


Abstract
N-Glucuronidation in vitro of six 4-arylalkyl-1H-imidazoles (both enantiomers of medetomidine, detomidine, atipamezole, and two other closely related compounds) by rat, dog, and human liver microsomes and by four expressed human UDP-glucuronosyltransferase isoenzymes was studied. Human liver microsomes formed N-glucuronides of 4-arylalkyl-1H-imidazoles with high activity, with apparent Vmax values ranging from 0.59 to 1.89 nmol/min/mg of protein. In comparison, apparentVmax values for two model compounds forming the N-glucuronides 4-aminobiphenyl and amitriptyline were 5.07 and 0.56 nmol/min/mg of protein, respectively. Atipamezole showed an exceptionally low apparent Kmvalue of 4.0 μM and a high specificity constant (Vmax/Km) of 256 compared with 4-aminobiphenyl (Km, 265 μM;Vmax/Km, 19) and amitriptyline (Km, 728 μM;Vmax/Km, 0.8).N-Glucuronidation of medetomidine was highly enantioselective in human liver microsomes; levomedetomidine exhibited a 60-fold Vmax/Kmvalue compared with dexmedetomidine. Furthermore, two isomeric imidazole N-glucuronides were formed from dexmedetomidine, but only one was formed from levomedetomidine. Dog liver microsomes formed N-glucuronides of 4-arylalkyl-1H-imidazoles at a low rate and affinity, with apparent Vmax values ranging from 0.29 to 0.73 nmol/min/mg of protein and apparentKm values from 279 to 1640 μM. Rat liver microsomes glucuronidated these compounds at a barely detectable rate. Four expressed human UDP-glucuronosyltransferase isoenzymes (UGT1A3, UGT1A4, UGT1A6, and UGT1A9) were studied for 4-arylalkyl-1H-imidazole-conjugating activity. Only UGT1A4 glucuronidated these compounds at an activity of about 5% of that measured for 4-aminobiphenyl. The observed activity of UGT1A4 does not explain the high efficiency of glucuronidation of 4-arylalkyl-1H-imidazoles in human liver microsomes.
Glucuronidation is a phase II conjugation reaction catalyzed by a family of UDP-glucuronosyltransferase isoenzymes (UGTs1; EC2.4.1.17) (Clarke and Burchell, 1994). Compounds possessing a nucleophilic O or N atom in their structure are the most common substrates for UGTs. Glucuronidation to a hydroxyl group (usually formed in a phase I reaction) is probably the best-characterized phase II reaction of drug metabolism. In contrast,N-glucuronidation represents a less examined pathway. Various nitrogen-containing functional groups, which are extremely common in drug chemistry, are susceptible to direct glucuronidation without any phase I modification. Compounds that formN-glucuronides include aliphatic and aromatic amines and various heterocycles. N-Glucuronidation constitutes a major detoxification reaction in the metabolism of some drugs and other xenobiotics (Hawes, 1998); for example, 63% of the oral dose of lamotrigine is excreted in human urine as a quaternary triazine-linkedN-glucuronide (Cohen et al., 1987; Sinz and Remmel, 1991). Significant, substrate-dependent differences between species in their ability to form N-glucuronides have been observed (Chiu and Huskey, 1998).
The new drug discovery paradigm emphasizing early prediction of absorption-distribution-metabolism-excretion properties has created a great interest in understanding the metabolism potential of different chemical structures. The imidazole ring is one of the common structural motifs in drug molecules susceptible to direct glucuronidation. Imidazole N-glucuronide was detected as a metabolite of croconazole isolated from rabbit urine and as a major metabolite of tioconazole isolated from human urine (Takeuchi et al., 1989; Macrae et al., 1990). These glucuronides, in addition to glucuronides of nafimidone alcohol and imiloxan isolated from human urine (Rush et al., 1990, 1992), are representatives ofN+-glucuronides (i.e., quaternary ammonium glucuronides carrying a permanent positive charge). QuaternaryN+-glucuronides are formed from imidazole compounds, which carry the original substituent in one of the imidazole ring nitrogens. In contrast, tertiaryN-glucuronides are formed from N-unsubstituted imidazoles [e.g., from methylbiphenyl-C4-imidazole (Huskey et al., 1994)].
Human UGT isoform(s) responsible for the glucuronidation of imidazoles have not yet been identified. Several isoforms of the UGT1A subfamily, including 1A3, 1A4, 1A6, 1A9 (Orzechowski et al., 1994; Green and Tephly, 1996, 1998; Green et al., 1998), and also UGT2B7 (Stevens et al., 2001), have been reported to be involved inN-glucuronidation of xenobiotics in humans.
4-Arylalkyl-1H-imidazoles belong to a class of pharmacologically active C4-imidazole compounds, which may form tertiary imidazole N-glucuronides. The pharmacological effect of these drugs is based on agonism of both pre- and postsynaptic α2-receptors (MacDonald and Virtanen, 1992). Two of these compounds, detomidine and medetomidine (Fig.1), are therapeutically used as analgesic sedatives for animals. Dexmedetomidine (Precedex; Abbott Laboratories, Abbott Park, IL) was recently approved for clinical use for sedation of patients in intensive care. Biotransformation is the most important pathway in the elimination of these compounds (Salonen et al., 1988,1991; Salonen and Eloranta, 1990). When the metabolism of levomedetomidine was first studied in human liver microsomes, direct glucuronidation to the imidazole ring nitrogen was observed (Lehtonen and Salonen, 1997). According to preliminary results,N-glucuronidation is an important route in the metabolism of dexmedetomidine in humans in vivo.
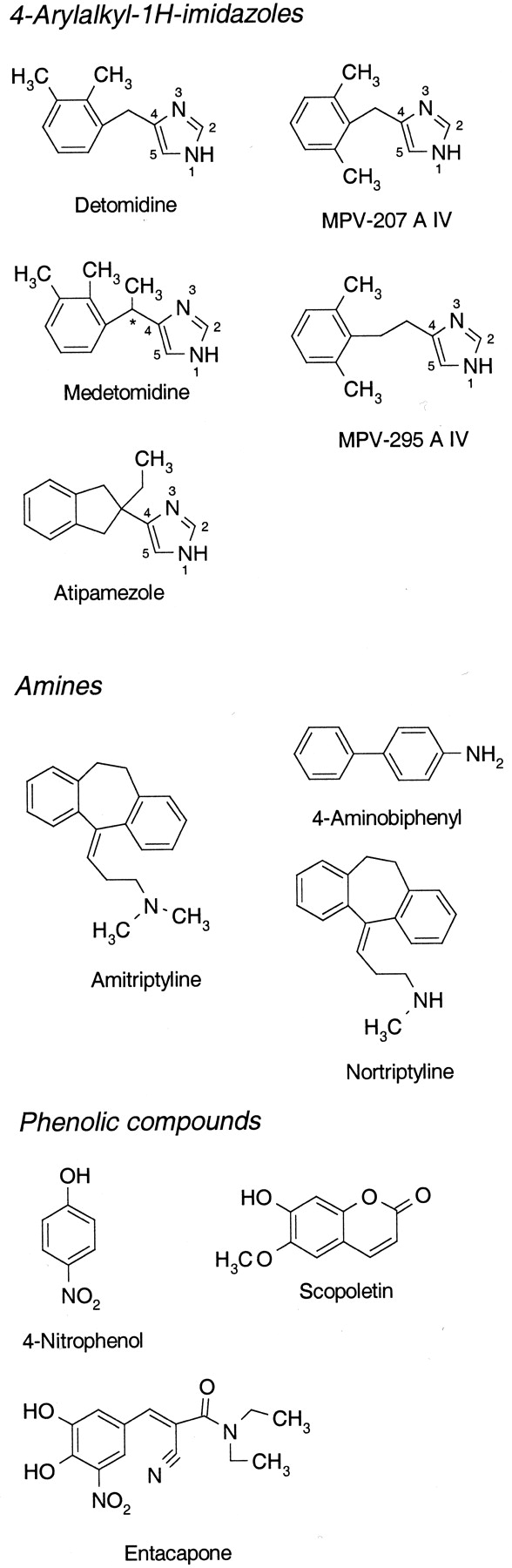
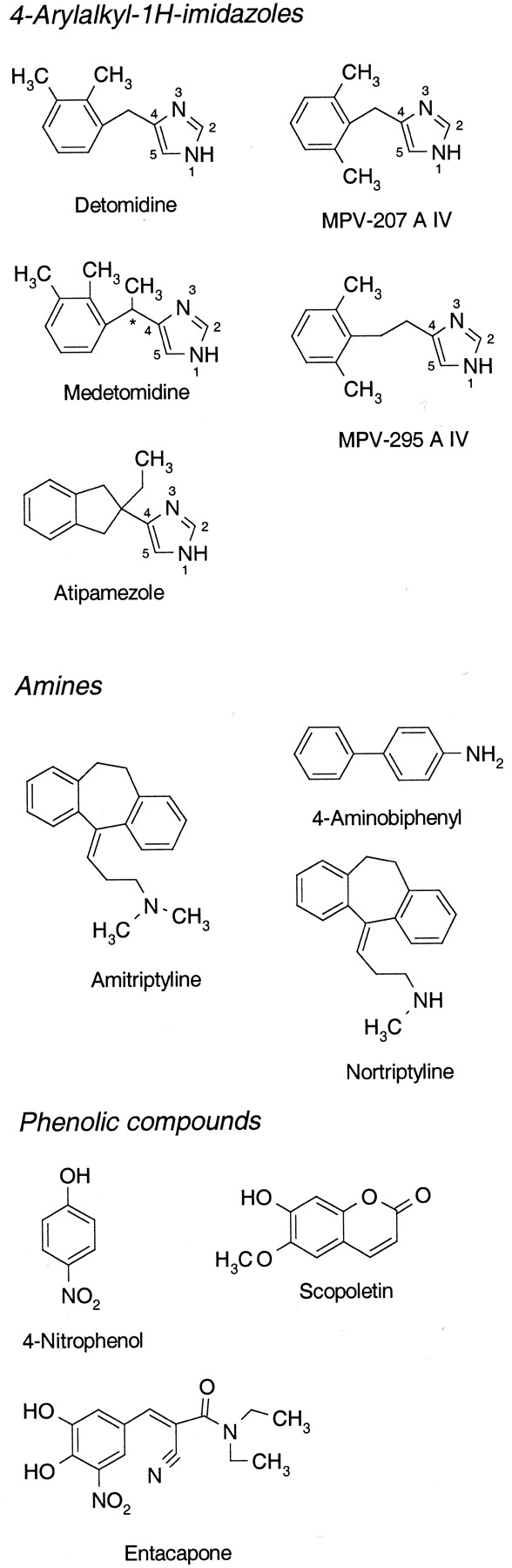
Chemical structures of substrates.
Chiral center marked with ★.
In our study, species differences in N-glucuronidation of detomidine, both enantiomers of medetomidine, atipamezole, and some other structurally related 4-arylalkyl-1H-imidazoles, between rat, dog, and human liver microsomes were determined. The effects of minor differences in the chemical structures of these compounds on their glucuronidation rates were observed. UGT activities for these 4-arylalkyl-1H-imidazoles were elucidated also in expressed human UGT isoforms 1A3, 1A4, 1A6, and 1A9.
Materials and Methods
Chemicals.
Saccharolactone, 4-nitrophenol, amitriptyline, nortriptyline, 4-aminobiphenyl, and scopoletin were purchased from Sigma (St. Louis, MO). UDPGA was obtained from Roche Molecular Biochemicals (Mannheim, Germany) and [14C]UDPGA from PerkinElmer Life Sciences (Boston, MA). 4-Arylalkyl-1H-imidazoles levomedetomidine [(−)-R-medetomidine], dexmedetomidine [(+)-S-medetomidine], detomidine, atipamezole, MPV-207 A IV, MPV-295 A IV, and [3H]levomedetomidine (Fig. 1) were kindly provided by Orion Pharma (Espoo, Finland). Entacapone was also provided by Orion Pharma. All other reagents were purchased from commonly used suppliers and were of the highest grade available.
Assays for Liver Microsomal UGTs.
All reaction mixtures contained 50 mM phosphate buffer, pH 7.4, 10 mM MgCl2, 5 mM saccharolactone, and depending on the aglycon, 0.5 to 10,000 μM substrate in a total volume of 100 μl. 4-Nitrophenol, scopoletin, entacapone, and 4-aminobiphenyl were added in 4 μl of dimethyl sulfoxide and amitriptyline in 5 μl of methanol to increase the solubility of these substrates. For each substrate and enzyme preparation, an optimal concentration [0–0.05 mg/ml corresponding to 0–1 (mg/mg) detergent/protein ratio] of Triton X-100 was used as a detergent. Pooled human liver microsomes were purchased from Human Biologics (Scottsdale, AZ). Dog liver microsomes from a male beagle were prepared as previously described (Salonen, 1991), and pooled rat liver microsomes from six male Wistar rats were prepared as described in Luukkanen et al. (1997). Microsomes were added at a protein concentration of 0.02 to 0.3 mg/ml. Preincubation was carried out at 37°C for 10 min, and incubation at 37°C for 10 to 60 min was initiated by the addition of 5 mM UDPGA. A blank sample was prepared and incubated in the same way, but substrate or UDPGA was replaced with incubation buffer. All reactions were shown to be linear with respect to protein concentration and incubation time. Reactions were terminated with 100 μl of ice-cold methanol (4-aminobiphenyl) or with 10 μl of 4 M perchloric acid (other substrates) while maintaining the reaction mixture in an ice bath. Reaction mixtures were centrifuged at 14,000 rpm for 5 min, and 80 μl of the supernatant was injected into the HPLC column.
Assays for Expressed Human UGT Isoenzymes.
All reaction mixtures contained 50 mM phosphate buffer, pH 7.4, 10 mM MgCl2, 5 mM saccharolactone, and 500 μM substrate in a total volume of 100 μl. Human UGT1A3 expressed in baculovirus-infected insect SF-9 cells was purchased from Panvera (Madison, WI), and UGT1A4 expressed in human B-lymphoblastoid AHH-1 cells was purchased from GENTEST (Woburn, MA). Human UGT1A6 and UGT1A9 were expressed in Chinese hamster lung fibroblast V79 cells using Semliki Forest virus vector, as previously described (Forsman et al., 2000). A protein concentration of 0.02 to 2 mg/ml and an incubation time of 45 min were used in conditions in which less than 5% of substrate was consumed. Preincubation was carried out at 37°C for 10 min, and the incubation at 37°C was initiated by the addition of 5 mM UDPGA. A blank sample was prepared and incubated in the same way, but the substrate or UDPGA was replaced with incubation buffer. Reactions were terminated with 100 μl of ice-cold methanol (4-aminobiphenyl) or with 10 μl of 4 M perchloric acid (other substrates) while maintaining the reaction mixture in an ice bath. Reaction mixtures were centrifuged at 14,000 rpm for 5 min, and 80 μl of the supernatant was injected into the HPLC column.
HPLC Conditions.
Glucuronides of various substrates were analyzed on a model 1100 or 1090 HPLC instrument (Hewlett Packard, Waldbronn, Germany), as described earlier (Kaivosaari et al., 2001). Briefly, glucuronides were separated on a Symmetry 150 × 3.9-mm C18 column (Waters, Milford, MA) or a Hypersil BDS 250 × 4-mm C18 column (Hewlett Packard). The mobile phase consisted of 50 mM phosphate buffer, pH 3.0, and methanol, with the exception of acid-labile 4-aminobiphenyl glucuronide, which was analyzed by an application of the method by Babu et al. (1996) using a mixture of 20 mM phosphate buffer, pH 6.8, and methanol. Glucuronides were detected by a model 1100 UV detector (Hewlett Packard).
Glucuronide Quantitation Using [14C]UDPGA.
Since reference N-glucuronides of the studied compounds were not available as quantitation standards for UV, radioactivity detection was used for quantitation of the formed N-glucuronide products. Separate quantitation samples were prepared to quantify glucuronide conjugates of each substrate, as described earlier (Kaivosaari et al., 2001). Briefly, quantitation was performed by adding [14C]UDPGA (0.1–0.4 μCi) and a variable amount (2.5–500 μM) of unlabeled UDPGA to the UGT incubation mixture. A substrate concentration of 500 μM and a protein concentration and incubation time of up to 0.5 mg/ml and 60 min, respectively were used. Glucuronide products were quantified using a model 9701 radioactivity detector (Reeve Analytical, Glasgow, UK) equipped with a heterogeneous 200-μl flow cell packed with silanized cerium-activated lithium glass (GS1/TSX; Reeve Analytical) positioned after a model 1100 UV detector (Hewlett Packard). A standard curve was attained for the UV detector in which the peak area of the glucuronide on the UV detector (mAU×s) was presented as a function of the quantified amount of glucuronide (picomoles) attained from the radioactivity detector. This curve was used to quantify glucuronide products formed in UGT assay samples containing 5 mM UDPGA but no14C-labeled UDPGA.
Glucuronide Quantitation Using [3H]Levomedetomidine.
Levomedetomidine glucuronidation in human liver microsomes was determined by adding [3H]levomedetomidine (0.5–1.5 μCi) to the UGT assay mixture. Glucuronide conjugates were quantified by a model 150TR radioactivity monitor (Packard, Meriden, CT) equipped with a homogeneous flow cell (500 μl), using Monoflow 3 scintillation fluid (National Diagnostics, Atlanta, GA) at a flow rate of 3 ml/min.
Calculation of the Apparent Kinetic Parameters.
To determine glucuronidation kinetics, UGT activities were measured at a minimum of seven substrate concentrations with two to four replicates at each concentration level. Apparent kinetic parametersKm andVmax were estimated using a nonlinear least-square fit to the Michaelis-Menten equation by the Leonora enzyme kinetics program version 1.0 by Cornish-Bowden (1995).
Results
Kinetics of Glucuronidation in Human Liver Microsomes.
All studied 4-arylalkyl-1H-imidazoles were substrates for human liver microsomal UGTs, with apparentVmax values ranging from 0.59 to 1.89 nmol/min/mg of protein (Table 1). These capacities were comparable with theVmax values measured for the model primary and tertiary amine substrates of N-glucuronidation (i.e., 4-aminobiphenyl and amitriptyline). The secondary amine nortriptyline, which is the demethylated metabolite of amitriptyline, was not glucuronidated at a detectable level. 4-Nitrophenol, the model phenolic compound forming an O-glucuronide, was glucuronidated at clearly the highest capacity (Vmax, 30.6 nmol/min/mg of protein).
Kinetics of glucuronidation in rat, dog, and human liver microsomes
In general, 4-arylalkyl-1H-imidazoles were approximately 10-fold higher affinity substrates than 4-nitrophenol, 4-aminobiphenyl, and amitriptyline. Atipamezole showed the highest affinity for human liver microsomal UGTs, with a very low apparentKm of 4.0 μM. The glucuronidation rate of atipamezole decreased when substrate concentration exceeded 25 μM, evidently due to substrate inhibition. This type of inhibition was not observed at substrate concentrations below 1000 μM for other 4-arylalkyl-1H-imidazoles.
Atipamezole was the best substrate also in terms of theVmax/Kmratio, which describes the efficiency of the enzymatic reaction, with a ratio of 256. Differences in theVmax/Kmratios between 4-arylalkyl-1H-imidazoles were striking, ranging from 256 (atipamezole) to 2.0 (dexmedetomidine). AlthoughVmax/Kmratios of similar magnitude were attained for the best 4-arylalkyl-1H-imidazole substrates and 4-nitrophenol, this was a result of 4-arylalkyl-1H-imidazoles being higher affinity (lower Km) yet lower capacity (lower Vmax) substrates compared with 4-nitrophenol.
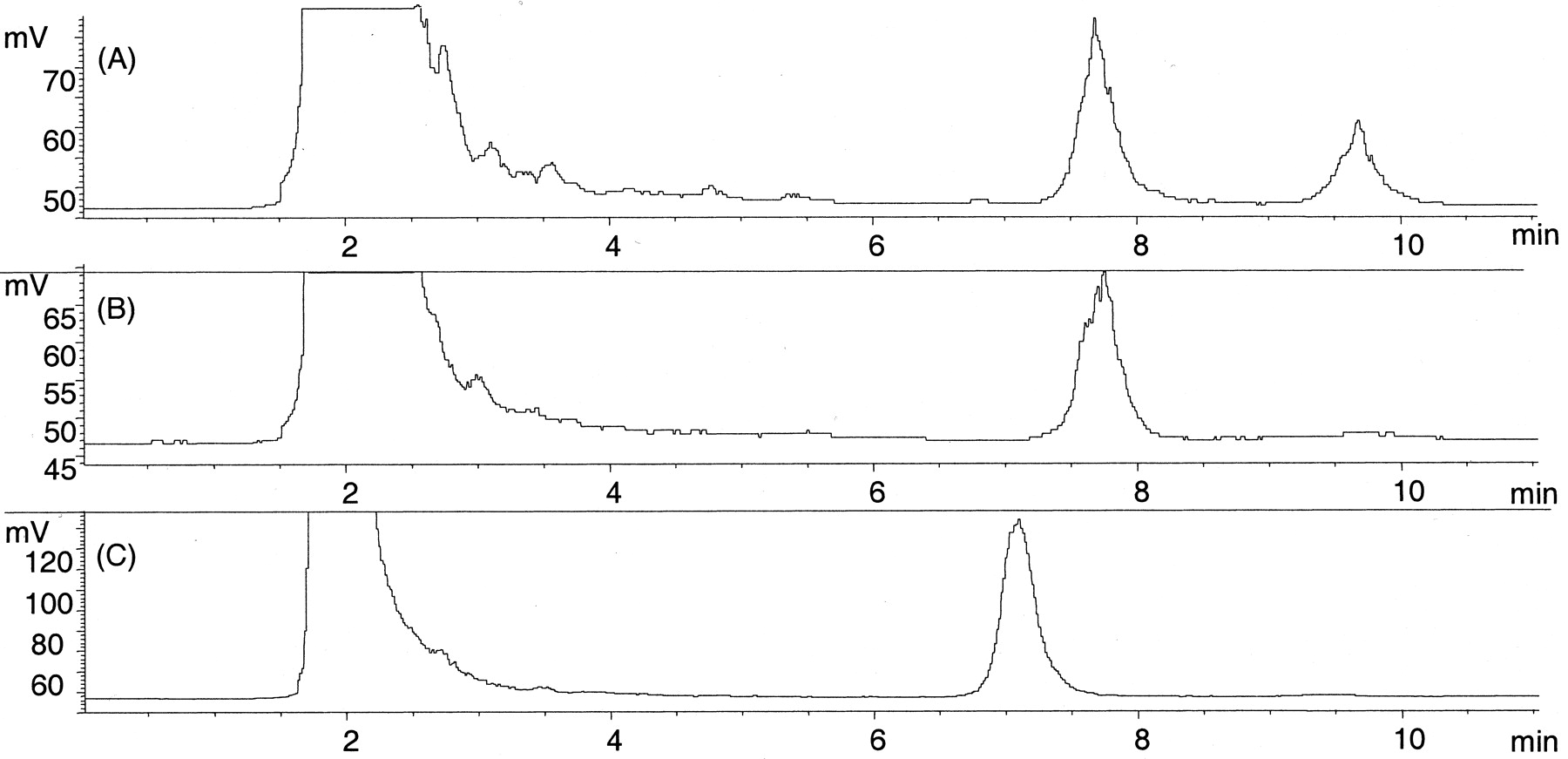
Some notable differences were apparent in the glucuronidation rates of various 4-arylalkyl-1H-imidazoles by human liver microsomes despite the very similar chemical structures of these compounds. First, compared with the other enantiomer levomedetomidine, dexmedetomidine was a poor substrate, with 23-fold higherKm value and 2.6-fold lowerVmax value. Two distinct glucuronides with different retention times (7.7 and 9.7 min) in HPLC were formed from dexmedetomidine (Fig. 2a), representing 70 and 30% of the total glucuronidation at 2000 μM substrate concentration, respectively. The apparent kinetic parameters were determined only for the glucuronide eluting at atR value of 7.7 min. In contrast, only one glucuronide conjugate (tR, 7.1 min) was formed from levomedetomidine (Fig. 2c).
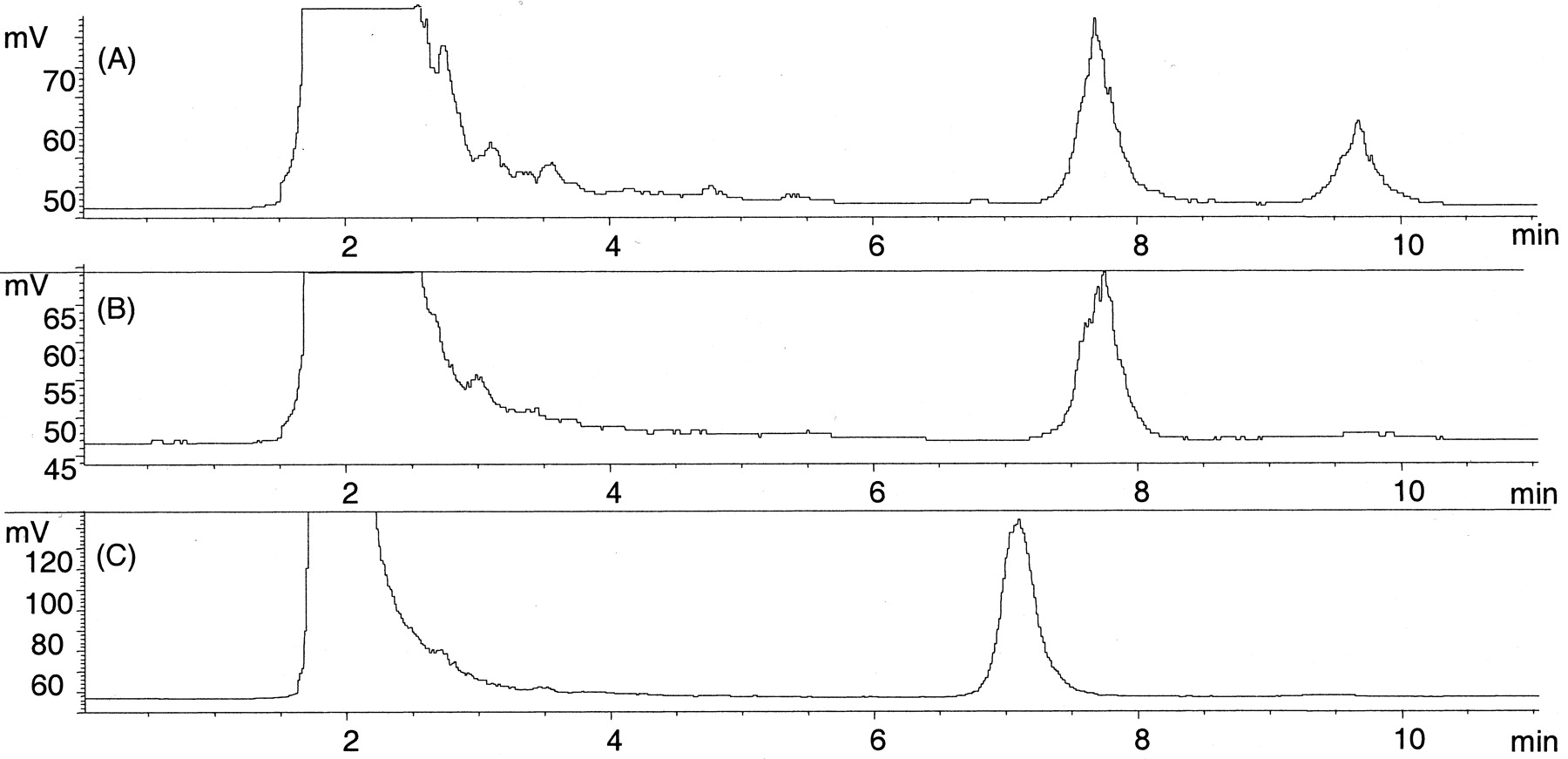
14C radiochromatograms (model 9701; Reeve Analytical) of UGT assays of medetomidine enantiomers.
A, two glucuronides were formed from dexmedetomidine in human liver microsomes with a tR/sof 7.7 and 9.7 min (unreacted [14C]UDPGAtR, 1.8 min); B, only one glucuronide (tR, 7.7 min) was formed from dexmedetomidine in dog liver microsomes. C, human liver microsomes formed only one glucuronide (tR, 7.1 min) from levomedetomidine, the other enantiomer of medetomidine. UGT assays were conducted as described under Materials and Methodsusing 0.5 mg/ml microsomal protein, 2000 μM dexmedetomidine, or 500 μM levomedetomidine and 50 μM (0.2 μCi) UDPGA.
Second, detomidine, a homolog of medetomidine, showed 4.9-fold lower affinity than levomedetomidine, although theVmax values were of similar magnitude. MPV-207 A IV and MPV-295 A IV, differing structurally by just one methylene group, showed very similarKm values. Yet, a 2.8-fold difference in Vmax in favor of the compound with a shorter alkyl chain (MPV-207 A IV) was observed.
Kinetics of Glucuronidation in Dog Liver Microsomes.
All studied 4-arylalkyl-1H-imidazoles were substrates for dog liver microsomal UGTs, with apparentVmax values ranging from 0.29 to 0.73 nmol/min/mg of protein (Table 1). The glucuronidation capacity of 4-aminobiphenyl (Vmax, 0.52 nmol/min/mg of protein) was comparable to that of 4-arylalkyl-1H-imidazoles. Amitriptyline and nortriptyline were not glucuronidated at detectable levels. 4-Nitrophenol was glucuronidated at a very high capacity, withVmax being >250-fold compared with the substrates forming N-glucuronides.
In general, N-glucuronidation of 4-arylalkyl-1H-imidazoles in dog liver microsomes occurred at a lower affinity than in human liver microsomes, with apparentKm values ranging from 279 to 1640 μM. Reliable estimation of Km andVmax values was difficult because for an unknown reason glucuronidation rates decreased when millimolar substrate concentrations were reached. The solubility of 4-arylalkyl-1H-imidazoles prevented the use of >5 mM substrate concentrations, but glucuronidation rates decreased even before solubility limits were exceeded, at >1 mM concentrations. When all data points, including the inhibition part of the curve, were included in the calculation of kinetic parameters and the “substrate inhibition equation” was used, the data points did not fit into the equation. Thus, the kinetic parameters given in Table 1 were obtained excluding the data points in the inhibition part of the plot and using the “conventional nonlinear Michaelis-Menten equation” of the Leonora enzyme kinetics program.
4-Arylalkyl-1H-imidazoles, of which levomedetomidine showed the highestVmax/Kmratio of 1.9, showed considerably lower efficiency for dog liver microsomal UGTs compared with 4-nitrophenol and 4-aminobiphenyl, withVmax/Kmratios of 725 and 27, respectively.
Glucuronidation of medetomidine was enantioselective also in dog liver microsomes, with theVmax/Kmvalue of levomedetomidine being 8-fold higher compared with dexmedetomidine. Unlike with human liver microsomes, only one glucuronide product of dexmedetomidine (, 7.7 min) was detected (Fig.2b). Similarly, only one glucuronide conjugate (tR, 7.1 min) was formed from levomedetomidine.
Kinetics of Glucuronidation in Rat Liver Microsomes.
Rat liver microsomes formed N-glucuronides of 4-arylalkyl-1H-imidazoles at a very low rate (Table 1); only MPV-295 A IV and atipamezole glucuronides were formed at a detectable level. Amitriptyline and nortriptyline were not glucuronidated at detectable levels. The only substrate forming anN-glucuronide at a sufficient rate to determine kinetic parameters was 4-aminobiphenyl, but it was not a very good substrate compared with 4-nitrophenol, which was a 61-fold better substrate in terms ofVmax/Kmratio.
Detergent Activation of Liver Microsomal UGTs.
Triton X-100 was added to the incubation mixture to fully activate liver microsomal UGTs. The optimal detergent concentration was determined separately for each enzyme source and substrate before determining the kinetic parameters. Optimal Triton X-100 concentrations varied between 0 to 0.05 mg/ml, corresponding to Triton/protein ratios (mg/mg) between 0 to 1 (data not shown). Generally, glucuronidation of 4-arylalkyl-1H-imidazoles in human liver microsomes was strongly activated, up to 5-fold, by the detergent (Fig.3a). In contrast, Triton X-100 had no or only a minor effect on the glucuronidation of 4-arylalkyl-1H-imidazoles in dog liver microsomes (Fig. 3b).

Triton X-100 strongly activated glucuronidation of MPV-207 A IV in (A) human liver microsomes (protein concentration, 0.1 mg/ml; [S], 100 μM) but had a minor effect on (B) dog liver microsomes (protein concentration, 0.2 mg/ml; [S], 500 μM).
Glucuronidation Activity in Expressed Human UGT Isoenzymes.
Scopoletin, 4-aminobiphenyl, 4-nitrophenol, and entacapone were used as model substrates to measure glucuronidation activity in expressed human UGT1A3, UGT1A4, UGT1A6, and UGT1A9 isoenzymes, respectively (Table2). The 4-arylalkyl-1H-imidazoles studied (levomedetomidine, atipamezole, and detomidine) were not substrates for human UGT1A3, UGT1A6, or UGT1A9. Human UGT1A4 formed N-glucuronides of these 4-arylalkyl-1H-imidazoles, but the glucuronidation rates were low, below 0.015 nmol/min/mg of protein.
Glucuronidation activity in expressed human UGT isoenzymes
Discussion
Human liver microsomes glucuronidated 4-arylalkyl-1H-imidazoles at a very high efficiency; theVmax/Kmratios (e.g., for atipamezole and levomedetomidine) were 256 and 117, respectively, compared with aVmax/Kmratio of 106 for 4-nitrophenol, a well characterized UGT substrate forming an O-glucuronide (Table 1). From the high efficiencies and high affinities (lowKm), it can be predicted thatN-glucuronidation is a major metabolic route in the elimination of these 4-arylalkyl-1H-imidazoles in humans. This is an important finding because these compounds are glucuronidated without any prior phase I metabolism. Preliminary in vivo studies with dexmedetomidine support the importance of this metabolic route in humans.
Dog liver microsomes also formed glucuronides of 4-arylalkyl-1H-imidazoles. TheVmax/Kmratios were 0.5 and 1.9 for atipamezole and levomedetomidine, respectively, compared with 726 for 4-nitrophenol (Table 1).
Rat liver microsomes formed glucuronides of 4-arylalkyl-1H-imidazoles at a very low rate; only atipamezole and MPV-295 A IV glucuronides were formed at detectable levels. Our results with 4-arylalkyl-1H-imidazoles, which are C4-imidazole compounds, are in good agreement with the findings ofHuskey et al. (1993, 1994), who studied species differences inN-glucuronidation of methylbiphenyltetrazole, -triazole, and -imidazole compounds. They concluded that human UGT(s) responsible forN-glucuronidation preferentially conjugated methylbiphenyl-C4-imidazole (and methylbiphenyl-1,2,4-triazole), whereas rat UGTs glucuronidated methylbiphenyl-C4-imidazole at a very low rate.
Among 4-arylalkyl-1H-imidazoles, atipamezole showed the highest affinity for human liver microsomal UGTs, with an exceptionally low apparent Km of 4.0 μM, and levomedetomidine showed the highest capacity, with an apparentVmax of 1.89 nmol/min/mg of protein (Table 1). Dog liver microsomes formed N-glucuronides of 4-arylalkyl-1H-imidazoles at 5- to 100-fold lower affinity (higher Km) than human liver microsomes. Levomedetomidine was the best substrate with aKm of 279 μM, and MPV-207 AIV showed a Vmax of 0.73 nmol/min/mg of protein (Table 1). At high substrate concentrations (>1 mM), glucuronidation of 4-arylalkyl-1H-imidazoles and 4-aminobiphenyl in dog liver microsomes showed substrate inhibition, making the reliable estimation of kinetic parameters difficult.
In principle, either of the imidazole ring nitrogens (Fig. 1) of the studied 4-arylalkyl-1H-imidazoles could be glucuronidated. However, N-glucuronidation of these compounds seemed to be regiospecific, and only a single glucuronide product of each substrate was observed, with one exception (dexmedetomidine). Huskey et al. (1994) discovered in their study using NMR spectroscopy that in human liver microsomes the favored N-glucuronidation site in C4-methylbiphenyl imidazole was the nitrogen located two bonds away from the substituted carbon (i.e., N1). The nitrogen located next to the substituted carbon (i.e., N3 was glucuronidated at a lower rate). Moreover, C2-methylbiphenyl imidazole, in which both nitrogens are immediate neighbors of the substituted carbon, was not glucuronidated. The 4-arylalkyl-1H-imidazoles in our study are C4-substituted imidazoles; therefore, it is possible that their glucuronidation occurs preferentially in the N1 nitrogen (Fig. 1). A plausible explanation for this is that the orientation leading toN3-glucuronidation is sterically hindered, resulting in highKm binding in the active site of the enzyme. The formation of both N1- andN3-glucuronides of these 4-arylalkyl-1H-imidazoles cannot, however, be excluded since the C18 column used in our HPLC system may have not separated them. Confirming the exact position of glucuronidation would require NMR spectroscopy.
The glucuronidation of medetomidine by human liver microsomes was highly stereospecific; levomedetomidine showed a 60-fold apparent specificity constantVmax/Km(which gives the relative rates of the reactions in an equimolar mixture) compared with dexmedetomidine. The lower efficiency of dexmedetomidine glucuronidation was mainly due to higher aKm value; dexmedetomidine showed the lowest binding affinity of all 4-arylalkyl-1H-imidazoles. Dexmedetomidine produced two N-glucuronide products when it was incubated with human liver microsomes (Fig. 2), whereas all the other 4-arylalkyl-1H-imidazoles produced only one. The lack of regioselectivity in the case of dexmedetomidine glucuronidation may be explained by comparable (low) affinity for both binding modes (N1 and N3). The regioselectivity of N-glucuronidation for all the other 4-arylalkyl-1H-imidazoles may be caused by one high-affinity (N1) and one low-affinity (N3) binding mode, leading to the detection of only one glucuronide product (N1).
Triton X-100 was used as a detergent to activate liver microsomal UGTs of the three species. If too low of a detergent concentration is used, the active sites of UGTs located in the lumen of the endoplasmic reticulum are not fully revealed, whereas excessively high concentrations tend to denature the protein. Therefore, an optimal detergent concentration was determined separately for each microsome preparation and substrate before kinetic characterization. The optimal Triton X-100 concentration for 4-arylalkyl-1H-imidazoles to activate liver microsomal UGTs of the three species varied between 0 to 0.05 mg/ml (Triton/protein ratio (mg/mg) 0–0.5), which was in good agreement with previous studies, the optimum normally being between 0.2 to 1 Triton/protein ratio (Winsnes, 1969; Bansal and Gessner, 1980; Liu and Franklin, 1984; Lett et al., 1992).
The human UGT1A subfamily isoforms reported to catalyzeN-glucuronidation are UGT1A3, UGT1A4, UGT1A6, and UGT1A9 (Orzechowski et al., 1994; Green and Tephly, 1996; Green et al., 1998). All of these isoenzymes were tested in our study for their ability to glucuronidate 4-arylalkyl-1H-imidazoles. Only UGT1A4 formed glucuronides of these compounds at a low rate, between 0.007 and 0.014 nmol/min/mg of protein (Table 2). Glucuronidation activity of human UGT1A4 for 4-aminobiphenyl was 25-fold higher compared with levomedetomidine, yet the difference was only 2.7-fold whenVmax values of these compounds were compared in human liver microsomes. Therefore, it seems likely that in addition to UGT1A4, some other UGT isoenzyme(s) contribute to the glucuronidation of 4-arylalkyl-1H-imidazoles in human liver. Detectable amounts of glucuronides were not formed from 4-arylalkyl-1H-imidazoles by human expressed UGT1A3, UGT1A6, or UGT1A9. However, the involvement of UGT1A3 in the glucuronidation of 4-arylalkyl-1H-imidazoles cannot be excluded since the activity for scopoletin in the insect cell-expressed UGT1A3 used in our study was very low compared with a previous study on UGT1A3 (Green et al., 1998).
In conclusion, N-glucuronidation of several structurally related 4-arylalkyl-1H-imidazoles was elucidated by rat, dog, and human liver microsomes. Significant species differences were observed; glucuronidation occurred at high rate and at the highest affinity in human liver microsomes, but the dog also formed glucuronide conjugates of these compounds, albeit at a lower affinity. The rat formed N-glucuronides of 4-arylalkyl-1H-imidazoles at a very low rate. Further studies are needed to reveal which human UGT isoenzymes, in addition to UGT1A4, are responsible for the N-glucuronidation of these compounds.
Footnotes
-
This work was presented in part at the Drug Metabolism Workshop and International Society for the Study of Xenobiotics Meeting, June 11–16, 2000, St. Andrews, Scotland. The abstract was published inDrug Metab Rev32:26.
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- UDPGA
- UDP-glucuronic acid
- MPV-207 A IV
- 4-(2,6-dimethylphenyl)methyl-1H-imidazole
- MPV-295 A IV
- 4-(2,6-dimethylphenyl)ethyl-1H-imidazole
- HPLC
- high-performance liquid chromatography
- tR
- total retention time in HPLC
- Received June 7, 2001.
- Accepted December 5, 2001.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}