsomal pellets were resuspended in 50 mM potassium phosphate buffer (0.4 ml/g liver) and stored at -80°C. Microsomal P450 content was quantified spectrophotometrically using a Cary 400 UV-visible spectrophotometer (Varian, Inc., Palo Alto, CA) from the CO difference spectra of dithionite-reduced samples using an extinction coefficient of 91 mM/cm between 450 and 490 nm (Omura and Sato, 1964). Protein was quantified using a scaled-down version of the original method by Lowry et al. (1951), and bovine serum albumin was used as a standard.
S-Mephenytoin Hydroxylation. The metabolism of S-mephenytoin to its major CYP2C19-dependent metabolite 4′-hydroxymephenytoin was studied using liver microsomes from 9-week-old heterozygous transgenic and wt mice. The rates of mephenytoin 4′-hydroxylation were determined in incubations containing liver microsomes (100 μg of microsomal protein) and S-mephenytoin (10, 25, 50, 100, and 250 μM from a stock solution in ethanol, giving a final ethanol concentration of 1% in each incubation) in a total volume of 200 μl of 50 mM potassium phosphate buffer, pH 7.4. Reactions were incubated at 37°C for 3 to 5 min before the addition of NADPH (1 mM final concentration) to initiate metabolism. Incubations were performed in duplicate for 0, 5, 10, and 20 min. Additional incubations without NADPH or microsomal protein were used as negative controls. Reactions were terminated by the addition of 1 volume of ice-cold acetonitrile. Precipitated proteins were removed by centrifugation (12,000g, 10 min, 4°C) followed by liquid chromatography/tandem mass spectrometry analysis of the supernatant.
Michaelis-Menten kinetics were assumed, and apparent Km and Vmax were estimated for each animal using Lineweaver-Burke plots. Incubations containing S-mephenytoin at the two lowest concentrations (10 and 25 μM) were excluded from analysis because the observed levels of the 4′-OH metabolite were below the limit of quantification in several instances. The apparent Km, Vmax, and Clint values from the majority of 9-week-old mice could be calculated using both the total amount of microsomal protein and nanomoles of total P450. In the three instances in which hepatic microsomal protein yields were insufficient for the spectral quantification of P450 content, kinetic constants were instead determined solely based on the microsomal protein content.
High-Performance Liquid Chromatography and Mass Spectroscopy for Detection of Mephenytoin Metabolism. Chromatographic separations were performed using a three-column liquid chromatography system described previously (Lindqvist et al., 2004). Briefly, three high-purity 5-μm, 50 × 2.1-mm i.d. columns were used with a binary gradient consisting of a mixture of acetonitrile/water/glacial acetic acid (2:98:0.1 and 80:20:0.1) at a flow rate of 0.4 ml/min. The injection volume was 20 μl. Fractions were analyzed by tandem mass spectrometry using electrospray ionization with multiple-reaction monitoring on a Quattro Micro (Micromass UK Limited, Manchester, UK). The dwell time for each transition was 0.1 s. The desolvation and source temperatures were 250 and 120°C, respectively. The cone and desolvation gas flows were 130 and 920 l/h, respectively. Nitrogen and argon were used as cone and collision gases, respectively. Transitions, mode, cone voltage, and collision energy used for S-mephenytoin and its metabolite were 217.07 > 188.26, ESP, CV = 30, CE = 22 and 233.14 > 161.05, ESP, CV = 28, CE = 22, respectively.
Analysis of Omeprazole Metabolism.R-Omeprazole was used as an additional probe drug for CYP2C19 activity in microsomes from male transgenic mice. The rate of 5′-hydroxyomeprazole generation was determined essentially as previously published (Tybring et al., 1997). Liver microsomes for R-omeprazole studies and Western blotting (described below) were prepared from 12-week-old male wt mice or from age-matched transgenic male mice that had previously been control animals sham-operated 8 weeks before sacrifice and isolation of microsomes. The sham-operated mice were subjected to 30 min of general anesthesia and were used to minimize the total number of transgenic mice used in the present study. Before sample analysis, the linearity of metabolite formation versus both the amount of microsomal protein and incubation time were evaluated. The incubation mixture (200 μl) contained 50 μg of protein, 10 μM R-omeprazole, 1 mM freshly made NADPH, and 50 mM potassium phosphate buffer, pH 7.4. Incubations were carried out for 15 min and were stopped with the addition of 800 μl of dichloromethane/acetonitrile, 9:1, and an internal standard (H 259/36, AstraZeneca). All the samples were extracted for 10 min using a vortex mixer followed by centrifugation (2000g, 10 min) and aspiration of the plasma/aqueous phase. The organic phase was transferred to a new tube and evaporated to dryness at 60°C. Samples were reconstituted in 100 μl of 10 mM disodium phosphate buffer, pH 9.3. Chromatography was performed using a Varian Prostar reverse-phase high-performance liquid chromatography system, consisting of a model 240 solvent delivery module, model 310 UV-visible detector, and a model 410 autosampler, combined with a LiChrospher 60 RP-select B 125 × 4 mm, 5-μm particle column and an identical 4 × 4 mm precolumn (Merck, Darmstadt, Germany). The mobile phases were 62.5 mM ammonium acetate, pH 7.0 (A), and acetonitrile (B). The solvent gradient was as follows: initially 80% A/20% B with a linear ramp to 55% A/45% B from 2 min to 18 min, a linear return to initial condition from 23 min to 27 min followed by a 5-min equilibration before injection of the next sample. The flow rate was 1 ml/min. Metabolites and internal standard were monitored via absorbance at 302 nm, and peak areas were determined using the Varian Star version 5.51 software package.
Expression of CYP2C18 in Cultured Cells. A pCMV6-XL5 construct of CYP2C18 purchased from Origene (Rockville, MD) was transiently transfected into Huh-7 and COS-7 cells. In brief, cells were maintained at 37°C in an atmosphere of 5% CO2 in modified Dulbecco's modified Eagle's medium supplemented with fetal bovine serum, sodium pyruvate, and antibiotics. For transfection experiments, cells (1.5 × 106 cells/plate) in 100-mm dishes (50–90% confluency) were transfected using Lipofectamine 2000 (Invitrogen). After 24 h, microsomal protein was prepared from the transfected cells. Following centrifugation, the cell pellet was resuspended in buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 10% glycerol) followed by sonication and centrifugation (9000g, 20 min, 4°C). The supernatant was additionally centrifuged (105,000g, 1 h). The pellet was then dissolved in 100 to 200 μl of cold buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM KCl, 20% glycerol, and protease inhibitor mixture).
Western Blot Analyses. Western blot analysis was also performed to determine the expression of CYP2C18 protein in liver and kidney microsomes from the transgenic mice along with Huh-7 and COS-7 cells transiently transfected with recombinant CYP2C18. Either 30 μg of mouse liver microsomal protein, 40 μg of mouse kidney microsomal protein, or 25 μg of microsomes from transfected cells was separated via SDS-polyacrylamide gel electrophoresis on a 10% gel followed by wet-transfer to a nitrocellulose filter. The filters were blocked with 5% milk in Tris-buffered saline (TBS) buffer and then incubated with the anti-CYP2C18 antibody diluted in TBS (1:500). This was followed by incubation with a 1:2000 dilution of a goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (Dako Cytomation, Glostrup, Denmark) and detection using SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL).
To detect CYP2C19 in liver microsomes from transgenic mice, the α-hCYP2C antipeptide antiserum was used. Microsomes corresponding to 20 μg of protein were subjected to SDS-polyacrylamide gel electrophoresis using a 10% gel. After transfer, the Hybond-C extra membrane (Amersham Pharmacia Biotech, Uppsala, Sweden) was blocked in TBS containing 0.05% (v/v) Tween 20 and 5% fat-free milk and incubated with a 1:1000 dilution of the CYP2C antisera and further processed as described above.
Statistical Methods. Genotype-associated differences in S-mephenytoin and R-omeprazole metabolism within the male and female mouse groups were compared using the Mann-Whitney test.
Results
Chromosomal Integration. Mice were made transgenic using a BAC clone fragment containing both the human CYP2C18 and CYP2C19 genes (Fig. 1). Transgenic mice were identified by isolating gDNA from tail or ear tissue followed by transgene-specific amplification using conventional PCR (Table 1). The primer specificity was evident because of the absence of any amplification product using gDNA from wt C57BL/6 mice (data not shown).
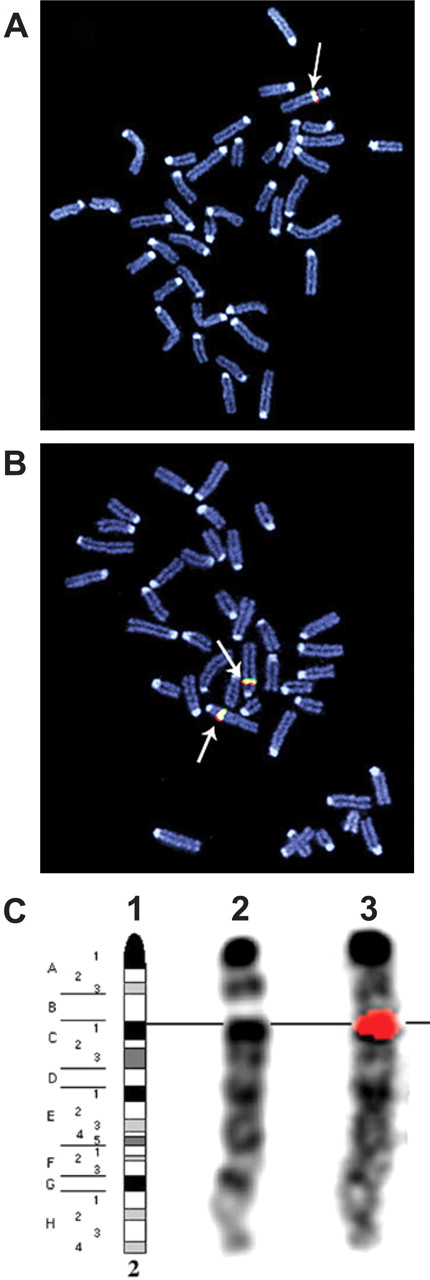
The chromosomal integration of the human clone was analyzed using FISH. As evident from Fig. 2, the genes were localized to region C1 of murine chromosome 2. The transgene copy number was determined using real-time PCR and CYP2C19-specific primers in combination with either mIL2 or hIL2 as endogenous reference genes. The amplification plots for CYP2C19, hIL2, and mIL2 were linear throughout the tested concentration range (Fig. 3), and the determined efficiencies were used in the subsequent relative copy number calculations. Comparison of four mouse gDNA samples with four human gDNA samples revealed the transgenic mouse genome to contain 6.1 ± 0.4 times more copies of the CYP2C19 gene compared with the reference human gDNA samples. Because the human genome contains two copies of the CYP2C19 gene, the heterozygous mice would therefore contain 12.1 ± 0.9 copies, which would equate to an estimate of ∼11 to 13 copies. Similar results were obtained in identical experiments using an alternative reference gene (PCBP2) instead of IL2 (data not shown).
Expression at the mRNA Level. The expression of CYP2C18 and CYP2C19 mRNA was determined by real-time PCR and isoform-specific primers. Total RNA was isolated from the liver, small intestine, kidney, heart, lung, and brain from 26- to 31-week-old male and female mice. As shown in Fig. 4, there was a very abundant expression of CYP2C18 mRNA in liver of male mice with female expression levels approximately 10-fold less, a trend also observed in the kidney. Similarly, CYP2C19 mRNA was highly expressed in male livers and kidneys with much lower amounts observed in females. By contrast, no such sex difference was apparent in the small intestine, although this tissue expressed very high levels of both transcripts. Indeed, CYP2C19 mRNA expression was higher in the small intestine than in liver and was also approximately 10-fold higher in small intestine than observed for CYP2C18 mRNA. Limited transcript expression was observed in both the lung and brain with no appreciable differences between transcripts or sex. Although transgene mRNA expression levels were the lowest in the heart, male mice expressed statistically significant greater amounts of both transcripts in this tissue.

FISH analysis to determine the chromosomal location of the CYP2C18/19 transgene. A single hybridization signal is observed at region C1 of mouse chromosome 2. Nick-translated DNA fragments from the ∼196 kilobase pair DNA insert from BAC clone RP11-466J14 were used as hybridization probes. Chromosomal preparations from heterozygous mouse (A) and homozygous mouse (B). C1, reference DAPI banding pattern of mouse chromosome 2. C2, DAPI banding pattern before probe hybridization. C3, superimposed probe hybridization signal (red).
Expression at the Protein Level. Examination of the specificity of the CYP2C18 antiserum revealed that it did recognize recombinant CYP2C18 but not any CYP2C enzymes expressed in human liver microsomes (Fig. 5A). Furthermore, specific CYP2C18 immunoreactive protein of the appropriate apparent molecular weight was observed in microsomes isolated from COS-7 cells transfected with CYP2C18 cDNA but not in similar experiments using Huh-7 cells (Fig. 5B). Titration of recombinant CYP2C18 standards revealed a lower detection limit of 0.02 pmol (Fig. 5B). However, immunoreactive protein could not be detected using liver microsomes from either transgenic or wt mice (Fig. 5A) nor in microsomal preparations from kidney (not shown). This result indicates that the level of CYP2C18 protein in the transgenic mice is either very low (<0.8 pmol/mg) or nil.

CYP2C19 gene copy number determination in transgenic mice. Standard curves obtained from amplifying CYP2C19 (2C19), mIL2, or hIL2 from serial 2-fold dilutions of either mouse or human gDNA (2 to 0.25 ng). Data are plotted as cycle threshold versus log picogram DNA.
The expression of CYP2C19 protein was examined using the α-hCYP2C antipeptide antiserum directed toward the four C-terminal amino acids of CYP2C18, CYP2C19, CYP2C8, CYP2C9, and coincidentally CYP2C70 (the only murine CYP2C protein having this C-terminal sequence and thus this reactivity). Because human CYP2C8 and CYP2C9 are not present in mice and CYP2C18 was undetectable using the CYP2C18-specific antibody, the α-hCYP2C antiserum was used to quantitate CYP2C19 and CYP2C70 protein expression. As shown in Fig. 6A, Western analysis of liver microsomal proteins from male mice using the α-hCYP2C antiserum revealed a single immunoreactive protein band of the appropriate apparent molecular weight. Densitometric analyses revealed transgenic male mice to have 42% more immunoreactive protein than wt male mice (Fig. 6B).
Expression at the Catalytic Level. Assessment of CYP2C-dependent catalysis was done by examining the rate of hydroxylation of the CYP2C19 probes S-mephenytoin and R-omeprazole. In 9-week-old male mice, a statistically significant increase (p < 0.05) in intrinsic clearance (as microliter per nanomole of P450 per minute) was observed in the transgenic CYP2C18/19 mice compared with their wt controls (Table 2). A much smaller and statistically insignificant increase in intrinsic clearance could be seen in transgenic female mice compared with wt controls, consistent with the limited expression of CYP2C19 mRNA. There was also a statistically significant reduction in the total microsomal P450 content. Similar to the trend in intrinsic clearance, the difference was much greater and only statically significant in male mice. These observations could suggest a concomitant down-regulation of the endogenous murine P450 enzymes as a result of the transgene expression (Table 2).
Apparent enzyme kinetic parameters for S-mephenytoin metabolism in liver microsomes from 9-week-old wt and heterozygous CYP2C18/19 transgenic mice The table shows parametric and nonparametric parameters for each group. Statistical analyses were done using the Mann-Whitney test. p values <0.05 are shown in bold.
Liver microsomes from transgenic and wt 12-week-old males were incubated with R-omeprazole followed by quantitation of the 5′-hydroxylated metabolite. Linearity in the assay was ensured with respect to protein and time. The results presented in Fig. 7 show a statistically significant (p = 0.028) 40% increase in the median rate of omeprazole hydroxylation in male transgenic mice compared with wt mice. This difference is similar in magnitude to the increase in CYP2C19 (and potentially CYP2C70) immunoreactive protein (Fig. 6C).
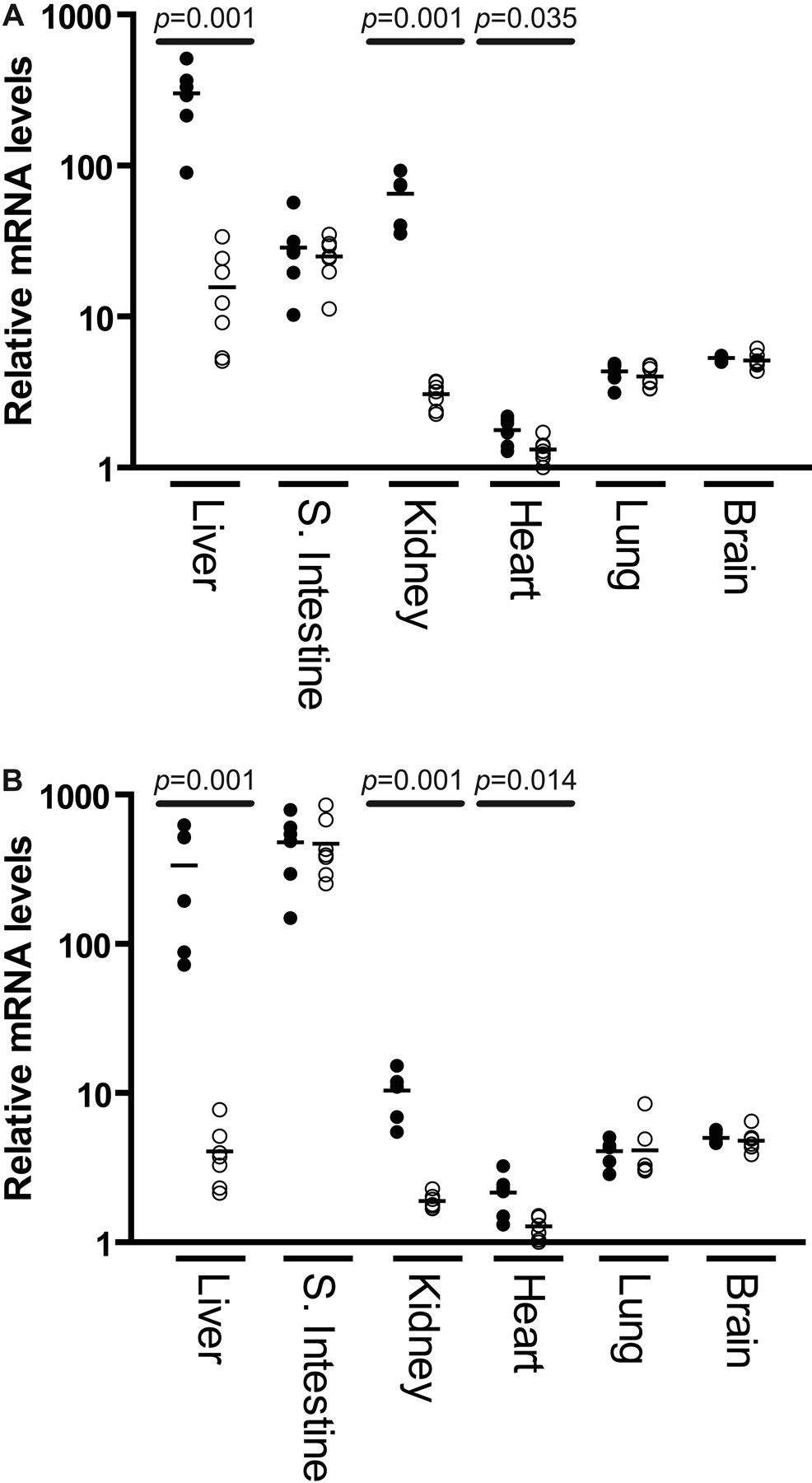
CYP2C18 (A) and CYP2C19 (B) mRNA expression in transgenic male (•) and female (○) mice 26 to 31 weeks of age using reverse-transcribed total RNA from liver, small intestine, kidney, heart, lung, and brain. Relative expression levels of each gene transcript were determined using real-time PCR and gene-specific primers (Table 1). The geometric mean of two reference transcripts (GAPDH and HPRT) was used to normalize the expression levels of CYP2C18 and CYP2C19. Bars denote mean values. Male and female mice were compared using the Mann-Whitney test.
Discussion
Mice were made transgenic using a DNA fragment representing a contiguous 196-kilobase pair region of the human genome containing the CYP2C18 and CYP2C19 genes and putative cis-acting regulatory regions. The transgene was found to have integrated into a single site composed of ∼12 tandem copies in region C1 of mouse chromosome 2 (Figs. 2 and 3). Obtaining such relatively high transgene copy numbers using BAC clones is not uncommon (Xing et al., 2007). Despite the high transgene copy number, Western blot analysis showed only an approximate 40% increase in protein immunoreactive with a CYP2C19 and CYP2C70 targeted antibody in transgenic male mice as compared with wt controls (Fig. 6). This relationship was also reflected in similar increases in rates of liver microsomal S-mephenytoin and R-omeprazole hydroxylation. Interestingly, there was also a reduction in total hepatic P450 content in the transgenic mice compared with wt. Mirroring the trend observed in mRNA expression, the difference was much greater in male mice. One possible explanation for the higher levels of CYP2C19 mRNA observed in the liver of the transgenic mice with only a modest increase in hepatic CYP2C immunoreactive protein could be that murine P450 enzymes are down-regulated in a feedback regulatory mechanism as a result of the expression of the CYP2C19 transgene. Because P450s have a critical role in regulating the levels of many endogenous endocrine factors, it could be suggested that the expression of the transgenic human CYP2C19 enzyme could play a compensatory role.

Western blot analysis of CYP2C18 expression using a CYP2C18-specific antibody. A, expression of CYP2C18 in liver microsomes (30 μg) from transgenic and wt mice. r2C18, recombinant CYP2C18 supersomes; HLM, human liver microsomes. B, expression of CYP2C18 in Huh-7 cells and in COS-7 cells (25 μg) after transfection with CYP2C18 cDNA. Amounts of recombinant CYP2C18 standards (supersomes) used to approximate lowest detectable amount of CYP2C18 protein. (+), transfected with CYP2C18 cDNA; (-), transfected with empty vector.

Western blot analysis of CYP2C19 and CYP2C70 protein expression in liver microsomes (20 μg) from transgenic and wt mice using the α-hCYP2C antipeptide antibody targeted against the four C-terminal amino acids of the four human CYP2Cs and murine CYP2C70 (representative blots shown). Tg, heterozygous transgenic. A, CYP2C19 and CYP2C70 expression levels in 12-week-old male transgenic and male wt mice. B, densitometric analyses reveals a 42% increase in CYP2C19 and/or CYP2C70 immunoreactive protein in liver microsomes from 12-week-old male transgenic (n = 4) compared with wt (n = 9) mice; p = 0.034 (Mann-Whitney test).

R-omeprazole 5′-hydroxylation in liver microsomes from 12-week-old male transgenic (n = 4) and male wt mice (n = 10). Mean values ± S.E.M. *, p = 0.028 (Mann-Whitney test). Tg, heterozygous transgenic.
Interestingly, real-time PCR analysis revealed that both the CYP2C18 and CYP2C19 genes were expressed in a sexually dimorphic manner in the liver, kidney, and heart but not in the small intestine, lung, or brain (Fig. 4). Although male mice expressed similar amounts of both CYP2C18 and CYP2C19 transcripts in liver, females expressed levels that were only about 1/10 to 1/100 of this level, respectively. A different relationship was observed in the kidneys, with females expressing roughly similar amounts of both human CYP2C transcripts, and males expressing 20- and 5-fold greater amounts of CYP2C18 and CYP2C19 mRNA, respectively. These results clearly indicate the human genes are subject to murine and tissue-specific regulatory factors.
Important sex differences are evident in the expression of specific CYP2C genes in rats. Sex difference in growth hormone (GH) secretory patterns results in differing levels of transcriptional activation, with transcriptional factors such as signal transducer and activator of transcription 5 playing a critical role (Park and Waxman, 2001). By contrast, no such sexually dimorphic CYP2C isoform expression is seen in human livers. Interestingly, our data from the transgenic mice suggest that the factors controlling the sexually dimorphic expression of rodent CYP2C genes can interact with the regulatory elements of the CYP2C18 and CYP2C19 genes, resulting in sexually dimorphic expression analogous to the male-specific CYP2C11 gene in the rat (Waxman and O'Connor, 2006; Thangavel and Shapiro, 2007) and the CYP2D genes in mouse liver (Sueyoshi et al., 1999). Thus, sexually dimorphic CYP gene expression in rodents, as opposed to humans, is not necessarily dependent on gene structure per se but rather the presence of sex- and tissue-specific factors for transcriptional regulation of CYP2C gene expression. Therefore, our findings and model system would be of great value in the future identification of the regulatory elements responsible for sexually dimorphic gene expression. Interestingly, Cheung et al. (2006) observed sexually dimorphic hepatic expression of CYP3A4 in transgenic mice; however, female mice were found to express significant amounts of CYP3A4 mRNA and protein, whereas neither CYP3A4 mRNA nor protein could be detected in adult males. Furthermore, by administering a constant GH infusion via an s.c. minipump to override the normally pulsatile male plasma GH profile, they were able to clearly show a feminization of the expression of CYP3A4 and of the endogenous murine Cyp2b and Cyp3a44 genes in male mice. It is plausible that GH could have a similar role in the tissue- and sex-preferential expression of CYP2C18 and CYP2C19 observed in the present study.
CYP2C19 mRNA was most abundant in the intestine and liver followed by kidney, with only moderate amounts being observed in lung and brain. The high level of intestinal and hepatic CYP2C19 mRNA expression is consistent with previous studies in humans showing similar levels of both CYP2C19-associated metabolic activity and mRNA expression in the liver and intestine (Läpple et al., 2003; Galetin and Houston, 2006; Bièche et al., 2007).
The relationship of mRNA levels correlating with levels of immunoreactive protein and catalytic activity determinations held true in studies of 12-week-old transgenic male mice, which showed a 42% increase in immunodetectable hepatic CYP2C19 and/or CYP2C70 compared with wt mice. A concomitant increase in the rate of hepatic R-omeprazole 5′-hydroxylation was seen (Fig. 7). In liver microsomes from 9-week-old transgenic males, S-mephenytoin intrinsic clearance was significantly increased on the order of 3-fold greater than wt controls (Table 2). Taken together, these data indicate that the human CYP2C19 mRNA expressed in the male mice is translated into functionally active CYP2C19 enzyme, although at a much lower level than expected from the high gene copy number (see above). By contrast, the absence of a statistically significant increase in S-mephenytoin hydroxylation in liver microsomes from female mice was in accordance with the observed very low levels of CYP2C19 mRNA (Fig. 4).
Whereas the above results show that the transgenic CYP2C19 gene was translated into functional protein, this clearly was not the case for CYP2C18, as evident using CYP2C18-specific antibodies. Indeed, the CYP2C18 protein has not been found in detectable amounts in any human tissues (Goldstein, 2001) despite high levels of CYP2C18 mRNA (Nishimura et al., 2003). The consistency of the events following transcription of the CYP2C18 gene along with the apparent insensitivity to any of the alterations associated with heterologous expression, as shown by the absence of CYP2C18 in the transgenic mice, was somewhat unexpected, and the underlying reasons remain unknown. Transient transfection studies were performed using cell lines derived from different tissue types to both further validate the CYP2C18-targeted antibody and to investigate any potential relationship between the translation or stability of CYP2C18 protein and any cell-type specific regulatory factors. It was shown that the CYP2C18 cDNA was effectively translated into detectable protein in the kidney cell-derived COS-7 cell line, whereas the efficiency was much less in the Huh-7 hepatoma cell line (Fig. 5). Despite these observations, CYP2C18 protein was not detected in kidney microsomes prepared from transgenic mice. It is conceivable these tissues contain factors, such as regulatory RNAs, which prevent translation of the CYP2C18 mRNA. It may also be possible that the enzyme is not properly folded because of the absence of required chaperone proteins and thus subject to proteolytic degradation. Further experiments may shed light on this phenomenon and help to identify the necessary components required for CYP2C18 translation and folding. Nevertheless, the results obtained here indicate that the alterations in CYP2C protein and catalytic activity levels observed in the transgenic mice are solely the result of the expression of a functional CYP2C19 enzyme and not CYP2C18.
In conclusion, our results show that the transgenic mice express CYP2C18 and CYP2C19 mRNAs at relatively high levels in liver, kidney, and small intestine and that both genes are subject to sexually dimorphic regulation in the liver, kidney, and heart. Future studies will attempt to identify the sex- and tissue-specific factors responsible for the transcriptional regulation of CYP2C gene expression. The significantly increased hydroxylation of S-mephenytoin and R-omeprazole hydroxylation observed in transgenic male mice suggests that this male mouse model has a more human-like metabolism of CYP2C19 substrates than wt C57BL/6 mice. This model could be potentially useful in early drug discovery and development as a tool for pharmacological and toxicological studies of drug candidates that are specific substrates for human CYP2C19.
Acknowledgments
We thank Charlotta Ungmann and Golpar Pirzadeh for help with preliminary animal characterization. We also thank Dr. Robert Edwards and Dr. Eric Johnson for their gifts of CYP2C antibodies. We thank Tommy B. Andersson, Kjell Andersson, and Lars Weidolf from AstraZeneca for assistance, expertise, and for providing analytical standards.
Footnotes
-
This research was in part supported by a grant from The Swedish Research council.
-
S.L. and R.M.B. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019349.
-
ABBREVIATIONS: P450, cytochrome P450; PCR, polymerase chain reaction; BAC, bacterial artificial chromosome; gDNA, genomic DNA; wt, wild-type; HPRT, hypoxanthine guanine phosphoribosyltransferase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DAPI, 4′,6-diamidino-2-phenylindole; bp, base pair; FISH, fluorescent in situ hybridization; hIL2, human IL2; mIL2, murine IL2; TBS, Tris-buffered saline; GH, growth hormone.
-
↵1 Current affiliation: Department of Clinical Pharmacotherapeutics, Tohoku Pharmaceutical University, Sendai, Japan.
-
↵2 Current affiliation: Medivir AB, Huddinge, Sweden.
- Received October 18, 2007.
- Accepted February 11, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}