


Abstract
The quantitative impact of excretory transport modulation on the systemic exposure to xenobiotics and derived metabolites is poorly understood. This article presents fundamental relationships between exposure and loss of a specific excretory process that contributes to overall clearance. The mathematical relationships presented herein were explored on the basis of hepatic excretory data for polar metabolites formed in the livers of various transporter-deficient rodents. Experimental data and theoretical relationships indicated that the fold change in exposure is governed by the relationship, 1/(1 – fe), where fe is the fraction excreted by a particular transport protein. Loss of function of a transport pathway associated with fe < 0.5 will have minor consequences (<2-fold) on exposure, but exposure will increase exponentially in response to loss of function of transport pathways with fe > 0.5. These mathematical relationships may be extended to other organs, such as the intestine and kidney, as well as to systemic drug exposure. Finally, the relationship between exposure and fe is not only applicable to complete loss of function of a transport pathway but also can be extended to partial inhibition scenarios by modifying the equation with the ratio of the inhibitor concentration and inhibition constant.
Quantitative methods to predict the change in drug exposure resulting from drug-drug interactions occurring at the level of excretory transport have not been fully developed. A fundamental understanding of the effect of excretory transport modulation on drug/metabolite exposure is presently lacking. Kinetic approaches originally used to examine issues related to drug metabolism, such as the relative activity factor method, are now being applied to transport processes (Hirano et al., 2004). In this article, the equation describing the effect of inhibition of drug metabolism on systemic concentrations (Rowland and Matin, 1973) is adapted to explore the effect of transport modulation on exposure.
Hepatobiliary excretion data for polar metabolites generated in perfused livers of various transporter-deficient animals are used primarily in this article as supporting evidence for the theoretical equations. However, the relationships presented are applicable to any organ such as the intestine and kidney in which the formed metabolites can be excreted into two distinct compartments. Most importantly, the relationship describing the fold change in exposure is applicable not only to an isolated organ but also to the systemic exposure of drugs and metabolites when systemic clearance is mediated by excretory transport. This article provides proof that the maximum fold change in exposure to a drug or metabolite cleared by active excretion is dictated by the relationship 1/(1 – fe), where fe is the fraction of total clearance mediated by the ablated transport protein. Although the equations presented may seem to be theoretically obvious, extensive experimental proof of these relationships currently does not exist in the literature. The relationship, 1/(1 – fe), forms the basis for predicting the importance of a given excretory transport pathway as a potential site for drug-drug interactions.
Results
Clearance-Exposure Relationship. The fold increase (fold Δ) in systemic drug exposure caused by inhibition of metabolism has been described by Rowland and Matin (1973) as (eq. 1) 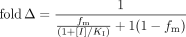 where fm is the fraction of total clearance mediated by the relevant enzyme, [I] is the inhibitor concentration, and KI is the inhibitor concentration at which enzyme activity is impaired 50% when substrate concentrations are below the Michaelis-Menten constant (KM).
where fm is the fraction of total clearance mediated by the relevant enzyme, [I] is the inhibitor concentration, and KI is the inhibitor concentration at which enzyme activity is impaired 50% when substrate concentrations are below the Michaelis-Menten constant (KM).
When [I] ≫ KI, the increase in systemic drug exposure is only a function of fm in the uninhibited condition:  This relationship is equally valid for complete genetic deficiency of an enzymatic pathway, where fm is determined at “normal” expression of the enzyme. Equation 2 indicates that at most, a 2-fold increase in systemic exposure will be observed when fm is ≤0.5, even when inhibition of the relevant enzyme is complete. In contrast, systemic exposure will increase exponentially in response to complete inhibition or knockout of the enzyme as fm increases from 0.5 to 1.
This relationship is equally valid for complete genetic deficiency of an enzymatic pathway, where fm is determined at “normal” expression of the enzyme. Equation 2 indicates that at most, a 2-fold increase in systemic exposure will be observed when fm is ≤0.5, even when inhibition of the relevant enzyme is complete. In contrast, systemic exposure will increase exponentially in response to complete inhibition or knockout of the enzyme as fm increases from 0.5 to 1.

Scheme depicting the bidirectional hepatic excretion of metabolites. All supporting data were generated in single-pass liver perfusion experiments in which the livers were perfused with parent compounds, and the recovery of relevant metabolites in bile and perfusate, as well as steady-state hepatic metabolite concentrations, were quantified directly.
In the case of drug transport, the relevant parameter is fe, the fraction of total clearance mediated by a particular excretory transport protein, as opposed to fm. However, the mathematical relationships are analogous. When a particular route of transport is inhibited completely or genetically ablated,  Metabolism and excretory transport function in concert to clear drugs from the body. In cases in which both metabolic and transport processes contribute to the overall clearance, the sum of fm and fe must equal 1, such that these fractions are calculated relative to total clearance and not metabolic or excretory clearance in isolation.
Metabolism and excretory transport function in concert to clear drugs from the body. In cases in which both metabolic and transport processes contribute to the overall clearance, the sum of fm and fe must equal 1, such that these fractions are calculated relative to total clearance and not metabolic or excretory clearance in isolation.
Note that all equations presented assume first-order kinetics. Transport proteins and drug-metabolizing enzymes are inherently capacity-limited and therefore nonlinear. All of the relationships presented herein assume approximate linear kinetic behavior, and thus all substrate concentrations are assumed to be below the KM of the relevant transport protein.
In the case of hepatic transport (i.e., excretion from the hepatocyte across the basolateral membrane into blood or across the canalicular membrane into bile) (Fig. 1), this fundamental relationship can be illustrated most clearly by considering the excretion of polar metabolites formed in the liver. In this case, the resultant influence of changes in excretory transport on systemic exposure is not confounded by the kinetics of drug uptake into the liver or passive membrane permeability. Furthermore, it is assumed that all probe substrates are not further metabolized, such that excretion is the only mechanism of clearance from the liver.
Unlike fm, the impact of fe on changes in exposure differs when the knocked-out or inhibited transport protein mediates or opposes the excretory route (apical or basolateral) of interest. For example, knockout or inhibition of a canalicular transport protein will result in decreased biliary metabolite recovery, whereas knockout or inhibition of a basolateral excretory transport protein will result in a higher driving force for biliary excretion (i.e., increased hepatic metabolite concentration) and therefore increased biliary metabolite recovery, assuming that a compensatory protein exists on the canalicular membrane with adequate affinity and capacity to transport the metabolite into bile.
Decrease in Excretion via Inhibition/Knockout of Direct Transport. When knockout or inhibition of a transport protein mediating metabolite excretory clearance in the direction of interest is considered, fe is calculated on the basis of the decrease in the excretory clearance across the relevant membrane: 
 where fe, Bile and fe, B/L refer to the fraction excreted by a canalicular or basolateral transport protein of interest, ClBile and ClB/L are the biliary and basolateral intrinsic excretory clearances, and WT and KO refer to the wild-type and transporter knockout/deficient animals, respectively. In other words, fe is a function of the fraction of vectorial flux mediated by the transport protein of interest.
where fe, Bile and fe, B/L refer to the fraction excreted by a canalicular or basolateral transport protein of interest, ClBile and ClB/L are the biliary and basolateral intrinsic excretory clearances, and WT and KO refer to the wild-type and transporter knockout/deficient animals, respectively. In other words, fe is a function of the fraction of vectorial flux mediated by the transport protein of interest.
Compensatory Increase in Excretion via Inhibition/Knockout of Opposing Transport. When a competing transport mechanism on the opposite plasma membrane domain is knocked out or inhibited, the increase in excretion in the opposite direction is a function of the fraction of total hepatic excretory clearance (i.e., the sum of ClBile and ClB/L) that is mediated by the impaired competing transport protein: 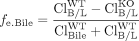
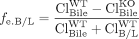 In this case, the change in fe is not caused directly by the impaired transport in the opposing direction but rather by the decrease in total excretory clearance, which elicits an increase in hepatic concentrations of the metabolite that serve as the driving force for increased excretion in the direction of interest.
In this case, the change in fe is not caused directly by the impaired transport in the opposing direction but rather by the decrease in total excretory clearance, which elicits an increase in hepatic concentrations of the metabolite that serve as the driving force for increased excretion in the direction of interest.
Effect of Excretory Transport Inhibition/Knockout on Organ Exposure. The increase in hepatic metabolite concentrations in response to transport inhibition is governed not by the fe of a particular transport pathway, but by the change in total hepatic excretory clearance. The relationship also would be governed by the fe (eq. 5) if other transport processes were not altered in the knockout animal or if inhibitors were specific enough not to affect other transporters. However, rodents genetically deficient in transporters may exhibit alterations in other pathways, and inhibitors often affect multiple processes (for review, see Zamek-Gliszczynski et al., 2006a). Thus, the change in hepatic exposure to a derived metabolite is governed by the total fraction excreted (fe, Total): 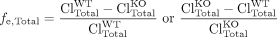 where ClTotal is the sum of the biliary and basolateral excretory clearances. Total excretory clearance in an animal genetically deficient in a transport protein may actually be higher than that in the corresponding wild type, and in this case the relationship on the right should be used to maintain the fe, Total between zero and unity.
where ClTotal is the sum of the biliary and basolateral excretory clearances. Total excretory clearance in an animal genetically deficient in a transport protein may actually be higher than that in the corresponding wild type, and in this case the relationship on the right should be used to maintain the fe, Total between zero and unity.
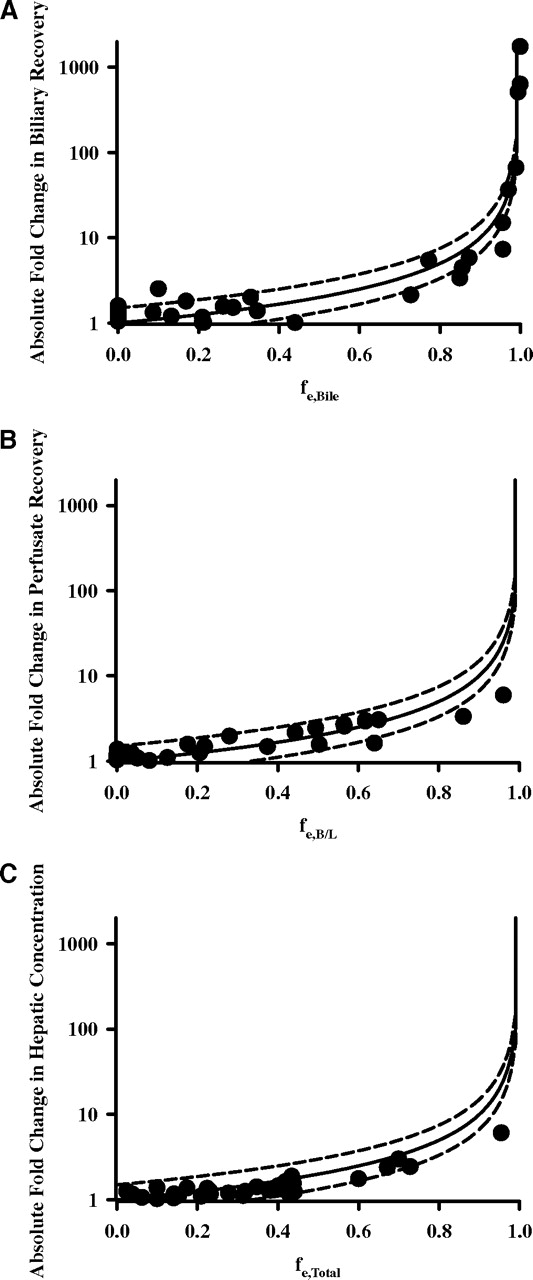
Figure 2 demonstrates the applicability of eq. 3 in predicting the fold change in biliary, systemic, or hepatic exposure to polar metabolites formed in the liver. These data were generated in single-pass liver perfusion experiments in which the biliary and perfusate (systemic) recovery of substrates could be quantified directly (Takenaka et al., 1995; Xiong et al., 2002; Nezasa et al., 2006; Zamek-Gliszczynski et al., 2006b,c). Rodents genetically deficient in a specific transport protein were used to examine the impact of complete abrogation of the transporter on exposure/cumulative recovery. In this experimental paradigm, biliary and perfusate recoveries serve as surrogates for intestinal and systemic metabolite exposure, respectively.

A, fold change in biliary recovery as a function of the fraction excreted (fe, Bile). B, fold change in perfusate recovery (a surrogate of systemic exposure) as a function of the fraction excreted (fe, B/L). C, fold change in hepatic metabolite concentration as a function of total fraction excreted (fe, Total). Solid line represents the theoretical relationship (eq. 3) ±50% represented by dashed lines. Data presented are summarized in Tables 1, 2, and 3.
Altered exposure/recovery of 4-methylumbelliferyl glucuronide (4MUG) in Mrp3 and Bcrp knockout mice (Tables 1, 2, 3) will be used to illustrate the application of the theoretical relationships. In mice, 4MUG is excreted into bile by Bcrp and across the hepatic basolateral membrane into blood by Mrp3 (Zamek-Gliszczynski et al., 2006a); therefore, biliary excretion of 4MUG would be expected to be impaired in Bcrp-knockout mice but would be elevated in mice lacking Mrp3, a transport pathway that opposes the excretory route of interest. Because Bcrp is directly involved in transporting 4MUG into bile, using eq. 4.1 and the excretory clearance values (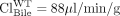 liver and
liver and 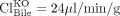 liver) reported by Zamek-Gliszczynski et al. (2006c), fe, Bile of Bcrp is calculated to be 0.73. Equation 3 predicts the decrease in biliary recovery of 4MUG to be 3.7-fold; experimentally a 2.1-fold decrease was observed. In contrast, Mrp3 functions in the opposite direction from biliary excretion, so using eq. 5.1 and excretory clearance values (
liver) reported by Zamek-Gliszczynski et al. (2006c), fe, Bile of Bcrp is calculated to be 0.73. Equation 3 predicts the decrease in biliary recovery of 4MUG to be 3.7-fold; experimentally a 2.1-fold decrease was observed. In contrast, Mrp3 functions in the opposite direction from biliary excretion, so using eq. 5.1 and excretory clearance values (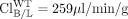 liver,
liver, 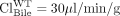 liver, and
liver, and  liver) reported by Zamek-Gliszczynski et al. (2006b), fe, Bile of Mrp3 is calculated to be 0.77. Equation 3 predicts the increase in biliary recovery of 4MUG to be 4.4-fold; experimentally a 5.4-fold increase was observed. By using eq. 6 and the same data set (
liver) reported by Zamek-Gliszczynski et al. (2006b), fe, Bile of Mrp3 is calculated to be 0.77. Equation 3 predicts the increase in biliary recovery of 4MUG to be 4.4-fold; experimentally a 5.4-fold increase was observed. By using eq. 6 and the same data set (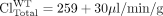 liver and
liver and 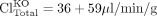 liver), fe, Total of Mrp3 is calculated to be 0.67. Equation 3 predicts that hepatic 4MUG exposure in Mrp3-knockout mice will be 3.0-fold higher relative to that of wild-type mice; experimentally an increase of 2.4-fold was observed.
liver), fe, Total of Mrp3 is calculated to be 0.67. Equation 3 predicts that hepatic 4MUG exposure in Mrp3-knockout mice will be 3.0-fold higher relative to that of wild-type mice; experimentally an increase of 2.4-fold was observed.
Fraction excreted (fe, Bile) by the ablated transport pathway in each animal model and the corresponding experimentally observed fold change in biliary recovery
Please note that fe, Bile values for canalicular transporters pumping directly into the bile were calculated using eq. 4.1, whereas fe, Bile for basolateral transporters pumping in the opposite direction (into perfusate) were calculated using eq. 5.1. Positive fold Δ values indicate an increase in exposure, whereas decreased exposure is denoted by a negative fold Δ value. All data points were calculated using mean values reported in the literature (Takenaka et al., 1995; Xiong et al., 2002; Nezasa et al., 2006; Zamek-Gliszczynski et al., 2006b,c).
Fraction excreted (fe, B/L) by the ablated transport pathway in each animal model and the corresponding experimentally observed fold change in perfusate recovery, a surrogate of systemic exposure
Please note that fe, B/L values for basolateral transporters pumping directly into perfusate were calculated using eq. 4.2, whereas fe, B/L for canalicular transporters pumping in the opposite direction (into the bile) were calculated using eq. 5.2. Positive fold Δ values indicate an increase in exposure, whereas decreased exposure is denoted by a negative fold Δ value. All data points were calculated using mean values reported in the literature (Takenaka et al., 1995; Xiong et al., 2002; Nezasa et al., 2006; Zamek-Gliszczynski et al., 2006b,c).
Total fraction excreted (fe, Total; eq. 6) by the ablated transport pathway in each animal model and the corresponding experimentally observed fold change in hepatic metabolite concentration in animal models lacking various transporter proteins
Positive fold Δ values indicate an increase in exposure, whereas decreased exposure is denoted by a negative fold Δ value. All data points were calculated using mean values reported in the literature (Takenaka et al., 1995; Xiong et al., 2002; Nezasa et al., 2006; Zamek-Gliszczynski et al., 2006b,c). Hepatic concentrations were not quantified for carboxydichlorofluorescein (Nezasa et al., 2006) and were not reported for E3040 sulfate and E3040 glucuronide (Takenaka et al., 1995).
Discussion
The fold change in exposure to polar metabolites in response to modulation of hepatic excretory transport is described by eq. 3, 1/(1 – fe). Although the change in metabolite exposure resulting from modulation of hepatic excretory transport can be predicted by complex physiologically based models of hepatobiliary pharmacokinetics (Chiba and Pang, 1993; Liu and Pang, 2006), the theoretical relationships presented in this article provide a simple and reliable prediction of the impact of excretory transport modulation on exposure. Hepatobiliary disposition data were used to support the relationship between exposure and fe, because of the existing wealth of data in the liver. However, as demonstrated below, the relationships presented herein are applicable to overall systemic drug/metabolite exposure as well as to any organ.
The relationship, 1/(1 – fe), is applicable to systemic exposure of parent drug. This point is exemplified by elevated pravastatin exposure in Mrp2-deficient rats. In normal bile duct-cannulated rats, 81% of the intravenous pravastatin dose was excreted in bile (Adachi et al., 1996). Mrp2 was shown to transport pravastatin in vitro (Sasaki et al., 2002). If it is assumed that Mrp2 entirely mediated the biliary excretion of pravastatin and assigned it an fe value of 0.81, then the expected increase in drug exposure in rats lacking this transporter would be approximately 5.3-fold (eq. 3). Relative to wild-type rats, Mrp2-deficient rats exhibited 4.7- and 6.1-fold higher drug exposure after intravenous and oral administration of pravastatin, respectively (Kivisto et al., 2005).
The role of Bcrp in renal excretion of the sulfate metabolite of E3040 was studied in mice (Mizuno et al., 2004). Renal excretory clearance of E3040 sulfate was decreased from 2.35 to 0.59 ml/min/kg in Bcrp-knockout mice. Thus, the fe of Bcrp in urinary excretion of E3040 sulfate is 0.75 (eq. 4), and so the maximal expected decrease in urinary recovery of this metabolite in knockout mice is 4-fold (eq. 3). Experimentally, urinary recovery of E3040 sulfate was decreased approximately 3-fold.
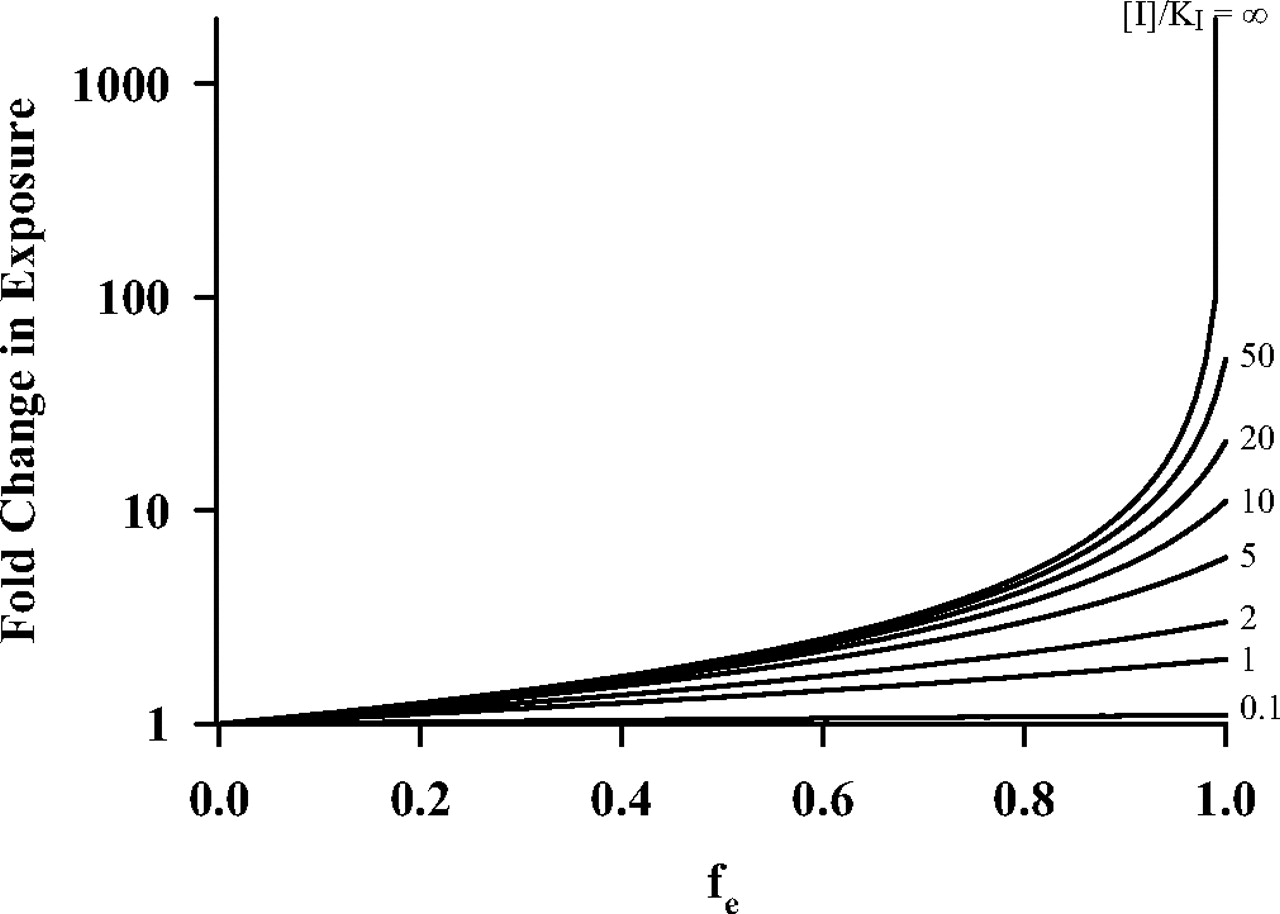
The relationships presented in this article are based on data generated in rodents genetically deficient in specific transport proteins, but eq. 3 could be modified as follows to describe transport inhibition: 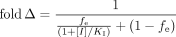 The change in exposure as a function of fe at various [I]/KI ratios is presented in Fig. 3. In the case of inhibition, the inhibitor concentration must exceed KI for a transport pathway representing fe > 0.5 to observe appreciable (>2-fold) changes in drug/metabolite exposure. Note that the fold change in systemic and biliary exposure in the presence of an inhibitor may be oversimplified by eq. 7, because many inhibitors impair multiple transport mechanisms on both plasma membrane domains (Zamek-Gliszczynski et al., 2006a). In contrast, fe, Total, which determines the fold change in hepatic exposure, accounts for the nonspecific effects of inhibitors.
The change in exposure as a function of fe at various [I]/KI ratios is presented in Fig. 3. In the case of inhibition, the inhibitor concentration must exceed KI for a transport pathway representing fe > 0.5 to observe appreciable (>2-fold) changes in drug/metabolite exposure. Note that the fold change in systemic and biliary exposure in the presence of an inhibitor may be oversimplified by eq. 7, because many inhibitors impair multiple transport mechanisms on both plasma membrane domains (Zamek-Gliszczynski et al., 2006a). In contrast, fe, Total, which determines the fold change in hepatic exposure, accounts for the nonspecific effects of inhibitors.

Fold change in exposure as a function of fe at various [I]/KI ratios as described by eq. 7.
Systemic clearance of methotrexate in rats is partially mediated by biliary excretion via Mrp2; the fe of this transport protein in systemic elimination of methotrexate is approximately 0.5 (Chen et al., 2003). In Mrp2-deficient rats, eq. 3 predicts a 2-fold increase in systemic exposure to methotrexate, which is in good agreement with the 1.9-fold increase observed by Chen et al. (2003). However, if Mrp2 function is not lost altogether, but instead inhibited to a limited extent, a less pronounced increase in exposure would be expected. The KI of probenecid for Mrp2 was determined to be 180 μM in vitro, and this inhibitor was intravenously infused to four different steady-state concentrations in wild-type rats to study its effect on methotrexate systemic exposure (Ueda et al., 2001). Equation 7 predicts 1.1-, 1.4-, 1.4-, and 1.5-fold increases in exposure at circulating probenecid concentrations of 243, 633, 704, and 905 μM, respectively. This prediction is in good agreement with the observed 1.2- and 1.3-fold increases in methotrexate exposure at 243 and 633 μM probenecid concentrations. However, 1.9- and 2.1-fold observed increases in drug exposure at 704 and 905 μM inhibitor concentrations exceeded the prediction, because at these higher concentrations, probenecid also was appreciably inhibiting hepatic uptake of methotrexate in addition to its biliary excretion (Ueda et al., 2001).
Theoretical relationships supported by experimental data indicate that a considerable change in drug or metabolite exposure will occur only when an appreciable excretory transport pathway (fe > 0.5) is inhibited substantially or knocked out. To elicit a considerable change in exposure with an inhibitor, the fe of the impaired pathway must exceed 0.5 and the [I]/KI ratio must exceed 1. As the fe of the extensively inhibited or knocked-out transport pathway increases above 0.5 and approaches unity, exposure will increase in an exponential manner.
Footnotes
-
This work was supported by the National Institutes of Health [Grant R01 GM41935] and by Eli Lilly and Company.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.023648.
-
ABBREVIATIONS: 4MUG, 4-methylumbelliferyl glucuronide; Mrp, multidrug resistance-associated protein; Bcrp, breast cancer resistance protein; WT, wild type; KO, knockout.
- Received July 30, 2008.
- Accepted November 13, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}