


Abstract
Rosiglitazone and pioglitazone are thiazolidinediones used for treatment of noninsulin-dependent diabetes mellitus. These compounds, along with troglitazone, were evaluated for the ability to induce cytochrome P450 enzymes (P450) in primary human hepatocyte cultures and to inhibit P450 in human microsomes. In induction studies, all three thiazolidinediones caused a dose-dependent increase in CYP3A4 activity and immunoreactive protein. While troglitazone was the most potent, rosiglitazone and pioglitazone generally exceeded troglitazone in absolute CYP3A4 activity achieved at concentrations ≥10 μM. A comparable concentration-dependent increase in CYP2B6 immunoreactive protein was observed with all three thiazolidinediones. Microarray analysis revealed rifampin > troglitazone > pioglitazone > rosiglitazone in terms of CYP3A4 mRNA induction potential with 10 μM compound. Inhibition studies conducted for CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP2A6, and CYP2E1 showed troglitazone to be the most nonselective and potent inhibitor followed by rosiglitazone and pioglitazone. In vitro, the thiazolidinediones were strong inhibitors of CYP2C8, withKi values between 1.7 and 5.6 μM, and of CYP3A4, with Ki values between 1.6 and 11.8 μM. Troglitazone, in addition, inhibited CYP2C9 (Ki 0.6 μM). Although the inhibitory effects of the thiazolidinediones have not been demonstrated clinically, our results suggest there is potential for interactions with CYP2C8 substrates. This is the first report of in vitro induction of P450 enzymes by rosiglitazone and pioglitazone. While only the induction of CYP3A4 by troglitazone has been demonstrated in vivo, these results suggest that other thiazolidinediones may have the potential to cause clinically significant drug interactions at sufficiently high doses.
The class of drugs known as the thiazolidinediones are widely used as part of antidiabetic treatments (Peters, 2001). These agents act by targeting insulin resistance instead of stimulating insulin secretion by interacting with the gamma subtype of the peroxisome proliferator-activated receptor (PPAR-γ2). PPAR-γ, a member of the nuclear receptor subfamily, stimulates gene expression of proteins involved in glucose metabolism (Lehmann et al., 1998). This results in an increase in insulin sensitivity in skeletal muscle, liver, and adipose tissues (Kumar et al., 1996). The first drug in this class to be approved in the United States was troglitazone in 1997. While troglitazone offered significant clinical benefits to many diabetic patients, it was associated with an elevation of serum alanine aminotransferase in approximately 1 to 2% of patients and in rare cases, hepatic failure and was therefore withdrawn from the market in 2000. Subsequent to the launch of troglitazone, two other thiazolidinedione replacement drugs entered the market, rosiglitazone and pioglitazone. Although limited, there are reports of liver toxicity associated with rosiglitazone and pioglitazone (Al Salman et al., 2000;Forman et al., 2000; Gouda et al., 2001; Maeda, 2001; May et al., 2002). A major distinguishing factor in the clinical regimen between these thiazolidinedione is the dose necessary for efficacy. While troglitazone was administered at 200 to 600 mg/day, the dose for rosiglitazone is 4 to 8 mg/day and that of pioglitazone is 45 mg/day.
Many clinically relevant drug interactions are the result of induction or inhibition of major drug-metabolizing enzymes. In vitro, the three thiazolidinediones are inhibitors of CYP2C enzymes (Yamazaki et al., 2000). However, there are no reports on in vitro induction of P450 enzymes with rosiglitazone and pioglitazone. It has been reported that drugs that clinically induce CYP3A4 are typically given at high doses (Smith, 2000), and this class of compounds seems to validate the hypothesis. Troglitazone has been associated with significant clinical drug interactions due to induction, particularly with compounds known to be substrates for CYP3A4 (Loi et al., 1998a,b, 1999). There are no reports on clinical induction of P450 enzymes by rosiglitazone or pioglitazone to date.
In these studies we examined both the P450 induction and inhibition potential for troglitazone, rosiglitazone, and pioglitazone. Primary cultures of human hepatocytes were used as the model system for assessing the induction potential because these cells remain differentiated and retain the major drug metabolizing enzymes for several days (LeCluyse et al., 2000; Sahi et al., 2000). For the present studies, in vitro induction of CYP3A4 and CYP2B6 was characterized by determining mRNA, protein levels, and enzyme activities in primary human hepatocytes. This is the first report on the in vitro induction of P450 enzymes by rosiglitazone and pioglitazone. Also in this paper, the results of a comprehensive analysis of the in vitro inhibition of troglitazone, rosiglitazone, and pioglitazone are reported. The enzyme inhibition potential of the three thiazolidinediones was characterized (IC50) for eight major human P450 isoforms and where substantial inhibition was observed, a Ki determination was performed. The relevance of these findings to published clinical results also is addressed.
Materials and Methods
Rosiglitazone, pioglitazone, and troglitazone were obtained from Parke Davis Pharmaceutical Research (Ann Arbor, MI). Human liver microsomes (pool of at least fifteen donors) were obtained from Xenotech, LLC (Kansas City, KS). ITS+ (insulin, transferrin, selenium, linoleic acid, and bovine serum albumin supplement), hepatostim culture media and matrigel were purchased from Collaborative Biomedical Research (Bedford, MA). Collagenase type IV was from Sigma-Aldrich (St. Louis, MO), and collagen, type I (Vitrogen), was obtained from CelTrix (Santa Clara, CA). Petri dishes (60 mm, LUX, Permanox) were purchased from NUNC (Naperville, IL). All other media and culture reagents were from Invitrogen (Grand Island, NY). Western blotting kits were from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). 5-Bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium phosphatase substrate was from Kirkegaard and Perry Laboratories (Gaithersburg, MD). Glucose 6-phosphate, glucose-6-phosphate dehydrogenase, 11β-hydroxy-testosterone, NADPH, testosterone, β-naphthoflavone, dexamethasone, phenacetin, acetominophen, coumarin, 7-hydroxycoumarin, paclitaxel, tolbutamide, and hydroxytolbutamide were purchased from Sigma-Aldrich. 6β-Hydroxytestosterone was from Steraloids, Inc. (Wilton, NH), 6α-hydroxypaclitaxel from BD Gentest Corporation (Woburn, MA), (S)-mephenytoin and 6-hydroxychlorzoxazone were from Ultrafine Chemicals (Manchester, UK), 4′-hydroxymephenytoin from Cedra Corporation (Austin, TX), dextromethorphan hydrobromide and dextrorphan D-tartrate from Research Biochemicals Inc. (Manchester, UK) and chlorzoxazone from RBI Chemical Co. (Manchester, UK). All solvents and other chemicals used were of HPLC grade or the highest purity available.
In Vitro Induction Studies.
Isolation of human hepatocytes
Hepatocytes were isolated from human liver tissue by the two-step collagenase digestion method of MacDonald et al. (2001). Encapsulated liver tissue (25–100 g) was perfused with calcium-free buffer and then digested with a buffer containing 1.5 mM calcium and collagenase (0.3–0.4 mg/ml). Hepatocytes were dispersed in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal calf serum, insulin, and dexamethasone and washed by low-speed centrifugation (70g, 4 min). Cell pellets were resuspended in supplemented DMEM and 90% isotonic Percoll (3:1 v/v) and centrifuged at 100g for 5 min. The resulting pellets were resuspended in supplemented DMEM. Viability was determined by trypan blue exclusion and was typically between 80 and 90%.
Primary cultures of human hepatocytes.
Hepatocytes were cultured according to the method of LeCluyse et al. (2000) and as described previously (Sahi et al., 2000). Briefly, 4 to 4.5 million hepatocytes in 3 ml of supplemented DMEM were added to 60-mm NUNC Permanox culture dishes coated with a collagen, type I, substratum. After an attachment period of 4 to 6 h, medium was aspirated and fresh ice-cold medium containing 0.25 mg/ml matrigel added to each dish. Cells were maintained in a humidified incubator at 37°C with 95%:5%, air/CO2, and medium changed daily. Primary cultures of human hepatocytes were maintained for 36 to 48 h before initiating treatment. Groups of hepatocyte cultures (n = 3 dishes per treatment) were treated for 3 consecutive days with vehicle alone (control) or with various concentrations of test compounds or prototypical inducers.
Cell harvest and microsome preparation.
At the end of the treatment period, primary human hepatocytes were rinsed twice with ice-cold Hanks' balanced salt solution, scraped, and sonicated in a buffer containing 50 mM Tris-HCl, pH 7.0, 150 mM KCl, and 2 mM EDTA with a Vibra-Cell probe sonicator (Sonics and Materials, Danbury, CT). Cell lysates were centrifuged at 9,000g for 20 min at 4°C and the resulting supernatants were centrifuged at 100,000g for 60 min at 4°C. The final microsomal pellets were resuspended in 0.2 to 0.4 ml of sucrose (0.25 M) in phosphate buffered saline and stored at −80°C.
Microarray analysis for mRNA.
RNA was extracted with Trizol reagent using the method recommended by the manufacturer (Invitrogen, Carlsbad, CA). The microarray was fabricated and used as described previously (Kane et al., 2001; Wen et al., 2002). Briefly, three oligonucleotides per gene were designed and amino-modified 50mer oligos were spotted onto SuModic slides using a Molecular Dynamic Gen III robotic spotter (Amersham Biosciences). Yeast control 100 to 600 expression plasmids from Incyte were chosen as spiking controls, and synthetic transcripts were generated by in vitro transcription (MEGAscript, Ambion, Austin, TX). A mixture of synthetic transcripts and each mRNA at a specific copy per cell was spiked into experimental RNA. Labeled cDNA target was generated by reverse transcription (Superscript II, Invitrogen) in the presence of random primers (3.75 μM) and the following fluorescent probes: Cy3 for control and Cy-CTP (0.16 mM) for treated samples. Two replicate hybridization reactions were carried out overnight at 42°C, and florescent cDNA hybridization signals were detected using a Molecular Dynamic Gen III scanner. Data were normalized based upon intensity values between the Cy3 and Cy5 channels of control transcripts spiked at a 1:1 ratio.
Western blot analysis and CYP3A4 enzyme activity.
The microsomal CYP3A4 and CYP2B6 content in hepatocyte cultures was determined using Western immunoblot analysis (Parkinson and Gemzik, 1991). Microsomal protein samples (10–30 μg) were resolved by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose membranes. Membranes were probed with specific polyclonal antibodies raised in rabbit to human CYP3A4 or CYP2B6 (Chemicon International, Temecula, CA), followed by an anti-rabbit Ig-biotinylated secondary antibody and streptavidin-alkaline phosphatase conjugate. Enzyme protein was visualized using 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium phosphatase substrate. CYP3A4 activity was determined by measuring testosterone 6β-hydroxylation activity in microsomal samples obtained from hepatocyte cultures using the methods of Pearce et al. (1996).
Enzyme Inhibition Studies.
Pooled human liver microsomes from at least 15 donors were used for all inhibition assays. For IC50 determinations, the substrate probes were employed at their approximate in vitroKm values. All incubations were performed with 100 mM potassium phosphate buffer (pH 7.4) and 1 mM NADPH. Organic solvents were used to prepare stock solutions. Reactions were terminated with ethyl acetate for CYP3A4 and acetone for all other enzymes. For all Ki determinations, five substrate concentrations and at least five inhibitor concentrations were used. The concentration ranges for the substrate probes were approximately 0.2- to 3-times the in vitroKm values. Inhibitor concentrations were chosen based on the results from the IC50determinations (Table 2). Either a Micromass Quattro II or LC tandem quadrupole mass spectrometer was used to monitor ions of the respective P450 marker metabolites.
IC50 values for rosiglitazone, pioglitazone, and troglitazone against drug oxidation activities catalyzed by human hepatic microsomes
CYP1A2 activity.
Incubations (30 min) were performed with 0.1 mg/ml microsomal protein, 10 μM phenacetin, and inhibitor. Reactions were terminated and internal standard added (betatxolol or d4-acetaminophen), samples extracted, and analyzed according to a validated liquid chromatography-tandem mass spectrometry analytical method. The marker metabolite, acetaminophen, was quantitated from 1.00 to 200 ng/ml.
CYP2A6 activity.
Incubations (5 min) were performed with 0.025 mg/ml microsomal protein, 0.4 μM coumarin, and inhibitor. Reactions were terminated, and the marker metabolite, 7-hydroxycoumarin, was quantitated from 0.250 to 100 ng/ml.
CYP2C8 activity.
Incubations (20 min) were performed with 0.075 mg/ml microsomal protein, 4 μM paclitaxel, and inhibitor. Reactions were terminated, and the marker metabolite, 6α-hydroxypaclitaxel, was quantitated from 10.0 to 600 ng/ml.
CYP2C9 activity.
Incubations (20 min) were performed with 0.1 mg/ml microsomal protein, 140 μM tolbutamide, and inhibitor. Reactions were terminated and the marker metabolite, hydroxytolbutamide, was quantitated from 10.0 to 2000 ng/ml.
CYP2C19 activity.
Incubations (20 min) were performed with 0.1 mg/ml microsomal protein, 50 μM (S)-mephenytoin, and inhibitor. Reactions were terminated and the marker metabolite, 4′-hydroxymephenytoin, was quantitated from 5.00 to 1000 ng/ml.
CYP2D6 activity.
Incubations (30 min) were performed with 0.1 mg/ml microsomal protein, 20 μM dextromethorphan, and inhibitor. Reactions were terminated and the marker metabolite, dextrorphan, was quantitated from 5.00 to 1000 ng/ml.
CYP2E1 activity.
Incubations (20 min) were performed with 0.1 mg/ml microsomal protein, 50 μM chlorzoxazone, and inhibitor. Reactions were terminated, and the marker metabolite, 6-hydroxychlorzoxazone quantitated from 5.00 to 1000 ng/ml.
CYP3A4 activity.
Incubations (7 min) were performed with 0.05 mg/ml microsomal protein, 50 μM testosterone, and inhibitor. Reactions were terminated and the marker metabolite, 6β-hydroxytestosterone, was quantitated from 50.0 to 2000 ng/ml.
Statistical Analysis.
For the induction studies, results are expressed as mean ± S.D. of three separate hepatocyte preparations. Within each experiment, assays were performed in triplicate. All data were processed and graphed with Microsoft Excel 97 (Redmond, WA). For the inhibition studies, IC50 values were estimated from plots of remaining activity (percent relative to 0 μM inhibitor) versus the thiazolidinediones on a logarithmic scale. ForKi value estimation, the data were reviewed graphically using Lineweaver-Burke and Dixon analyses to establish the most appropriate inhibition model. TheKi value was subsequently estimated by nonlinear regression analysis of the appropriate model using Systat 6.0.1 (SPSS Science Inc., Chicago, IL).
Results
Effect of Rosiglitazone, Pioglitazone, and Troglitazone on mRNA Levels of CYP3A4 and CYP2B6.
The induction potential of the thiazolidinediones was studied by microarray analysis of RNA isolated from human hepatocytes treated with 10 and 50 μM concentrations of the compounds for three days. Rifampin (50 μM), dexamethasone (10 μM), and phenobarbital (1 mM) were used as positive controls. As shown in Table1, at the concentrations tested, rifampin was the most potent inducer of CYP3A4, increasing activity by 14.6-fold over control. Troglitazone at 10 μM induced CYP3A4 mRNA to similar levels as 1 mM phenobarbital (9.6- and 8.1-fold increase over control, respectively), whereas rosiglitazone at 50 μM and pioglitazone at 10 μM increased mRNA to similar levels as dexamethasone (5.5-fold). The largest increase in CYP2B6 mRNA was caused by phenobarbital (6.5-fold), followed by 50 μM rosiglitazone (3.9-fold). Pioglitazone (50 μM) and troglitazone (10 μM) increased CYP2B6 activity to similar levels as 50 μM rifampin (2.3- to 2.6-fold).
Effect of rosiglitazone, pioglitazone, troglitazone, and prototypical inducers on mRNA in primary cultures of human hepatocytes
Effect of Rosiglitazone, Pioglitazone, and Troglitazone on P450 Protein Expression.
The capacity of the three compounds to increase CYP3A4 and CYP2B6 protein concentrations was studied by Western immunoblotting. The effects of treating primary human hepatocyte cultures for three consecutive days with rosiglitazone, pioglitazone, and troglitazone at different concentrations (0.5–100 μM) were examined on microsomal CYP3A4 (Fig. 1) and CYP2B6 (Fig.2) immunoreactive protein. Rifampin (10 μM) was used as a positive control and caused a 2.6- and a 2.4-fold increase in CYP3A4 immunoreactive protein in HL092 and HL096, respectively. By comparison, troglitazone (10 and 50 μM) increased CYP3A4 immunoreactive protein 1.2- and 1.8-fold in HL092 and 1.3- and 1.2-fold in HL096, respectively. Rosiglitazone did not increase CYP3A4 protein at 10 μM, whereas at 50 and 100 μM, protein concentrations increased by 1.5- and 1.9-fold in HL092 and 2.1 and 2.0 in HL096, respectively. Pioglitazone caused an increase in immunoreactive protein at 100 μM in HL092 (1.8-fold) and at 50 μM (1.5-fold) and 100 μM (2.0-fold) in HL096. With few exceptions, Western blots probed with CYP2B6-specific antibodies also showed induction of CYP2B6 immunoreactive protein that paralleled that of CYP3A4 (Fig. 2). In HL092, rifampin caused a 4.2-fold increase in CYP2B6 immunoreactive protein. Marked increases were observed with troglitazone at 50 μM (2.7-fold), rosiglitazone at 100 μM (2.7-fold), and pioglitazone at 50 (2.6-fold), and 100 μM (3-fold). In HL096, major increases in CYP2B6 immunoreactive protein were found with all compounds: 4.4-fold by rifampin; 3.1- and 5.7-fold by 50 and 100 μM troglitazone; 3.3- and 4.4-fold by 50 and 100 μM rosiglitazone; and 3- and 4.6-fold by the same concentrations of pioglitazone, respectively.

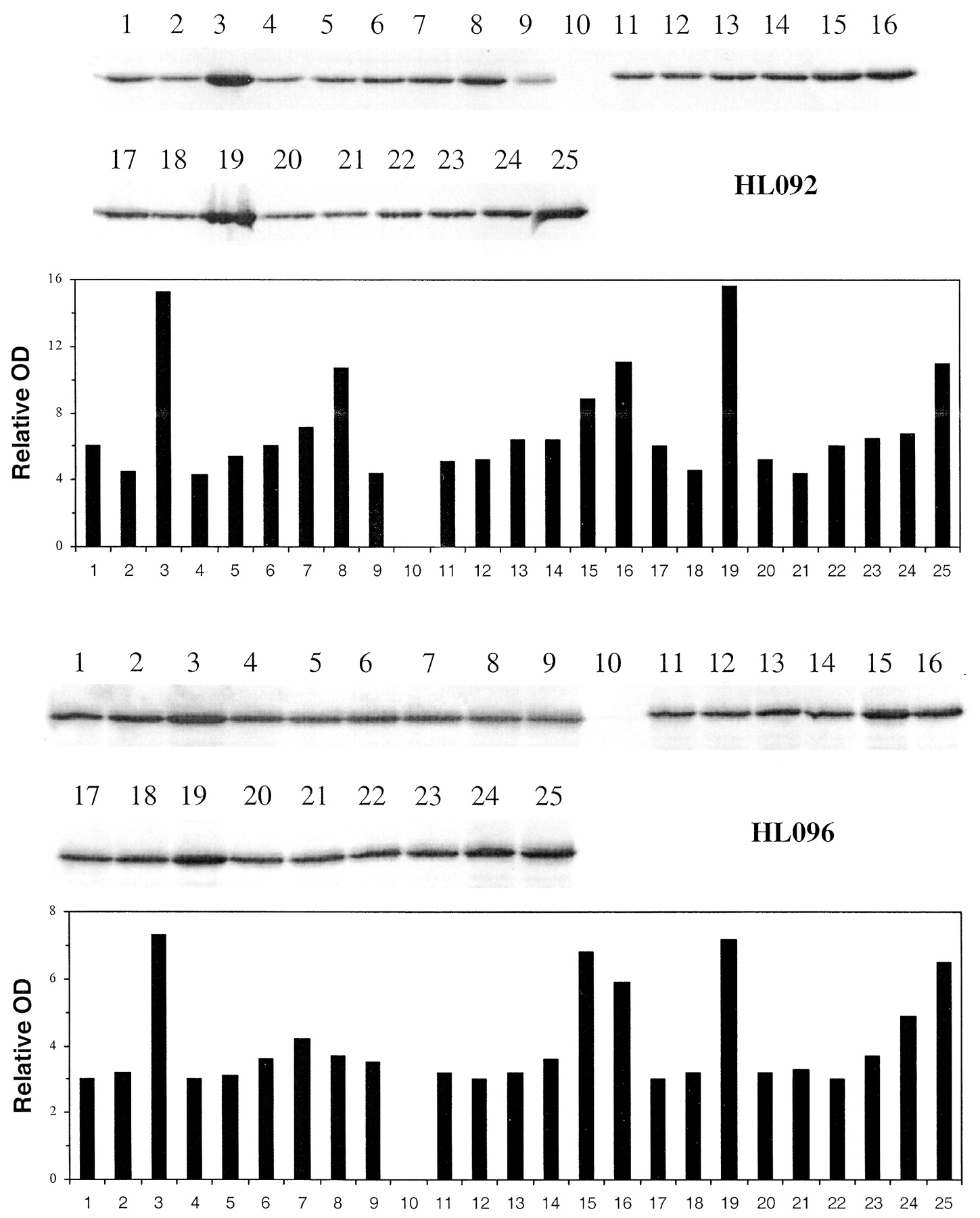
Induction of CYP3A4 immunoreactive protein by troglitazone, pioglitazone, and rosiglitazone in primary cultures of human hepatocytes.
Freshly isolated hepatocytes prepared from human liver were placed in primary culture for 36 to 48 h prior to treating with 0.1% DMSO controls, 10 μM rifampin, and troglitazone, pioglitazone and rosiglitazone at 0.5 to 100 μM. Seventy-two hours post treatment cells were harvested. Microsomes were isolated and analyzed by Western blot hybridization for CYP3A4 as described under Materials and Methods and resultant bands quantitated by densitometry. HL092 and HL096 represent hepatocytes from two different donors. Lane 1, control (0.1% DMSO); 2, control (0.2% DMSO); 3, rifampin (10 μM); 4 to 9, troglitazone (0.5, 1, 5, 10, 50, and 100 μM); 10, blank; 11 to 16, rosiglitazone (0.5, 1, 5, 10, 50, and 100 μM); 17, control (0.1% DMSO); 18, control (0.2% DMSO); 19, rifampin (10 μM); 20 to 25, pioglitazone (0.5, 1, 5, 10, 50, and 100 μM).
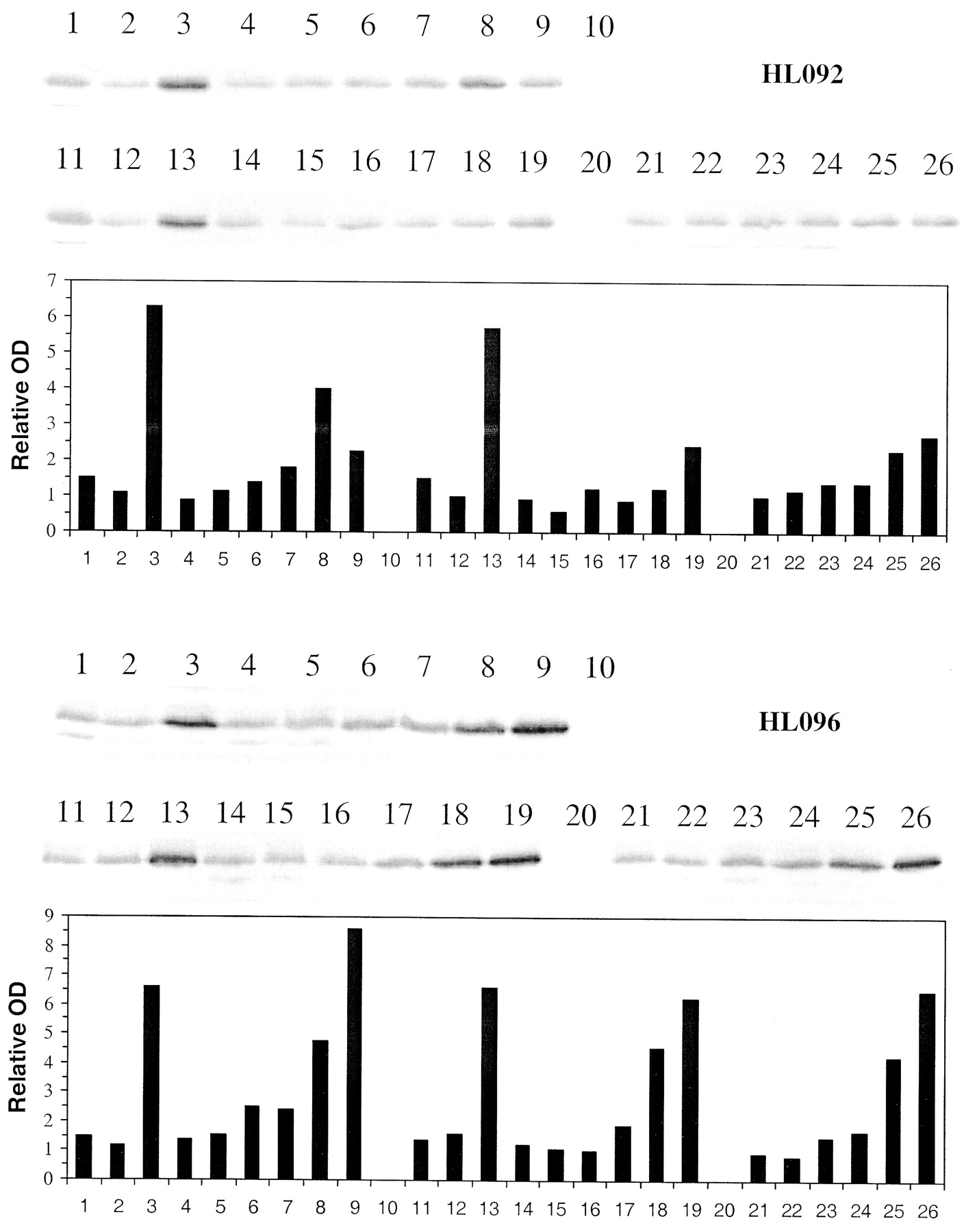
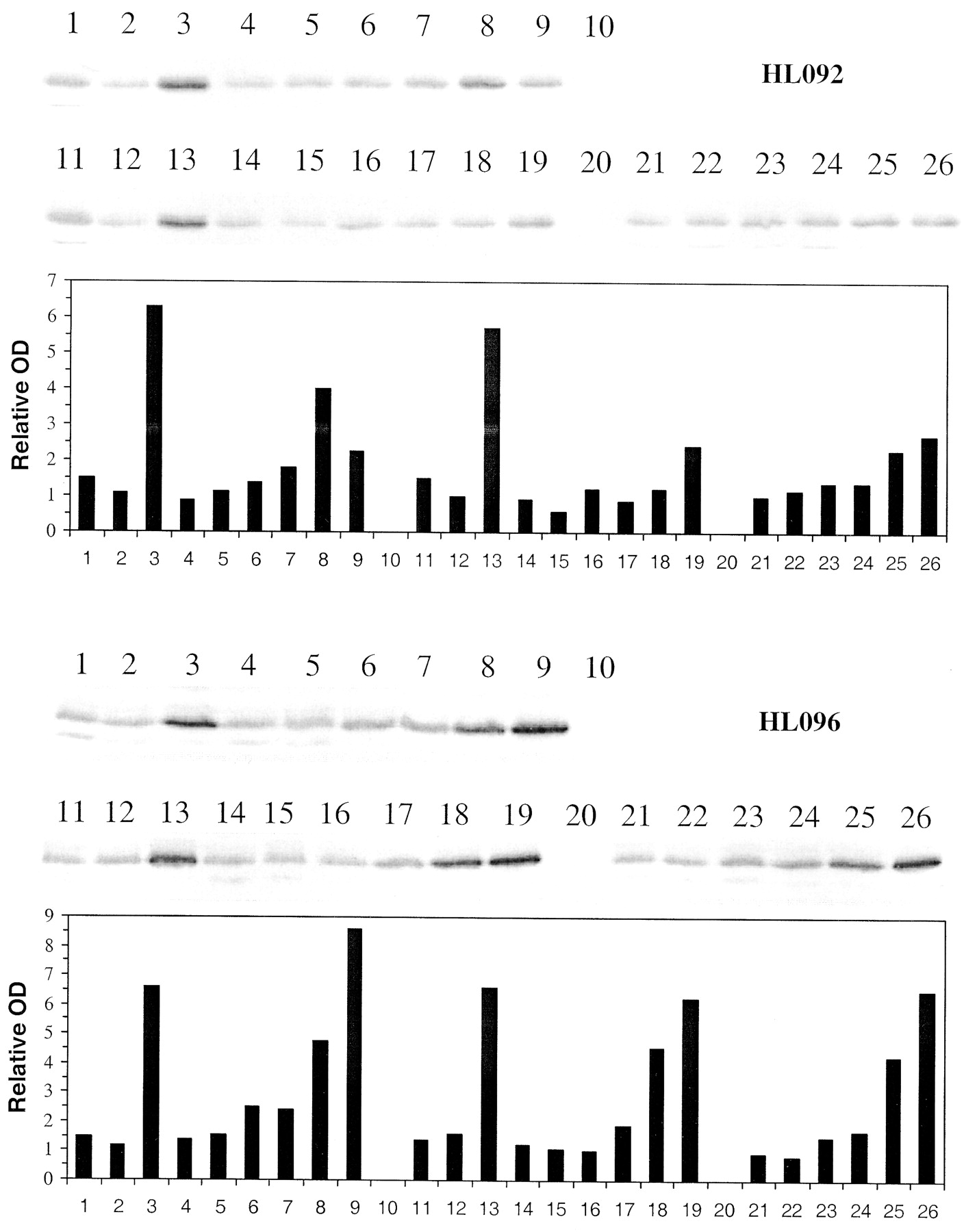
Induction of CYP2B6 immunoreactive protein by troglitazone, pioglitazone, and rosiglitazone in primary cultures of human hepatocytes.
Freshly isolated hepatocytes prepared from human liver were placed in primary culture for 36 to 48 h prior to treating with 0.1% DMSO controls, 10 μM rifampin, and troglitazone, pioglitazone, and rosiglitazone at 0.5 to 100 μM. Seventy-two hours post treatment cells were harvested. Microsomes were isolated and analyzed by Western blot hybridization for CYP2B6 as described under Materials and Methods and resultant bands quantitated by densitometry. HL092 and HL096 represent hepatocytes from two different donors. 1, control (0.1%DMSO); 2, control (0.2% DMSO); 3, rifampin (10 μM); 4 to 9, troglitazone (0.5, 1, 5, 10, 50, 100 μM); 10, blank; 11, control (0.1% DMSO); 12, control (0.2% DMSO); 13, rifampin (10 μM); 14 to 19, rosiglitazone (0.5, 1, 5, 10, 50, 100 μM); 20, blank; 21 to 26, pioglitazone (0.5, 1, 5, 10, 50, 100 μM).
Effect of Rosiglitazone, Pioglitazone, and Troglitazone on CYP3A4 Catalytic Activity.
The effects of treating three different primary human hepatocyte cultures with rosiglitazone, pioglitazone, and troglitazone for 72 h at three concentrations (0.5, 5, and 50 μM) were examined on microsomal testosterone 6β-hydroxylase activity (Fig.3). The positive controls rifampin, dexamethasone, and phenobarbital caused an increase in testosterone 6β-hydroxylation activity that was reflective of their potency and concentration. Rifampin was the strongest of the three controls, with a 3- to 5-fold increase in basal CYP3A4 activity, followed by phenobarbital (2-fold increase) and dexamethasone (1.3-fold increase). None of the thiazolidinedione tested markedly increased CYP3A4 activity over control levels at the lowest concentration tested (0.5 μM). All three compounds caused a significant increase in CYP3A4 activity at 50 μM. There was variability in the extent of induction in the three different preparations, with rosiglitazone being the most effective inducer at 50 μM in HL096 and troglitazone being the strongest inducer at 50 μM in HL092. The three compounds exhibited somewhat equal induction in HL091.

Concentration-dependent induction of CYP3A4 activity by rosiglitazone, pioglitazone, troglitazone, and prototypical inducers in primary cultures of human hepatocytes.
Freshly isolated hepatocytes prepared from the human liver were placed in primary culture for 36 to 48 h prior to initializing treatment with DMSO for control (Con, open bars), 50 μM rifampin (hatched bars), 50 μM dexamethasone (stippled), 2 mM phenobarbital (horizontal bars) or troglitazone (Trz), rosiglitazone (Rosi), and pioglitazone (Pio) at 0.5, 5, and 50 μM concentrations. Seventy-two hours post treatment; cells were harvested, microsomal membranes prepared, and testosterone 6β hydroxylation activity measured, as per procedures under Materials and Methods. HL091, HL092, and HL096 represent preparations of human hepatocytes from three different donor livers.
Cytochrome P450 Inhibition by Troglitazone, Pioglitazone, and Rosiglitazone.
IC50 determinations illustrate the overall inhibition profile as a function of compound concentration. All initial velocity measurements were compared with samples that contained only substrate at the approximate Km value along with the inhibitor dissolution solvent (100% activity). As shown in Table 2, based on IC50 measurements, the most potent CYP2C inhibitor was troglitazone with IC50 values of 2.3 and 2.7 μM against CYP2C8 and CYP2C9, respectively. Rosiglitazone and pioglitazone both inhibited CYP2C8 with similar IC50 values (9.6 and 9.4 μM, respectively). However, both rosiglitazone and pioglitazone were far less inhibitory against CYP2C9 as compared with troglitazone, with IC50 values of 83, 100, and 2.7 μM, respectively. CYP2C19 was only inhibited by troglitazone (IC50 25.7 μM), and CYP2D6 was slightly inhibited by rosiglitazone (IC50 42.1 μM). All three glitazone compounds demonstrated some degree of CYP3A4 inhibition. Troglitazone and pioglitazone had similar IC50 values of 14.5 and 12.3 μM, respectively. Rosiglitazone was comparatively less inhibitory against CYP3A4 with an IC50 value of 28 μM.
Inhibition mechanisms of selected interactions were further evaluated for Ki determinations and the data fit using appropriate equilibrium-based inhibition models. For graphical evaluation, the activity data were plotted in both Lineweaver-Burke and Dixon formats. After graphical and statistical evaluation, the data set was fit using the selected inhibition model. Inhibition constants were determined against CYP2C8, CYP2C9, and CYP3A4 for all three compounds (Table 3). Rosiglitazone, pioglitazone, and troglitazone exhibited similar inhibition constants against CYP2C8 with Ki values of 5.6, 1.7, and 2.6 μM, respectively, and were all found to be predominantly competitive inhibitors of CYP2C8. A Lineweaver-Burke plot of the most potent inhibitor of CYP2C8, pioglitazone, is shown in Fig.4A. Troglitazone and pioglitazone were competitive inhibitors against CYP2C9 withKi values of 0.6 and 32 μM. The inhibition of CYP2C9 by troglitazone is shown in Fig. 4B in the form of a Lineweaver-Burke plot. These same trends were mirrored with CYP3A4, where troglitazone was found to be more inhibitory than either rosiglitazone or pioglitazone. The Kivalue for troglitazone against CYP3A4 was 1.6 μM (Fig. 4C) as compared with 36 and 12 μM for rosiglitazone and pioglitazone, respectively. All three compounds were found to be competitive inhibitors against CYP3A4. In addition, troglitazone inhibited CYP2C19 with a Ki of 10.2 μM, and rosiglitazone inhibited CYP2D6 with aKi of 29.5 μM.
Ki values for rosiglitazone, pioglitazone, and troglitazone against drug oxidation activities catalyzed by human hepatic microsomes
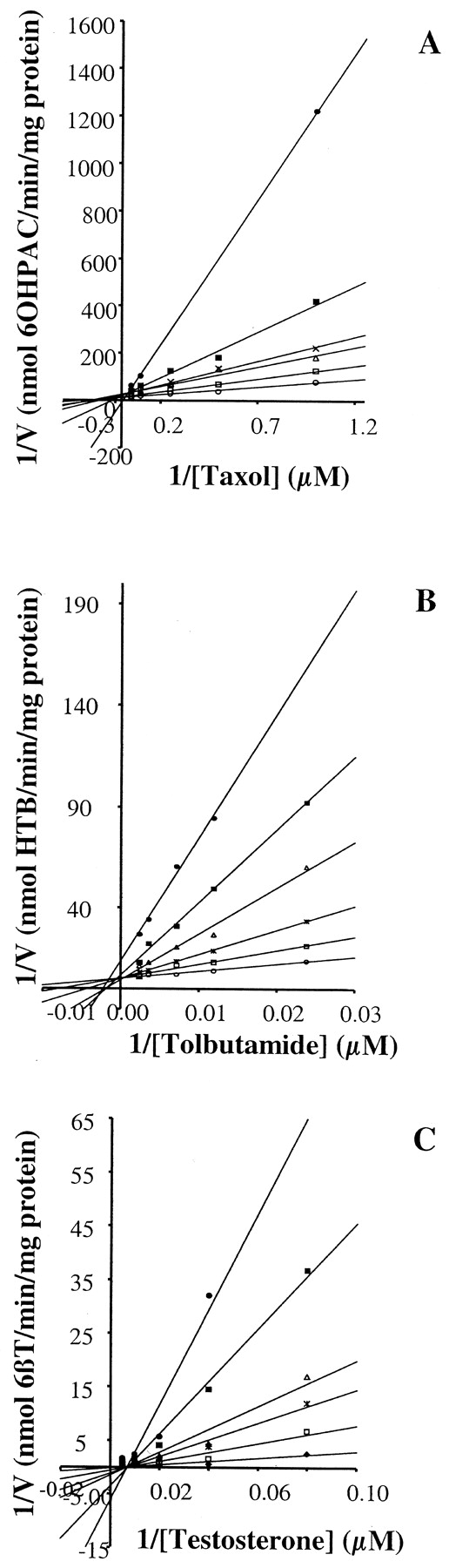
Concentration-dependent inhibition of CYP2C8 activity by pioglitazone (A) CYP2C9 activity by troglitazone (B) and CYP3A4 activity by troglitazone in human liver microsomes (C).
These Lineweaver-Burke plots illustrate the extent of equilibrium-based inhibition of pioglitazone against paclitaxel metabolism (A), troglitazone against tolbutamide metabolism (B), and troglitazone against testosterone metabolism (C). Incubations were conducted with pooled microsomes (n = 15) under the conditions outlined under Materials and Methods. A Micromass Quattro II or an LC tandem quadrupole mass spectrometer was used to monitor product ions. A, pioglitazone at 0 (open circles), 2.5 (open squares), 5 (open triangles), 8 (crosses), 14 (filled squares), and 25 μM (filled circles). 6OHPAC, 6α-hydroxypaclitaxel. B, troglitazone at 0 (open circles), 1 (open squares), 2 (crosses), 4 (open triangles), 6 (filled squares), and 10 (filled circles) μM. HTB, hydroxytolbutamide. C, troglitazone at 0 (filled diamonds), 4 (open squares), 8 (crosses), 15 (open triangles), 25 (filled squares), and 40 (filled circles) μM. 6βT, 6β-hydroxytestosterone.

Troglitazone.

Rosiglitazone.

Pioglitazone.
Discussion
Thiazolidinediones are a new class of drugs for the treatment of Type 2 diabetes mellitus. Troglitazone (withdrawn from market), pioglitazone, and rosiglitazone represent the first compounds in this therapeutic class and the future certainly holds new and improved PPAR-γ agonists. Understanding the biotransformation of these compounds and the in vitro effects of enzyme inhibition or induction will be useful in interpreting and predicting pharmacokinetics and potential drug interactions of future drug candidates. The oxidative metabolism of troglitazone and pioglitazone are through CYP3A4 and CYP2C8, whereas rosiglitazone is metabolized by CYP2C8 and to a minor extent by CYP2C9 (Baldwin et al., 1999; Yamazaki et al., 2000; Kassahun et al., 2001 and internal data). Changes in the activity of these enzymes could result in drug interactions of administered or coadministered drugs. Unfortunately, limited information is available on the in vitro inhibition and induction effects of these compounds, albeit there are few known drug interactions reported for this class of compounds.
To date, there are no reports of significant clinical drug interactions with either rosiglitazone or pioglitazone. Rosiglitazone did not change the pharmacokinetics of the CYP3A4 substrates ethinylestradiol or norethindrone, or the CYP2C9 substrates glyburide or warfarin in clinical trials (Inglis et al., 2001 and rosiglitazone package insert). An interaction study with rosiglitazone and nifedipine (CYP3A4 substrate) indicated a minor decrease in the area under the curve of nifedipine (13%) after chronic rosiglitazone administration, suggesting that rosiglitazone was a weak inducer of CYP3A4 (Harris et al., 1999). Similarly, pioglitazone does not alter the pharmacokinetics of the coadministered drugs simvastatin, ethinylestradiol, or norethindrone (CYP3A4 substrates) or warfarin, glyburide, or glipizide (CYP2C9 substrates) (Carey and Liu, 2000; Gillies and Dunn, 2000;Hanefeld, 2001). These data indicate that at the doses administered, rosiglitazone and pioglitazone are not significant inducers or inhibitors of CYP3A4 or CYP2C9. Troglitazone induced CYP3A4 activity as exemplified by clinical interactions with atorvastatin, simvastatin, oral contraceptives, cyclosporine, and terfenadine (Kaplan et al., 1998; Loi et al., 1999) and did not affect the CYP2C9-mediated metabolism of warfarin or glyburide (Rezulin package insert).
The human induction caused by troglitazone was unexpected, as preclinical studies did not demonstrate changes in CYP1A, CYP2B, or CYP3A activities in livers of rats (100 mg/kg daily for 10 days) or monkeys (800 mg/kg daily for three months) (Parke Davis, unpublished data). These data indicate that the rat and monkey are not predictive models of the human P450 induction caused by troglitazone. It is known that there are marked species differences in the effects of xenobiotics on CYP3A expression (Kocarek et al., 1995; Jones et al., 2000). For example, pregnenolone 16α-carbonitrile and dexamethasone are potent CYP3A inducers in rats and comparatively mild inducers in human, and rifampin markedly induces CYP3A in humans but not in rats. This highlights the importance of using human hepatocytes to complement animal data, especially where marked species differences are noted in the nuclear receptor activation profiles. Recent studies have shown that both the pregnane X receptor and the constitutively active receptor exhibit significant species-specific activation by many xenobiotics (Jones et al., 2000; Moore et al., 2000).
Our in vitro studies with primary human hepatocytes demonstrated induction of CYP3A4 and CYP2B6 with all three compounds. Treatment of hepatocytes with rosiglitazone or pioglitazone increased the mRNA levels of CYP3A4 similar to dexamethasone (a relatively mild inducer of CYP3A4), indicating mild induction of CYP3A4. CYP2B6 mRNA was induced by these two compounds to similar levels as found with troglitazone and rifampin, again suggesting weak induction. There was a marked increase in the immunoreactive protein with both rosiglitazone and pioglitazone, indicating an increase in translation of these proteins. Troglitazone was a more potent inducer of both enzymes although the capacity to induce maximum CYP3A4 expression (Emax) was greater for pioglitazone and rosiglitazone than for troglitazone in most experiments. Activity assays revealed that these compounds are strong inducers of CYP3A4 in vitro at a concentration of 50 μM and troglitazone causes a significant change in CYP3A4 activity at a concentration of 5 μM. These results indicate that all three compounds have the potential to cause drug-drug interactions through induction of CYP3A4 if sufficient concentrations are achieved in the liver. One reason for the lack of clinical induction by rosiglitazone and pioglitazone could be the lower doses administered as compared with troglitazone. Typically, drugs that induce CYP3A4 are administered at high doses and this induction can be reduced or eliminated at lower doses (Smith, 2000). While troglitazone was administered at 200 to 600 mg/day, the dose for pioglitazone is 45 mg/day and rosiglitazone, 4 to 8 mg/day. The corresponding clinical peak plasma concentrations are as follows: troglitazone 1 to 3 μg/ml (2.3–6.8 μM), pioglitazone 0.7 to 1.7 μg/ml (2.0–4.8 μM), and rosiglitazone 0.2 to 0.6 μg/ml (0.6–1.7 μM) (product package inserts and Gillies and Dunn, 2000).
Our interpretation of the inhibition data employed the total therapeutic plasma concentration and the ratio of inhibitor concentration to the Ki (Tucker et al., 2001). The Ki values for troglitazone inhibition of CYP2C8, CYP2C9, and CYP3A4 fall within the therapeutic window of troglitazone (2.3–6.8 μM): 2.6, 0.6, and 1.6 μM, respectively. Based on these data, the in vitro inhibition results indicate that in vivo, troglitazone has the potential to inhibit CYP2C8, CYP2C9, and CYP3A4, where in each case the ratio of plasma concentration to Ki is ∼1.0 or greater. Pioglitazone also has the potential to inhibit CYP2C8 whereas rosiglitazone would not be expected to inhibit the P450 enzymes tested. Due to the extremely low Kifor CYP2C9 inhibition, troglitazone was expected to cause detectable clinical interactions with coadminstered CYP2C9 substrates, such as warfarin ([I]/Ki ∼4–11). However, the prothrombin time for patients taking warfarin in conjunction with troglitazone did not increase, indicating that troglitazone was not inhibiting the major pathway of warfarin elimination [i e., the CYP2C9-mediated 7-hydroxylation of S-warfarin (Plowman and Morreale, 1998)]. This suggests that the in vitro inhibition by troglitazone is not reflective of the actual in vivo situation, perhaps due to an overestimation of the actual liver concentration using the systemic plasma concentration, or in vivo or in vitro protein binding effects. As all three compounds are >99% protein bound, the thiazolidinediones could represent a situation where the unbound plasma concentration (∼2–28 ng/ml) results in a better prediction of in vivo inhibition. In regards to the expected inhibition of CYP2C8 by either troglitazone or pioglitazone, to date no clinical studies have addressed the issue of CYP2C8 inhibition, and there are no literature reports of such an interaction in vivo.
Inhibition of P450 by thiazolidindiones has previously been reported (Yamazaki et al., 2000) and compared at 5 and 50 μM using cDNA-expressed enzymes and troglitazone IC50determinations were also examined in human liver microsomes along with an evaluation of mechanism of inhibition in cDNA-expressed enzymes. Troglitazone significantly inhibited CYP2C8, CYP2C9, CYP2C19, and CYP3A4, in good agreement with our results on mechanism of inhibition and inhibition potency. Yamazaki et al. (2000) reported that pioglitazone and rosiglitazone only slightly inhibited CYP2C isoforms and CYP2B6, whereas no other P450s were inhibited. These percent inhibitions are similar to the IC50 data presented here except our data shows greater CYP3A4 inhibition with pioglitazone and rosiglitazone (IC50 = 12.3 and 28.0 uM, respectively), whereas no inhibition was observed by Yamazaki (2000). This discrepancy in results between our labs could be due to the different matrices or experimental conditions used.
Our IC50 and Kidata and that of Yamazaki et al. (2000) are similar and indicate that troglitazone is a competitive inhibitor of CYP3A4 with an IC50 value between 15 and 20 μM and aKi value of ∼1.6 μM. In addition,Kassahun et al., 2001 determined that troglitazone was a NADPH- and time-dependent inhibitor in human liver microsomes, indicating a combination of irreversible and reversible mechanisms of inhibition. Since the major metabolites of troglitazone are not inhibitors of CYP3A4 (Yamazaki et al., 2000), the preincubation data suggests that an irreversible mechanism of inhibition is occurring with troglitazone in human liver microsomes in addition to some degree of reversible inhibition. Although these inhibition effects are observable in the in vitro systems and perhaps occur in vivo, the overwhelming capacity of troglitazone to induce CYP3A4 would be expected to mask any clinical inhibition effects.
In summary, we have provided a background of in vitro inhibition and induction on three thiazolidinediones and have made comparisons to reported clinical drug interactions. Clearly all three thiazolidinediones have the potential to induce CYP3A4, and we suggest that the resulting clinical outcome of the CYP3A4 induction may be related to the dose and subsequent pharmacokinetics of each drug. The in vitro inhibition data indicates that in general troglitazone is the most potent P450 inhibitor of the three compounds. However, the clinical effects of P450 enzyme inhibition have not been demonstrated either due to a lack of clinical data (CYP2C8), overwhelming P450 induction (CYP3A4), or for reasons we do not yet understand (CYP2C9). It is the last of these scenarios that require additional research to better use in vitro inhibition data to predict potential drug-drug interactions of these and future thiazolidinediones.
Acknowledgments
The authors thank Dr. Steve Madore for help with the microarray work, Dr. Birong Liao for designing the oligos for the microarray, and Dr. Rebecca Boyd for reviewing this manuscript.
Footnotes
-
↵1 M. W. Sinz is presently at Bristol-Myers Squibb, Wallingford, CT 06422.
- Abbreviations used are::
- PPAR
- peroxisome proliferator activated receptor
- P450
- cytochrome P450
- DMEM
- Dulbecco's Modified Eagle's medium
- Received August 19, 2002.
- Accepted December 20, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}