


Abstract
Paroxetine, a selective serotonin reuptake inhibitor, is a potent inhibitor of cytochrome P450 2D6 (CYP2D6) activity, but the mechanism of inhibition is not established. To determine whether preincubation affects the inhibition of human liver microsomal dextromethorphan demethylation activity by paroxetine, we used a two-step incubation scheme in which all of the enzyme assay components, minus substrate, are preincubated with paroxetine. The kinetic parameters of inhibition were also estimated by varying the time of preincubation as well as the concentration of inhibitor. From these data, a Kitz-Wilson plot was constructed, allowing the estimation of both an apparent inactivator concentration required for half-maximal inactivation (KI) and the maximal rate constant of inactivation (kINACT) value for this interaction. Preincubation of paroxetine with human liver microsomes caused an approximately 8-fold reduction in the IC50 value (0.34 versus 2.54 μM). Time-dependent inhibition was demonstrated with an apparent KI of 4.85 μM and an apparent kINACT value of 0.17 min−1. Spectral scanning of CYP2D6 with paroxetine yielded an increase in absorbance at 456 nm suggesting paroxetine inactivation of CYP2D6 via the formation of a metabolite intermediate complex. This pattern is consistent with the metabolism of the methylenedioxy substituent in paroxetine; such substituents may produce mechanism-based inactivation of cytochrome P450 enzymes. In contrast, quinidine and fluoxetine, both of which are inhibitors of CYP2D6 activity, did not exhibit a preincubation-dependent increase in inhibitory potency. These data are consistent with mechanism-based inhibition of CYP2D6 by paroxetine but not by quinidine or fluoxetine.
Although CYP2D6 constitutes a relatively minor fraction of the total hepatic P4501 content (Shimada et al., 1994), the contribution of this isoform is significant due to its role in the metabolism and clearance of many therapeutic agents that target the cardiovascular and central nervous system. In addition, clinically significant polymorphisms in the CYP2D6 gene have been identified in a variety of populations with altered metabolic activity. The majority of poor metabolizers, or individuals with impaired enzyme function, are accounted for by frame shift deletions, substitutions resulting in splicing defects, or gene deletions (van der Weide and Steijns, 1999;Bertilsson et al., 2002). Extensive metabolizers, or individuals with normal enzyme function, are heterozygous or homozygous for the wild-type allele. In vivo clearance of CYP2D6 substrates in poor metabolizers is generally much lower than in extensive metabolizers, leading to higher plasma concentrations and the potential for clinical toxicities with therapeutic doses (Bertilsson et al., 2002).
Paroxetine is a selective serotonin reuptake inhibitor with nonlinear kinetics that is both a substrate for and an inhibitor of CYP2D6 (Greenblatt et al., 1999; Belpaire et al., 1998; Otton et al., 1996;Bloomer et al., 1992; Sindrup et al., 1992a,b). Paroxetine is metabolized by CYP2D6 via demethylenation of the methylenedioxy group, yielding a catechol metabolite and formic acid (Haddock et al., 1989;Bloomer et al., 1992). Paroxetine inhibits CYP2D6 activity at IC50 concentrations ranging from 150 nM to 2.0 μM, depending on the substrate (Crewe et al., 1992; von Moltke et al., 1995; Fogelman et al., 1999). Although not investigated directly, paroxetine has been regarded as a competitive, reversible inhibitor of CYP2D6 (Otton et al., 1996).
We previously observed that the in vitroKi for paroxetine versus desipramine hydroxylation yielded an underestimate of the degree of desipramine clearance inhibition when desipramine was coadministered with paroxetine in a clinical study (von Moltke et al., 1995; Alderman et al., 1997). We accounted for this discrepancy on the basis of extensive partitioning of paroxetine from plasma into hepatic tissues such that intrahepatic concentrations substantially exceeded total plasma concentrations (von Moltke et al., 1995). Hemeryck et al. (2000, 2001) observed similar discrepancies, in which in vitro inhibition of metoprolol hydroxylation by paroxetine yielded an underestimate of the in vivo inhibition of metoprolol clearance by cotreatment with paroxetine. They suggest that the discrepancy could be explained by mechanism-based inhibition (MBI) of CYP2D6, leading to an in vitro determination of Ki that underestimates the actual inhibitory potency in vivo.
MBIs differ from reversible inhibitors in that they require enzymatic activation by the target protein prior to exerting an inhibitory effect. This initial activation step leads to the formation of active inhibitor, often referred to as the metabolite intermediate complex (MIC). The MIC can then exert its inhibitory effect by 1) forming a direct covalent interaction, 2) forming a noncovalent tight binding complex, or 3) forming an inactive enzyme product that is released from the inhibitor (Silverman, 1995). In vitro models of MBIs typically exhibit the following characteristics: 1) time-dependent loss of enzyme activity, 2) saturation kinetics, 3) inhibition that is dependent on initial enzyme activity, and 4) a 1:1 stoichiometry for inhibitor and enzyme (Ito et al., 1998; Venkatakrishnan et al., 2001). Moreover, by comparing the effect of preincubation on IC50values, in vitro studies investigating MBIs have demonstrated preincubation-dependent increase in inhibitor potency (Jones et al., 1999; Mayhew et al., 2000). However, other types of inhibition, via the production of nonspecific toxic intermediates or potent inhibitory metabolites, may show similar time-dependent characteristics. Two principal kinetic constants that are specific for MBIs are the maximal rate constant of inactivation (kINACT) and the inactivator concentration required for half-maximal inactivation (KI) (Silverman, 1995). Although these values are experimentally derived, their definitions are based on rate constants for each enzymatic sequence of inhibitor activation and enzyme inactivation (Silverman, 1995). Note that theKI is not equivalent to the inhibition constant (Ki) that is applicable to reversible inhibitors.
In this study, we investigated paroxetine as a mechanism-based inhibitor of CYP2D6 using a two-step incubation scheme to measure the effect of preincubation with paroxetine on dextromethorphan demethylation activity in human liver microsomes. Dextromethorphan was selected as the substrate because CYP2D6 is the dominant isoform in the formation of dextrorphan via O-demethylation reaction (Kerry et al., 1994; von Moltke et al., 1998a). Results were compared with those obtained for quinidine and fluoxetine, two potent CYP2D6 inhibitors that do not demonstrate mechanism-based inhibition (von Moltke et al., 1994; Cheer and Goa, 2001).
Materials and Methods
Materials.
Paroxetine hydrochloride was kindly provided by GlaxoSmithKline (Research Triangle Park, NC). All other reagents were obtained from commercial sources or were kindly provided by their manufacturers. Liver samples were obtained from either the National Disease Research Interchange (Philadelphia, PA) or the Liver Tissue Procurement and Distribution Services (Minneapolis, MN). Human liver microsomes (HLMs) were prepared as previously described and stored at −80°C until used (von Moltke et al., 1994). HLMs used for all experiments were selected due to their high metabolism of CYP2D6 substrates, but CYP2D6 genotype determination was not done.
Preincubation-Dependent Inhibition Using Human Liver Microsomes.
To investigate the effect of preincubation on paroxetine inhibition of dextromethorphan demethylation activity in vitro, various concentrations of paroxetine (0.0, 0.5, 1.0, 2.5, 5.0, 10.0, and 25.0 μM final concentrations) were added to incubation vials containing the necessary cofactors (a reduced NADPH-regenerating system) and human liver microsomal protein (n = 4). Reaction mixtures were constructed such that one set of vials had enzyme and paroxetine present during the preincubation, while a separate set were constructed with enzyme alone during the preincubation. Aliquots of each preincubation reaction were then transferred in a 1:1 dilution to separate incubation vials containing fresh cofactors and dextromethorphan (final concentration 25 μM), thus allowing the detection of any effect on enzymatic activity as a consequence of preincubation with inhibitor. Reactions were initiated with the addition of human liver microsomal protein (final concentration 0.25 μg/μl) and were preincubated (without substrate) for 20 min in a rotating 37°C water bath. All incubations were performed in duplicate. Reactions were allowed to incubate for an additional 20 min as previously described and then terminated by the addition of cold acetonitrile and kept on ice. These experiments were compared with duplicate assays in which various concentrations of paroxetine (as described above) were added to the second incubation vial instead of the preincubation vial. The internal standard (25 μl of a 0.25 μg/μl pronethalol solution) was added to each reaction vial. Incubation vials were centrifuged at 14,000g for 10 min (Sorvall, Newton, CT), and then transferred to high performance liquid chromatography (HPLC) tubes. HPLC was performed using a 3000 × 3.9 mm μBondapak C18 column (Waters, Milford, MA) with a mobile phase of 70% 50 mM KH2PO4 and 30% acetonitrile (pH 6.0) at a flow rate of 1.5 ml/min. Fluorescence detection was performed with wavelengths of 280 and 310 nm (excitation and emission). All incubations were performed in duplicate. This experiment was replicated using quinidine (0, 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, and 25.0 μM) and fluoxetine (0, 0.25, 0.5, 1.0, 2.5, 5.0, 10.0, and 25.0 μM) as control inhibitors for each liver sample (n = 4).
Time-Dependent Incubations Using Human Liver Microsomes.
To investigate the possible time-dependent inactivation of CYP2D6 activity, we used a two-step incubation scheme to assay paroxetine inhibition of dextromethorphan demethylation. During the preincubation period, mixtures containing paroxetine (0, 0.25, 0.5, 1.0, or 2.0 μM) and human liver microsomes (2.5 μg/μl final concentration) were assembled as described above and incubated in a 37°C water bath (0, 2, 4, 6, 8, 10, and 20 min, n = 4). Aliquots of each reaction were then transferred in a 1:10 dilution to separate incubation tubes containing fresh cofactors and dextromethorphan (25 μM final concentration). Each concentration tested was performed in duplicate. Reactions were allowed to incubate for an additional 20 min as described above, then by addition of cold acetonitrile and kept on ice. The internal standard (pronethalol) was added to each reaction vial. All remaining sample processing and HPLC analysis were then completed as described above. This experiment was replicated using quinidine (2.5 μM) as a control inhibitor for each liver sample (n = 4).
Spectral Complex Formation.
Purified CYP2D6 (250 pmol in 16 μl) was reconstituted with NADPH/cytochrome P450 reductase (250 pmol in 4.3 μl) and dilaurylphosphatidycholine (1 mg/ml presonicated suspension; 5.0 μl). After approximately 5 min, a solution of paroxetine in potassium buffer, pH 7.4, was added. Final concentrations of paroxetine and phosphate buffer were 10 μM and 100 mM, respectively. The mixture was split into two 0.5-ml cuvettes of 1-cm path length. To the reference cuvette was added water and to the sample cuvette was added an NADPH regeneration system comprised of NADPH (0.5 mM), isocitric acid (6.2 mM), isocitrate dehydrogenase (5 U/ml) and MgCl2(9.0 mM). A difference spectrum was taken scanning between 500 and 400 nm at 0, 1.5, 3, 6, 9, 12, 15, 20, 25, and 30 min at ambient temperature. The amount of spectral complex formed was calculated using an extinction coefficient of 75 mM−1cm−1 for the difference between 456 and 490 nm.
Data Analysis.
For all HPLC analysis, peak heights of the metabolite (dextorphan) were expressed as a ratio to the internal standard (pronethalol) peak height for each concentration of inhibitor. These peak height ratios represent the remaining demethylation activity in the HLM and were expressed as a percentage of the time-matched control samples without inhibitor. This procedure takes into account the anticipated time-related loss of enzyme activity under the incubation conditions.
In the preincubation studies using a fixed concentration of dextromethorphan (25 μM) and varying concentrations of inhibitor, apparent IC50 values for fluoxetine, paroxetine, and quinidine were determined by nonlinear regression analysis of the data (Sigma Plot software, SPSS Science Inc. Chicago, IL) as previously described (Venkatakrishnan et al., 1998). Values were calculated using mean values for each concentration of inhibitor or by averaging IC50 values obtained from each separate liver sample.
The calculation of the kinetic constants, which describe mechanism-based enzyme inactivators, was performed as described (Kitz and Wilson, 1962; Palamanda et al., 2001). Briefly, the relative inhibition of dextrorphan formation was determined by comparing the means of peak height ratios of time-matched samples with paroxetine versus control samples without paroxetine. These logarithm of relative inhibition values were plotted versus preincubation time for each concentration of paroxetine used, and the slopes determined by linear regression. These slope values represent the observed inactivation rate constants, which were used to calculate the half-life of the inactivation reaction. A Kitz-Wilson plot was then constructed using the calculated half-life values (y-axis) and the reciprocal of the associated inhibitor concentration (x-axis) (Kitz and Wilson, 1962). The apparent KI andkINACT values were determined from the reciprocal of the y-axis intercept and the negative reciprocal of the x-axis intercept of the Kitz-Wilson plot, respectively.
Results
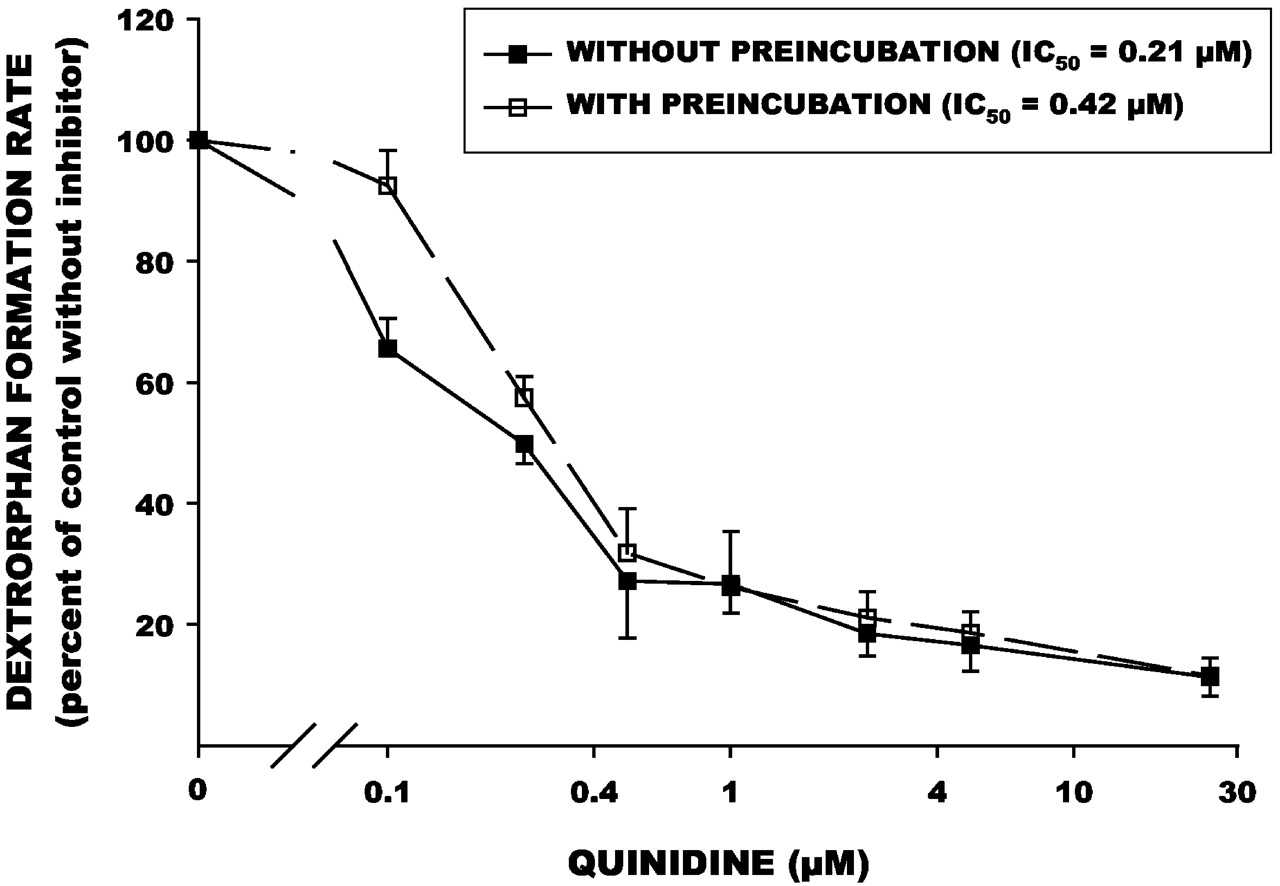
Paroxetine, fluoxetine, and quinidine were all potent inhibitors of dextromethorphan demethylation activity (Figs.1-3, Table 1). A mechanism-based component was evident for paroxetine, as the IC50 value was significantly reduced (i.e., more potent inhibition) when the inhibitor was preincubated with human liver microsomes (Fig. 1). In contrast, the IC50 values for quinidine and fluoxetine were not reduced by preincubation (Figs. 2 and 3), suggesting that this interaction is not mechanism-based. For three of the liver samples incubated with quinidine and fluoxetine, the IC50values were actually higher (i.e., less potent inhibition); this may be due to the consumption of inhibitor by other enzymes present in the HLM preparations.
Effect of preincubation on apparent IC50values for paroxetine, fluoxetine, and quinidine versus dextromethorphan O-demethylation in vitro

Effect of preincubation on inhibition of dextromethorphan O-demethylation by paroxetine in human liver microsomes in vitro.
Dextrorphan formation rates with inhibitor present are expressed as a percentage of the control rate without inhibitor. Each point is the mean (± S.E.) of values determined in four separate human liver samples. IC50 values were determined by nonlinear regression.

Effect of preincubation on inhibition of dextromethorphan O-demethylation by quinidine in human liver microsomes in vitro.
Dextrorphan formation rates with inhibitor present are expressed as a percentage of the control rate without inhibitor. Each point is the mean (± S.E.) of values determined in four separate human liver samples. IC50 values were determined by nonlinear regression.

Effect of preincubation on inhibition of dextromethorphan O-demethylation by fluoxetine in human liver microsomes in vitro.
Dextrorphan formation rates with inhibitor present are expressed as a percent of the control rate without inhibitor. Each point is the mean (± S.E.) of values determined in four separate human liver samples. IC50 values were determined by nonlinear regression.
Dextrorphan formation was inhibited by increasing the preincubation time of paroxetine with HLMs in a concentration-dependent manner (Fig.4). In contrast, inhibition of dextrorphan formation by quinidine was not influenced by preincubation time and showed a slight loss of inhibitory effect with increased preincubation time. The Kitz Wilson plot of mean data points (Fig.5) indicated aKI value of 4.85 μM, and akINACT value of 0.17 min−1. These were similar to the mean values of 6.6 (± 2.7) μM and 0.25 (± 0.09) min−1across the four liver samples.
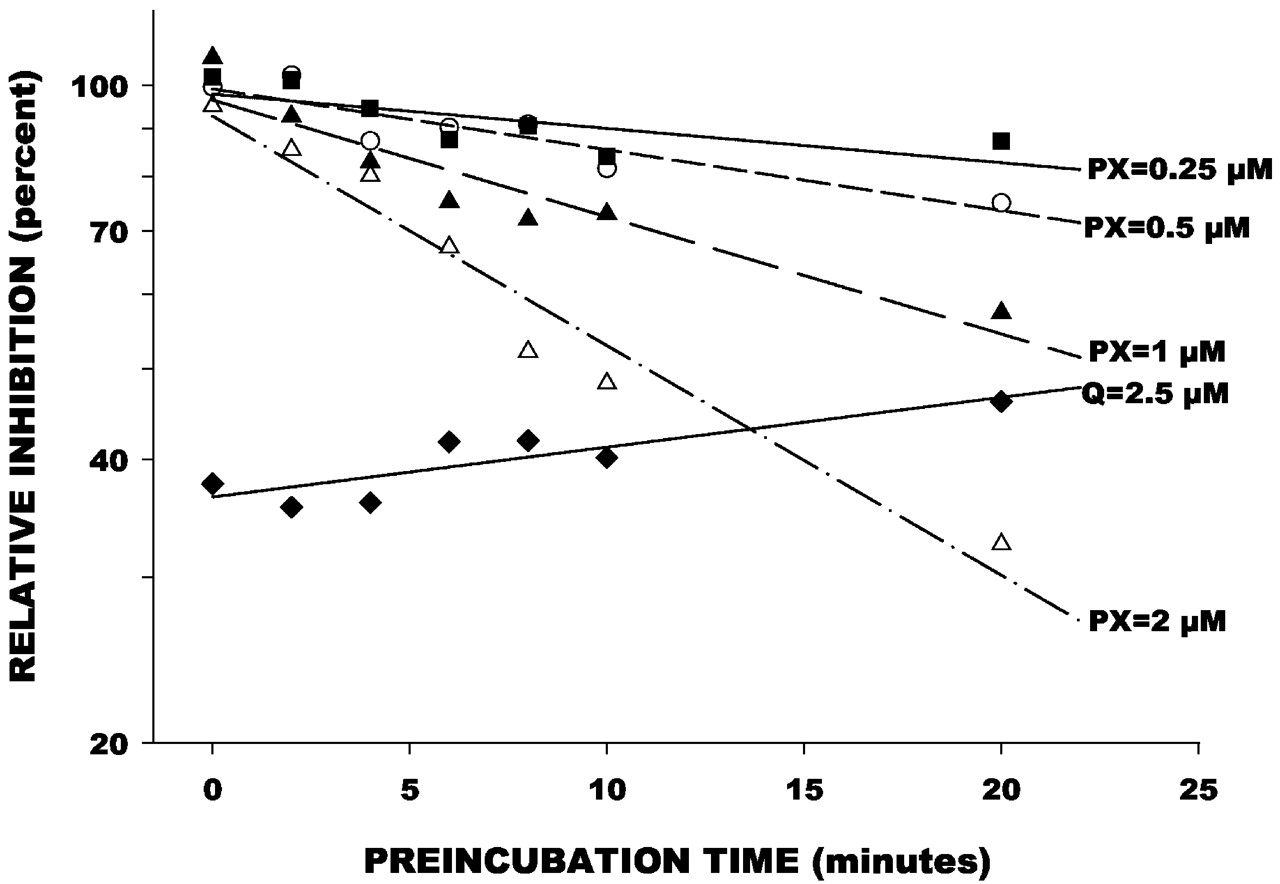
Effect of preincubation time and paroxetine (PX) concentration on inhibition of dextrorphan formation from dextromethorphan.
Data points represent mean (n = 4) reaction velocities with preincubation of paroxetine with microsomes, expressed as a percentage of the control reaction velocity determined when microsomes were not preincubated with paroxetine. Lines were determined by linear regression of time versus logarithm of reaction velocity ratio. Also shown are data from a parallel study of quinidine (Q) at 2.5 μM.
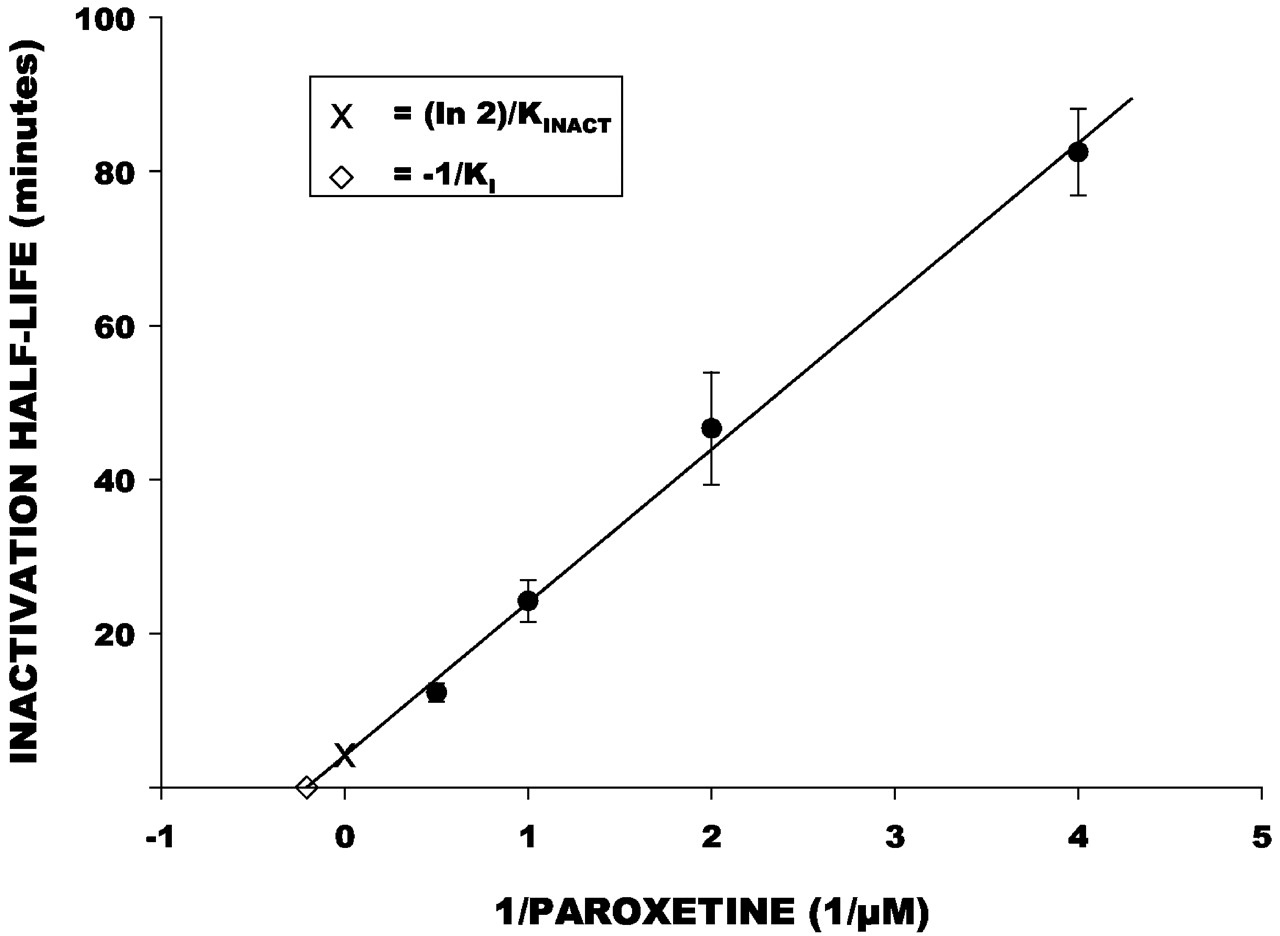
Kitz-Wilson plot of inactivation half-life (y-axis) versus reciprocal of paroxetine concentration (x-axis).
Each point (closed circles) is the mean (± S.E.) from four human liver microsomal preparations. The line represents the function determined by linear regression analysis (r-square = 0.99). The y-axis intercept (indicated by “x”) represents the estimated minimum inactivation half-life, which was used to calculate kINACT. Thex-axis intercept (indicated by an open diamond) represents the negative reciprocal of KI.
Spectral difference scanning of CYP2D6 upon incubation of paroxetine yielded an increase in absorbance at 456 nm over the incubation period, along with development of a shoulder peak at 430 nm (Fig.6A). Calculation of the concentration of MIC yielded the result that 95 pmol/ml was generated after 30 min. The rate of appearance of the 456 nm peak is shown in Fig. 6B. Regression of the data gathered over the first 15 min yielded a rate of MIC formation of 4.4 pmol/ml/min.

A, difference spectrum showing formation of an MIC generated upon incubation of paroxetine (10 μM) with human CYP2D6 (0.25 μM) reconstituted with NADPH/P450 reductase (0.25 μM) and phospholipid and NADPH (0.5 mM) at ambient temperature; scans were conducted at baseline, t = 0, 1.5, 3, 6, 9, 12, 15, 20, 25, and 30 min; the maximum was observed at 456 nm, with a shoulder at 430 nm; B, time course monitoring of the increase in absorbance at λ = 456 nm when paroxetine was incubated with CYP2D6 as above; points represent individual readings taken every 10 s.
Discussion
In these studies, we present data consistent with a mechanism-based component for the inhibition of CYP2D6 by paroxetine. Preincubation of paroxetine with HLMs increased the inhibitory potency of this interaction, which is strongly suggestive of a mechanism-based inactivation (Silverman, 1995). The methylenedioxy substituent of paroxetine may explain its capacity to form an inhibitory complex with CYP2D6 (Lin et al., 1996; Wu et al., 1997). The metabolism of methylenedioxy groups results in the generation of carbene intermediates, which can form a strong covalent complex with the iron center of the heme in P450 (Ortiz de Montellano and Correia, 1995). Alternatively, the catechol product of methylenedioxy demethylenation can be further oxidized to an ortho quinone, which can react with nucleophilic groups on macromolecules, as exemplified by metheylenedioxymethamphetamine (Wu et al., 1997) and safrole (Bolton et al., 1994). Our values for KI andkINACT for paroxetine in HLM are comparable to those of SCH66712 (KI = 4.8 μM, kINACT = 0.14 min−1), another reported mechanism-based inhibitor of CYP2D6 activity (Palamanda et al., 2001). There is no evidence to suggest that CYP2D6 inhibition by quinidine or fluoxetine is mechanism-based in nature, and our results using the two-step incubation scheme confirm this.
The methylenedioxyphenyl substituents can form MIC with cytochrome P450 enzymes (Franklin, 1971). Such complexes exhibit a characteristic absorbance peak at 456 nm, with a secondary peak at 430 nm, and are referred to as a type 3 binding spectra. P450-catalyzed metabolism of methylenedioxyphenyl groups results in initial hydroxylation of the methylene carbon. This unstable intermediate can partition between demethyleneation yielding a catechol metabolite and formaldehyde and dehydration to a carbene. The carbene intermediate can complex to the heme iron in P450 to yield the characteristic type 3 spectrum (Ortiz de Montellano and Correia, 1995). Paroxetine possesses a methylenedioxyphenyl substituent and is metabolized to a catechol metabolite (Haddock et al., 1989). This finding supports the mechanism of paroxetine inactivation of CYP2D6 as occurring via formation of a carbene-heme MIC.
Plasma concentrations of paroxetine with usual clinical doses generally are less than 0.5 μM (Sindrup et al., 1992a). Nonetheless usual doses and plasma levels of paroxetine can produce extensive inhibition of clearance of coadministered drugs that are substrates for CYP2D6 (Brøsen et al., 1993; Alderman et al., 1997; Özdemir et al., 1998; Alfaro et al., 1999). Approaches to predicting clinical pharmacokinetic drug interactions based on in vitro data continue to be controversial (Bertz and Granneman, 1997; Ito et al., 1998; von Moltke et al., 1998b; Venkatakrishnan et al., 2001). Plasma binding theoretically could limit drug access to hepatic tissue, thus limiting the overall inhibitory effect on hepatic P450s. Yet hepatic uptake of paroxetine, as well as other inhibitors, may greatly exceed unbound as well as total plasma concentrations (von Moltke et al., 1994, 1995,1998b; Greenblatt et al., 1996; Yamano et al., 1999). Mechanism-based inhibition may further complicate in vitro-in vivo scaling, and investigations of paroxetine and CYP2D6 activity that do not account for time-dependent inhibition may underestimate the inhibitory potency in vivo and underpredict the actual magnitude of pharmacokinetic drug interactions involving CYP2D6 substrates.
Footnotes
-
This work was supported by Grants MH-58435, DA-13209, DK/AI-58496, DA-05258, DA-13834, AG-17880, MH-34223, MH-01237, and RR-00054 from the Department of Health and Human Services.
- Abbreviations used are::
- P450
- cytochrome P450
- MBI
- mechanism-based inhibition
- MIC
- metabolite intermediate complex
- kINACT
- the maximal rate constant of inactivation
- KI
- the inactivator concentration required for half-maximal inactivation
- HLM
- human liver microsome
- HPLC
- high performance liquid chromatography
- Received September 3, 2002.
- Accepted November 27, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}