


Abstract
N-Hydroxylated amidines (amidoximes) can be used as prodrugs of amidines. The prodrug principle was developed in our laboratory for pentamidine and had been applied to several other drug candidates. One of these compounds is melagatran, a novel, synthetic, low molecular weight, direct thrombin inhibitor. To increase the poor oral bioavailability due to its strong basic amidine functionality selected to fit the arginine side pocket of thrombin, the less basic N-hydroxylated amidine was used in addition to an ethyl ester-protecting residue. The objective of this investigation was to study the reduction and the hydrolytic metabolism of ximelagatran via two mono-prodrugs (N-hydroxy-melagatran and ethyl-melagatran) to melagatran by in vitro experiments. New high-performance liquid chromatography methods were developed to analyze all four compounds. The biotransformation of ximelagatran to melagatran involving the reduction of the amidoxime function and the ester cleavage could be demonstrated in vitro by microsomes and mitochondria from liver and kidney of pig and human, and the kinetic parameters were determined. So far, one enzyme system capable of reducingN-hydroxylated structures has been identified in pig liver microsomes, consisting of cytochromeb5, NADH-cytochromeb5 reductase, and a P450 isoenzyme of the subfamily 2D. This enzyme system also reduces ximelagatran andN-hydroxy-melagatran. The participation of recombinant human CYP1A2, 2A6, 2C8, 2C9, 2C19, 2D6, and 3A4 with cytochromeb5 and b5reductase in the reduction can be excluded. In summary, ximelagatran and N-hydroxy-melagatran are easily reduced by several enzyme systems located in microsomes and mitochondria of different organs.
Melagatran is the active form of the novel, oral thrombin inhibitor ximelagatran (Gustafsson et al., 2001). Melagatran has suboptimal bioavailability because of the presence of a carboxylic acid, a secondary amine, and an amidine residue resulting in a charged molecule at physiological pH values. The prodrug ximelagatran was developed with a view to improving absorption. Ximelagatran comprises an ethyl ester group in place of the carboxylic acid and anN-hydroxyamidine group in place of the amidine. After protonation of amidines at the double-bonded nitrogen, cations are formed that are highly stabilized by mesomerism. Amidines are very strong bases (Albert et al., 1948) and protonated under physiological conditions. By introduction of an oxygen atom in the amidine functional group, the basicity is lowered by 5 pKa value. Amidoximes have been used as prodrugs for certain amidine-containing drugs, including pentamidine derivatives (Clement, 1993) and sibrafiban (Weller et al., 1996). It has been shown that certain amidoximes are rapidly reduced in vitro and in vivo to the amidines (Clement et al., 1988, 1992; Hauptmann et al., 1988). More recently, it has been demonstrated that the model compound benzamidoxime is reduced by microsomes and mitochondria from liver and other organs like kidney, lung, and even brain (Clement and Mau, 1999;Clement and Deters, 2000).
So far one enzyme system capable of reducing N-hydroxylated derivatives of strongly basic functional groups has been identified in pig liver microsomes consisting of cytochromeb5, NADH-cytochromeb5 reductase, and a P450 isoenzyme of the subfamily 2D (Clement et al., 1997).
Current orally active anticoagulants on the market are vitamin K antagonists, ADP antagonists, and the thromboxane A2 antagonist acetyl salicylic acid. These agents show some disadvantages and limitations in antithrombotic therapy (Hauptmann, 2002).
In particular, coumarins such warfarin or phenprocoumon, which achieve their anticoagulant effect by modulating the synthesis of vitamin K-dependent proteins resulting in the synthesis of defective coagulation factors without any coagulation activities (Hull et al., 1979), exhibit large variations in pharmacokinetics and a slow onset and offset of action (Hirsh et al., 1998). Additionally, drugs and food influence metabolism in particular in the case of changes in the vitamin K content of the diet. Consequently, extensive and expensive anticoagulation monitoring is necessary to maximize efficacy and safety of coumarins. The shortcomings of current available oral anticoagulants have stimulated great efforts to develop new oral drugs. One of these new anticoagulation drug candidates is melagatran, a new direct, low molecular weight thrombin inhibitor (Eriksson et al., 1999).
Melagatran has a strong basic amidine structure, a free carboxylic acid, and in addition a less basic amine function, implying that melagatran will be positively charged under physiological conditions. The oral bioavailability is only 3 to 7%. ByN-hydroxylating melagatran at the amidine function in addition to the inclusion of an ethyl group to protect the carboxylic acid functionality (Fig. 1), the bioavailability of melagatran is increased to 18 to 24% (Gustafsson et al., 2001). The resulting amidoxime has a lower pKa value than the amidine function, and the carboxylic ester group, which is uncharged, reduces the pKa of the secondary amine, too (Gustafsson et al., 2001).
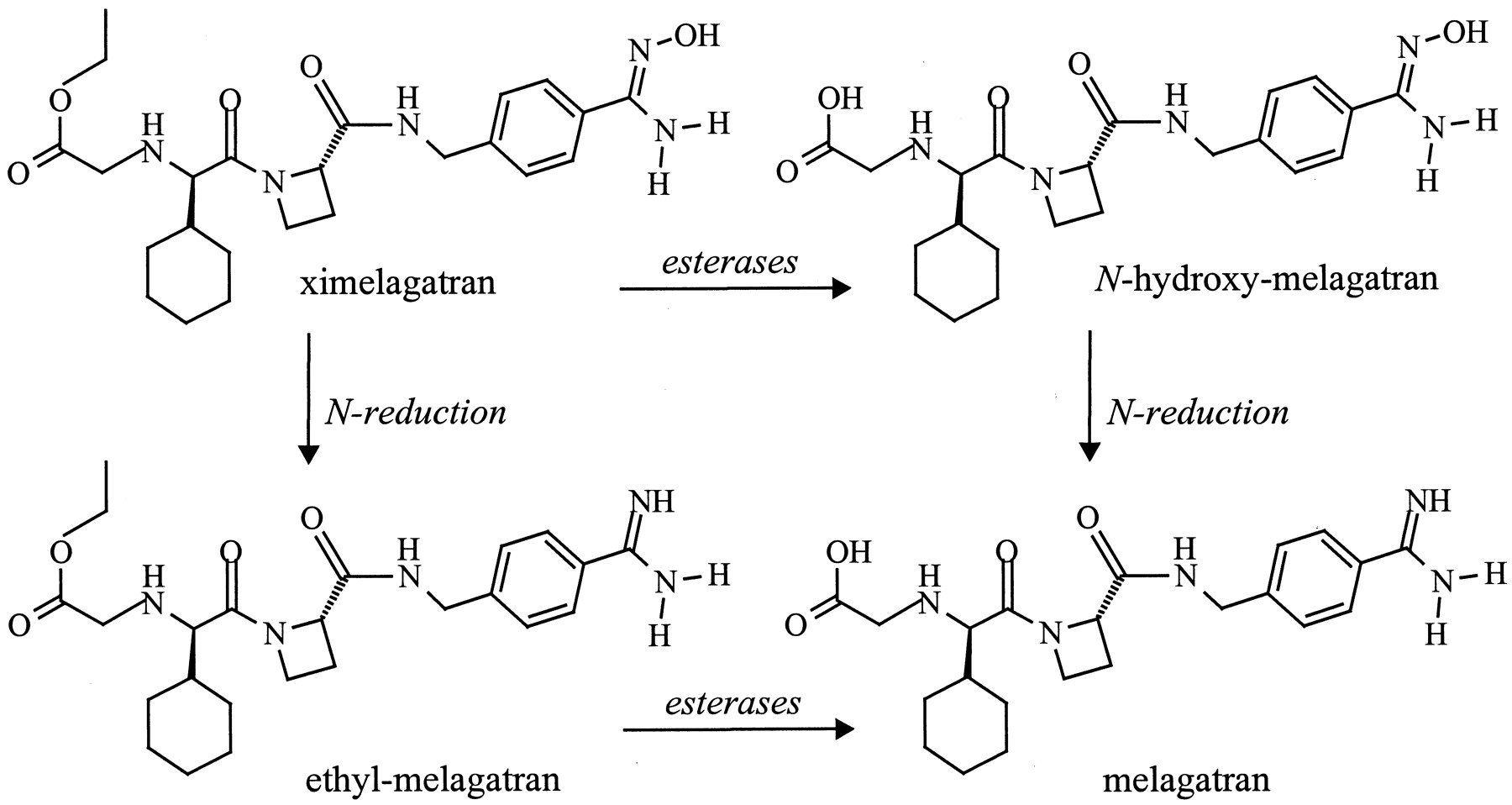
Activation of the double prodrug ximelagatran (H376/95) via two mono-prodrugs N-hydroxy-melagatran (H415/04) and ethyl-melagatran (H338/57) to melagatran (H319/68).
From in vivo studies, it is clear that ximelagatran is reduced and hydrolyzed very efficiently to the active principle melagatran. There is low intersubject variability of pharmacokinetic parameters and no signs of toxicity (Gustafsson et al., 1999). However, not much is known about the enzymes metabolizing prodrugs such as ximelagatran. This study is directed toward the elucidation of the enzymatic bases of ximelagatran bioactivation, which means the reduction of an amidoxime function and the cleavage of an ethyl ester. Because two protecting groups are present ximelagatran has properties of a double prodrug.
Materials and Methods
Ximelagatran and metabolites were obtained from Astra (Hässle, Mölndal, Sweden). NADH, NADPH, DLPC,1 and unspecific carboxyl esterases from pig liver were obtained from Sigma Chemie (Deisenhofen, Germany). All other chemicals were commercially available and of analytical grade, except acetonitrile and methanol, which were of HPLC grade.
Human liver samples were obtained from medicinal departments of several universities. They came from patients that were subjected to a partial hemihepatectomy because of secondary liver tumors. Prior consent of the local medical ethics committee and from the donors was obtained for these studies. Human and pig microsomes from liver were obtained by ultracentrifugation as described previously (Clement et al., 1996). The kidney microsomes were prepared analogously.
Human and pig mitochondria were prepared by differential centrifugation as described previously (Beattie, 1968; Kline et al., 1986) with slight modifications (Clement and Deters, 2000). To account for the biological variability liver samples from pigs or human organs were pooled (from at least three individuals per pool).
Mitochondria were checked for microsomal impurities by assessing rotenone-insensitive NADH cytochrome c reductase and succinate-cytochrome c reductase (Sottocasa et al., 1967). Microsomes were checked for mitochondrial impurities by assessing NADPH-cytochrome c reductase (Yasukochi and Masters, 1976).
Protein was assayed according to the method of Smith et al. (1985)using a bicinchoninic acid procedure, according to the manufacturer's directions (BCA reagent kit; Pierce Chemical, Rockford, IL).
Cytochrome P450 concentrations were determined by measuring the carbon monooxide difference spectra after reduction with dithionite (Omura and Sato, 1964). NADH-cytochrome b5reductase was purified from pig liver microsomes to homogeneity by affinity chromatography on 5′AMP-Sepharose 4B (Pharmacia, Freiburg, Germany) similar to the procedure described for the purification of NADPH-P450 reductase (Yasukochi and Masters, 1976) with modifications (Clement et al., 1997). Cytochrome b5was purified from pig liver microsomes according to a published method (Taniguchi et al., 1984). Pig benzamidoxime reductase was purified from liver microsomes described by Clement et al. (1997).
Recombinant CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, recombinant and purified CYP3A4, and NADPH-cytochrome P450 reductase were obtained from BD Gentest (Woburn, MA).
Calculation of Apparent Kinetic Parameters.
To determine N-hydroxy-melagatran reduction kinetics, activities were measured at a minimum of 0.05 mM substrate concentrations with two to four replicates at each concentration level. Apparent kinetic parameters Km andVmax were estimated using nonlinear regression analysis (SigmaPlot 5.0; SPSS Science, Chicago, IL).
Assay for Activation of Ximelagatran andN-Hydroxy-melagatran.
The incubation mixture consisted of 0.05 to 0.3 mg/ml microsomal or mitochondrial protein of liver or kidney (human or pig), 0.5 or 1 mM ximelagatran (H376/95), or 2 mM N-hydroxy-melagatran (H415/04) as substrate in 100 mM phosphate buffer pH 6.3, 7.0, and 7.4. A time course was run for all incubations. The reactions were linear for more than 30 min (up to 60 min). To obtain sufficient amounts of metabolites an incubation time of 30 min was chosen. After preincubation for 5 min at 37°C under aerobic conditions the reaction was started by the addition of NADH (final concentration 1 mM) to a total volume of 250 μl and maintained for 30 min. The reaction was terminated by the addition of 250 μl of cold methanol and vortexing. After centrifugation at 10,000 U/min (48g) (Mikroliterzentrifuge Hettich, Tuttlingen, Germany), 15 μl of the supernatant was analyzed by HPLC.
The standard incubation mixture with unspecific carboxylic esterases consisted of 0.5 U of esterases from pig liver and 2 mM ximelagatran in 100 mM potassium phosphate buffer pH 7.4. After a 3-min preincubation period at 37°C under aerobic conditions, the reaction was initiated by the addition of thermostated esterases. After 20 min, the reaction was stopped by the addition of 250 μl of cold methanol, followed by vortexing and centrifugation; 15 μl of the supernatant was analyzed by HPLC.
The incubation mixture of the reconstituted system consisted of 72.5 pmol of cytochrome b5, 0.35 U of NADH-cytochrome b5 reductase, and 5 μg of pig benzamidoxime reductase (CYP2D) or 5 μg of recombinant-expressed human CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4, 40 μM DLPC, 1 mM ximelagatran or 2 mMN-hydroxy-melagatran in 100 mM phosphate buffer pH 6.3. Ximelagatran was also incubated in reconstitution experiments with 5 μg of recombinant, purified CYP3A4 in combination with 50 μg of NADPH-cytochrome P450 reductase and 40 μM DLPC in phosphate buffer pH 6.3. After a 5-min aerobic preincubation period, the reaction was initiated by NADH (final concentration 1 mM). After 30 min, the reaction was stopped by adding 150 μl of cold methanol. Precipitated proteins were sedimented by centrifugation and 15 μl of the supernatant was analyzed by HPLC.
For HPLC analysis, a conventional system was used: Waters 616 pump, controller 600S, autosampler 717 plus, 486 TAD UV detector, and EZ Chrom integration software (Scientific Software Inc., San Ramon, CA). Solvents used in the analysis were filtered through a 0.45-μm Sartolon membrane filter (Sartorius AG, Göttingen, Germany) and degassed by bubbling with helium or sonication.
HPLC Method for Detection of Ximelagatran, Ethyl-Melagatran,N-Hydroxy-melagatran, and Melagatran.
The separation was carried out at room temperature by gradient elution by a LiChrospher RP-select B column (125 × 5 mm; Merck, Darmstadt, Germany) with an RP-select B precolumn (4 × 4 mm; Merck). The eluate was monitored at 238 nm. The first mobile phase contained phosphate buffer (20 mM, tetramethylammoniumchloride 10 mM, pH 4.5) and acetonitrile (92:8, v/v), and melagatran was eluted with a retention time of 7.4 ± 0.3 min andN-hydroxy-melagatran at 11.7 ± 0.6 min. The second eluent consisted of the same ingredients in the proportion of 50:50 (v/v), and the elution was started with a mixture of the first and second eluent 60:40 (v/v) at 14 up to 22 min. The retention times were 19.7 ± 0.7 min for ethyl-melagatran and 22.5 ± 0.3 min for ximelagatran.
Standard curves for each metabolite were constructed and found to be linear with correlation coefficients >0.99. Precision of the assays and accuracy were assessed by adding different concentrations (0–100 μM melagatran, 0–1000 μM N-hydroxy-melagatran, 0–600 μM ethyl-melagatran) of metabolites to standard incubation mixtures (without cosubstrate NADH). After the usual working up procedure, standard curves were constructed by linear regression analysis. The recoveries of melagatran, N-hydroxy-melagatran, and ethyl-melagatran from incubation mixture were 100 ± 5% of that obtained using samples that contained the same amount of each metabolite dissolved in phosphate buffer. The determination limits were about 0.15 μM for melagtran, 1.25 μM forN-hydroxy-melagatran, and 10 μM for ethyl-melagatran.
HPLC Method for the Reduction ofN-Hydroxy-Melagatran to Melagatran.
An isocratic separation was carried out by the first mobile phase described above, with the same retention times for melagatran andN-hydroxy-melagatran. Standard curves with 0 to 250 μM for melagatran were constructed and found to be linear over this range with a correlation coefficient >0.99. The recovery of melagatran from incubation mixture was 98.5 ± 3.8%. The determination limit of melagatran was about 0.125 μM.
HPLC Method for the Reduction of Ximelagatran to Ethyl-Melagatran.
The separation was carried out isocratically by phosphate buffer (20 mM, 10 mM tetramethylammoniumchloride, pH 4.5) and acetonitrile in the proportion of 80:20 (v/v) by a LiChrospher RP-select B column and a corresponding precolumn. The retention times were 8.3 ± 0.6 min (ethyl-melagatran) and 15.4 ± 0.5 min (ximelagatran).
Standard curves with 10 to 600 μM melagatran were constructed and found to be linear over this range with a correlation coefficient >0.98. The recovery of melagatran from incubation mixture was 102.5 ± 6.4%. The determination limit of melagatran was about 1.25 μM.
Results
Activation of Ximelagatran.
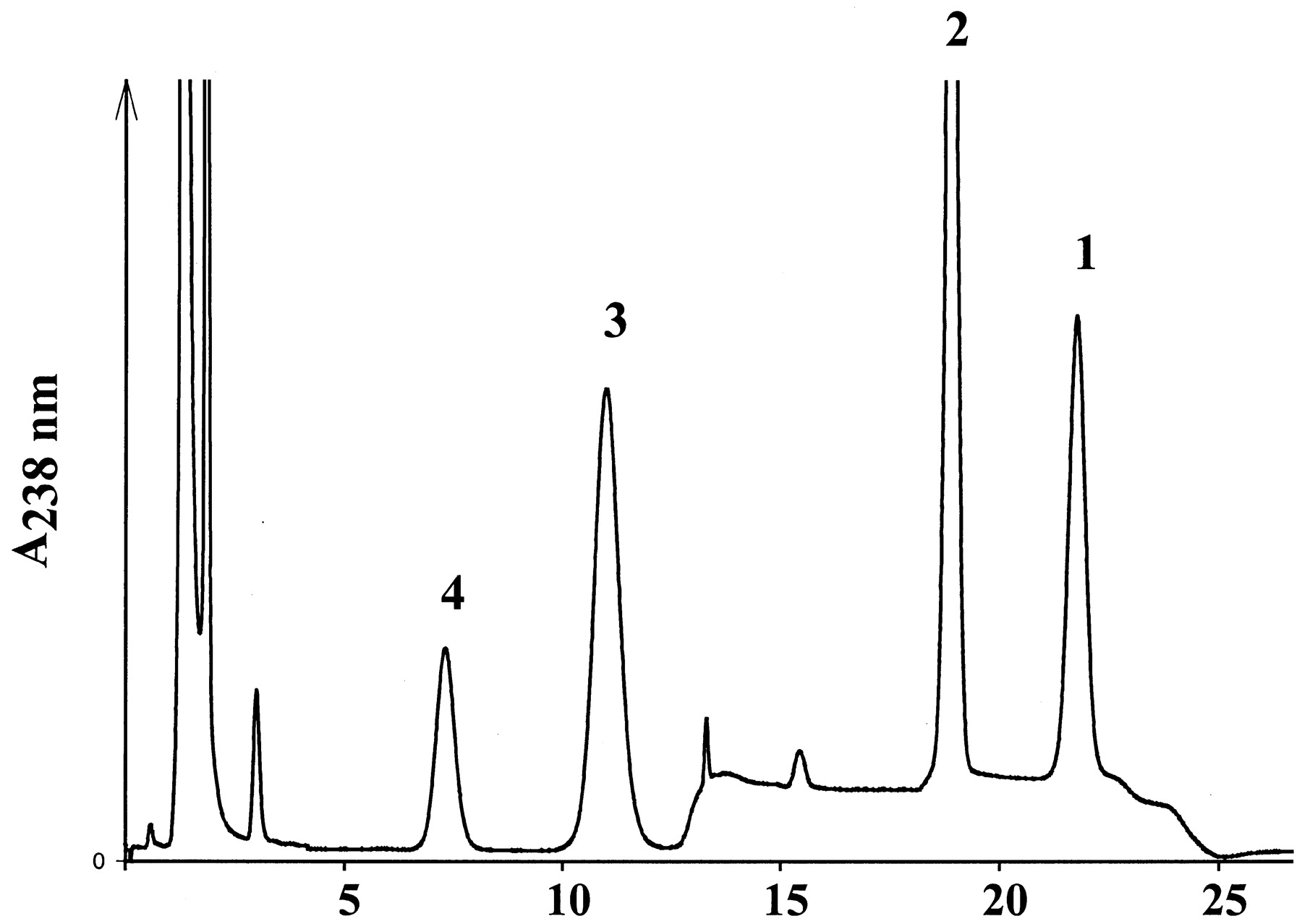
The in vitro activation of ximelagatran to melagatran via two intermediate mono-prodrugs, ethyl-melagatran (H338/57) andN-hydroxy-melagatran (H415/04), by liver and kidney microsomes and mitochondria from pig and human is demonstrated for the first time (Table 1). A new HPLC analytical method was developed to separate and quantify the metabolites of ximelagatran. A representative chromatogram, recorded after the incubation of ximelagtran with pig liver mitochondria, is shown in Fig. 2. The retention times of the metabolites are in accordance with those of the reference compounds: 7.4 ± 0.3 min (melagatran), 11.7 ± 0.6 min (N-hydroxy-melagatran), and 19.7 ± 0.7 min (ethyl-melagatran). The determination limits are 0.15 μM for melagatran, 1.25 μM for N-hydroxy-melagatran, and 10 μM for ethyl-melagatran. Both interstage mono-prodrugs were formed by different enzyme sources and cell compartments. In particular, human liver microsomes and mitochondria exhibited high rates for the ester cleavage. Although there was no ethyl-melagatran detectable when incubating the double prodrug ximelagatran with human liver microsomes, melagatran was formed. In addition, melagatran was undetectable after the incubation of ximelagatran with human kidney microsomes, whereas the reduction and the ester cleavage took place and the intermediate metabolites were formed. The activation of the double prodrug ximelagatran showed linearity over 60 min. The optimized pH value was pH 7.0, including both activation steps. The ester hydrolysis prefers a more basic pH and the N-reduction has its pH optimum at 6.0 (Fig. 3, a and b).
In vitro bioactivation of ximelagatran via ethyl-melagatran (H338/57) and N-hydroxy-melagatran (H415/04) to melagatran (H319/68) by liver and kidney microsomes and mitochondria of pig and human

Representative HPLC trace after incubation of ximelagatran (H376/95) 1 with mitochondrial preparations from pig liver homogenate; 2, ethyl-melagatran (H338/57); 3, N-hydroxy-melagatran (H415/04); 4, melagatran (H319/68).
For the incubation procedure, see Materials and Methods. For structures, see Fig. 1.
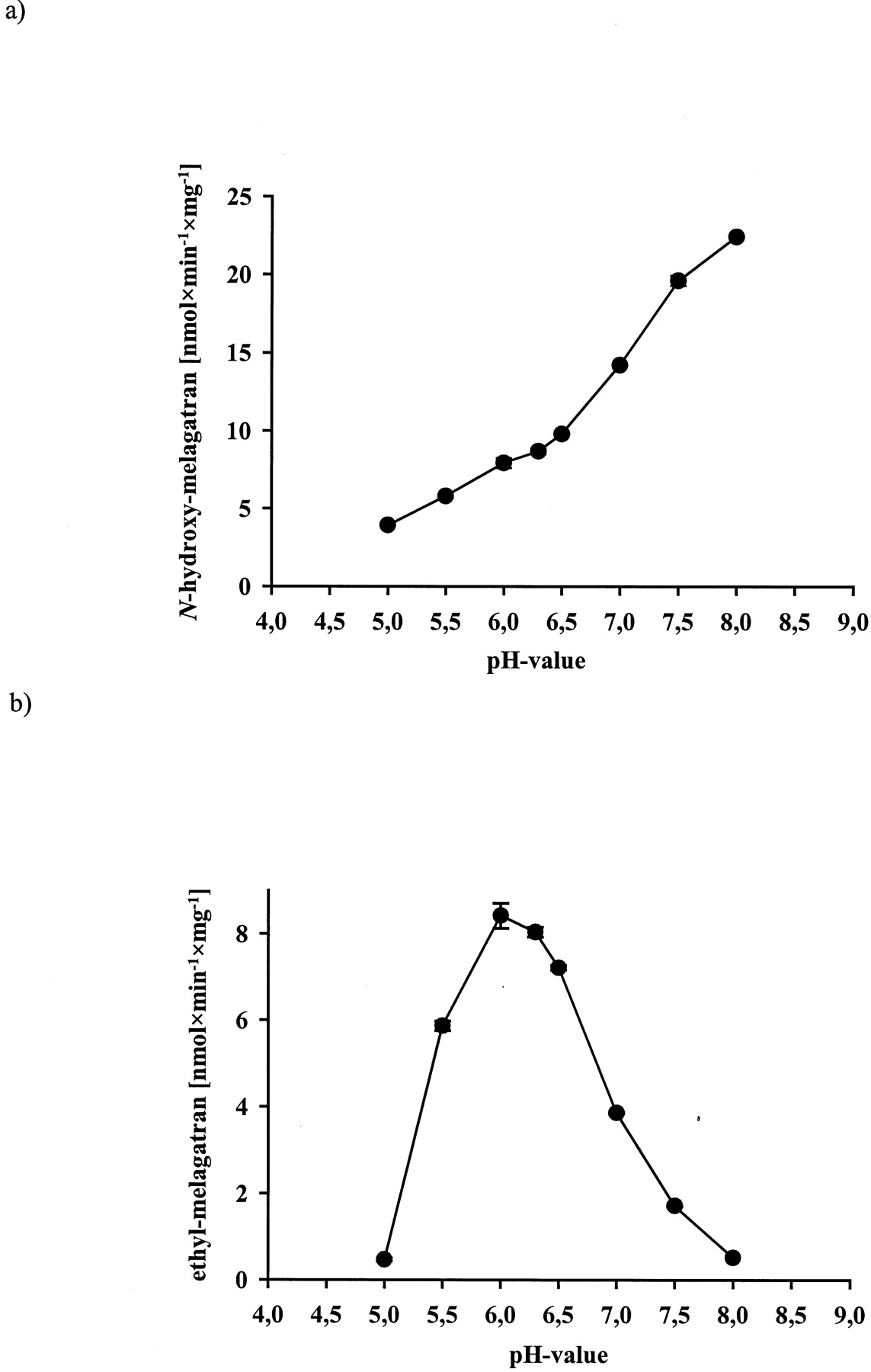
Effect of different pH values on the ester hydrolysis (a) and on the N-reduction (b) of ximelagatran to the corresponding metabolites with microsomal preparations from kidney microsomes.
The incubation mixture consisted of 0.5 mM ximelagatran as substrate and 0.2 mg/ml kidney microsomes in 100 mM potassium phosphate buffer of different pH values (5.0–8.0). After 5-min preincubation at 37°C under aerobic conditions the reaction was started by the addition of 1 mM NADH to a total volume of 250 μl. After 30 min, the reaction was stopped by the addition of cold methanol, and the supernatant was analyzed by HPLC. Data are means ± S.D. of four determinations.
The preferred cosubstrate for the reduction was NADH. If NADPH was used as cosubstrate, the rates were 50% lower. The ester cleavage was independent of any cosubstrate (NADH or NADPH). There was no significant effect of additional 3.3 mM MgCl2 on both reactions (data not shown).
Ester Cleavage of Ximelagatran toN-Hydroxy-melagatran.
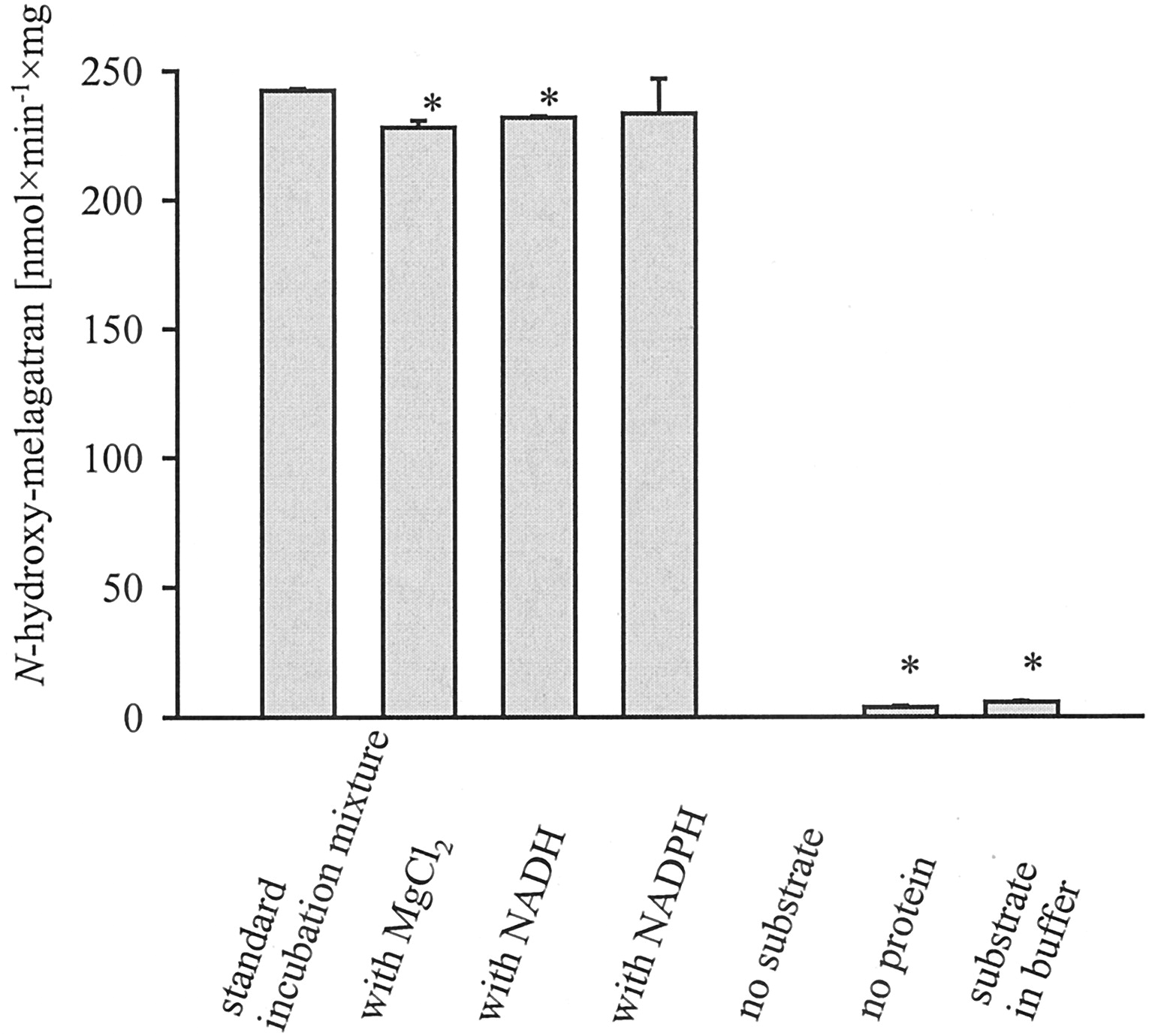
The in vitro ester hydrolysis of ximelagatran toN-hydroxy-melagatran could be shown by unspecific carboxylic esterases from pig liver (Fig. 4).

Characterization of the ester hydrolysis of ximelagatran to N-hydroxy-melagatran by unspecific carboxyl esterases from pig liver.
A standard incubation mixture consisted of 2 mM ximelagatran and 0.5 U of unspecific carboxylic esterases in 100 mM phosphate buffer pH 7.4. In the other experiments, 3.3 mM MgCl2 or 1 mM NADH or 1 mM NADPH was added. After a 3-min preincubation period at 37°C in a shaking water bath, the reaction was initiated by the addition of thermostated esterases. After 20 min, the reaction was stopped by the addition of cold methanol, and the supernatant was analyzed by HPLC. Each column represents the means ± S.D. of four determinations (∗, p < 0.05).
The activation of ximelagatran by esterases was linear over more than 100 min and followed Michaelis-Menten kinetics. Esterases/incubation mixture (0.5 U) was sufficient to obtain linear formations ofN-hydroxy-melagatran. The optimized substrate concentration was 2 mM, and the pH dependence shows higher product formation at higher pH values. A physiological pH of 7.4 was selected for further studies. The ester cleavage of ximelagtran toN-hydroxy-melagatran by unspecific carboxylic esterases does not require NADH or NADPH as cosubstrate, the addition of 3.3 mM MgCl2 had no significant effect. The kinetic parameters were determined: Vmaxamounts to 398 nmol of N-hydroxy-melagatran/min/mg esterases and Km is 2.16 mM. The catalytic efficiency was 1.84 × 10−4 l/min/mg protein.
To exclude the participation of CYP3A4 in the hydrolysis of the ester functionality of ximelagtran, it was tested on esterase activity. Recombinant, purified CYP3A4 in combination with NADPH-cytochrome P450 reductase was not capable of cleaving the carboxylic ester of ximelagatran to N-hydroxy-melagatran.
N-Reduction of N-Hydroxy-Melagatran to Melagatran.
The in vitro N-reduction of N-hydroxy-melagatran by microsomes and mitochondria of liver and kidney from pig and human was also demonstrated (Table 2) and showed very high rates. In particular, the activities for pig liver and kidney mitochondria were 2 to 4 times higher than for microsomes. An isocratic HPLC analytical method was used to separate and quantify the reduced metabolite melagatran of H415/04. The retention time of melagatran 7.4 ± 0.3 min also agreed with the retention time of the reference compound. The determination limit of melagatran was 0.125 μM. The reduction showed linearity over 60 min, followed Michelis-Menten-kinetics. The Kmvalues of all enzyme sources were in the same range (Table 2). The optimized pH value for the N-reduction was pH 6.3 and the addition of MgCl2 did not significantly increase the reductions (data not shown). The reaction required either NADH or NADPH as cosubstrate, whereas considerably higher conversion rates were detected in the presence of NADH (data not shown).
In vitro N-reduction of N-hydroxy-melagatran to melagatran by liver and kidney microsomes and mitochondria of different species and kinetic data
Reconstitution Studies with Ximelagatran andN-Hydroxy-melagatran.
To decrease the determination limit in the reconstitution experiments, a new HPLC method was developed for the reduction of the double prodrug ximelagatran to ethyl-melagatran. Ethyl-melagatran was eluted isocratically at 8.3 ± 0.6 min and ximelagatran at 15.4 ± 0.5 min. The reduction of N-hydroxy-melagatran to melagatran was measured with the same HPLC method as used for microsomes and mitochondria.
The reduction of the double prodrug ximelagatran and the mono-prodrugN-hydroxy-melagatran to their corresponding amidines ethyl-melagatran and melagatran could be demonstrated by pig purified cytochrome P450 isoenzyme of the subfamily 2D in the presence of cytochrome b5 and NADH-cytochromeb5 reductase (Table3). Omission of cytochrome P450 considerably reduced the conversion rates. Cytochromeb5 andb5 reductase alone were not capable of reducing the amidoximes. Furthermore, a chemical formation of amidine metabolites could be excluded by omitting of any protein.
N-Reduction of ximelagatran and N-hydroxy-melagatran, amidoxime prodrugs in the reconstituted system consisting of cytochrome b5, NADH-cytochrome b5reductase, and CYP2D
To exclude the participation of CYP3A4, the xenobiotic-metabolizing cytochrome P450 enzyme with the highest concentration in human liver, and other P450s in the reduction of N-hydroxylated amidines, reductase activity by a group of recombinant human P450s was tested. None of the recombinant P450s (CYP1A2, 2A6, 2C8, 2C9, 2C19, 2D6, and 3A4) tested together with cytochromeb5 and NADH-cytochromeb5 reductase was capable of reducing the melagatran prodrugs (data not shown).
Discussion
The introduction of modern technologies, such as combinatorial chemistry and high-throughput pharmacological screening in drug discovery, has resulted in a vast increase in the number of lead compounds identified. However, the compounds generated in high-throughput drug discovery programs very often possess properties that are not compatible with oral administration, which is desired because of the convenience of this administration route. So oral absorption, which means the transport of a drug molecule across the mucosal membrane, is one goal of drug development (van der Waterbeemd et al., 2001). In fact, the clinical development of new drugs is often terminated due to unfavorable pharmacokinetic characteristics, such as poor bioavailability of the drug after oral administration (Clement, 2002). Bioavailability can be improved by using amidoximes instead of amidines (Clement, 1993) as prodrugs. This principle has been applied to drug candidates such as sibrafiban (Weller et al., 1996) and melagatran (Eriksson et al., 1999), and further compounds under development (Kitamura et al., 2001; Schipper et al., 2001).
The enzymatic basis of the prodrug principle, reduction of the amidoxime to the amidine, was mainly studied with benzamidoxime as a model compound. This is the first study that clearly demonstrates that more complicated molecules such as the double-prodrug ximelagatran as well as the intermediate N-hydroxy-melagatran are metabolized in liver and kidney by microsomal and mitochondrial systems. Reduction of an amidoxime could be shown previously for model compounds such as benzamidoxime by pig liver and kidney microsomes (Clement and Mau, 1999) or mitochondria (Clement and Deters, 2000).
The formation of amidines by microsomal impurities in mitochondria and vice versa could be excluded by using established corresponding marker reactions (Sottocasa et al., 1967; Yasukochi and Masters, 1976) (data not shown). Both ximelagatran and N-hydroxy-melagatran were substrates for the reducing systems so that the activation follows the two pathways as shown in Fig. 1. It is clear that ethyl-melagatran and N-hydroxy-melagatran only represent intermediates that even in in vitro studies are sometimes not detectable (Table 1). The activation of the double prodrug ximelagatran to the active principle melagatran was catalyzed by all enzyme sources, except for human kidney microsomes. However, both mono-prodrugs were formed again. There were no indications for metabolites other than ethyl-melagatran,N-hydroxy-melagatran, and melagatran.
A pH of 7.0 and the use of NADH as cosubstrate constitute optimum incubation conditions for the complete activation of ximelagatran via two intermediate mono-prodrugs to melagatran. The preference for NADH has also been observed for the model compound benzamidoxime (Clement et al., 1997). This is in agreement with an electron transfer by NADH-cytochrome b5 reductase. The redox potential for transfer of electrons fromb5 to a P450 is unfavorable (Guengerich, 2001). The first reported reduction catalyzed by this system was thus unexpected and the mechanism needs further clarification. It is possible that by complexation, reduction potentials are changed, which has been reported for cytochromeb5 (Walker et al., 1988; Rivera et al., 1998). When using mechanisms of this new type of reduction involving P450 isoenzymes, it has to be taken into account that the reduction is not inhibited by oxygen (Clement et al., 1997; Clement, 2002). This unusual behavior might be explained by the formation of a complex between the N-hydroxylated structures and P450 iron in the ferric state. Electrons are donated viab5 reductase andb5 to the N-hydroxylated compound, which is reduced and the P450 enzyme in the resting state is regenerated. Oxygen cannot interfere because it is bound by ferrous P450 (Clement, 2002). It can also not be ruled out that the reduction is actually performed by b5 reductase and b5 and that the role of the P450 isoenzyme is to increase the interaction betweenb5 and its reductase (Clement, 2002).
The reduction in the presence of oxygen is in contrast to the known reductions performed by P450 enzymes alone or/and NADPH-cytochrome P450 reductase used, for example, for the bioactivation ofN-oxide prodrugs in tumor cells with low oxygen pressure (Patterson, 2002).
In particular microsomes were able to form higher concentrations of the intermediate N-hydroxy-melagatran than mitochondria, whereas mitochondria formed higher concentrations of the mono-prodrug ethyl-melagatran. This can be explained by higher concentrations of esterases in microsomal preparations than in mitochondria. On the other hand, the reducing enzymes are present in mitochondria more than in microsomes as evidenced by the higher activity of mitochondria in reducing N-hydroxy-melagatran (Tables 1 and 2).
The ester cleavage of ximelagatran to N-hydroxy-melagatran catalyzed by unspecific carboxylic esterases was independent of any cosubstrate such as NADH or NADPH. CYP3A4 in combination with NADPH-cytochrome P450 reductase was not able to catalyze the ester hydrolysis of ximelagatran. Thus, an oxidative ester cleavage as has been demonstrated, for example, for the ester hydrolysis of dihydropyridines such as nifedipin (Guengerich, 1987) can be excluded.
So far, one enzyme system from pig liver microsomes capable of reducing the amidoxime moiety has been identified that is composed of cytochromeb5, NADH-cytochromeb5 reductase, and a P450 isoenzyme of the subfamily 2D (Clement et al., 1997). This enzyme system apparently accepts a wide range of compounds as substrates and also reduces the melagatran prodrugs with very high rates (Table 3). It was not influenced by oxygen (data not shown) and thus is of high relevance for the in vivo situation.
To predict any possible drug interaction, it is very important to know the human enzyme system that is responsible for the reduction of suchN-hydroxylated amidines. CYP2D6 is not involved because microsomes from a human donor deficient in CYP2D6 were also capable of reducing amidoximes with high conversion rates (data not shown). It is known that a reaction that is catalyzed by a P450 enzyme of a certain subfamily in a distinct species is not necessarily performed by the isoenzyme of the same subfamily in another species. In this context, it is interesting to note that CYP2D25 can catalyze the 25-hydroxylation of vitamin D3, whereas CYP2D6 is not able to perform this reaction (Hosseinpour and Wikvall, 2000). Commercially available recombinant P450s expressed in lymphoblasts (CYP1A2, 2A6, 2C8, 2C9, 2C19, 2D6, and 3A4), which are involved in several xenobiotic metabolisms, were not capable of reducing the model compound benzamidoxime (data not shown) as well as the melagatran prodrugs ximelagatran and N-hydroxy-melagatran.
These experiments indicate that none of the known foreign compound-metabolizing human cytochrome P450 enzymes are involved in the reduction. Another explanation could be that the reductive activity cannot be reconstituted by the recombinant enzymes mentioned above because they contain inhibitors of the reduction and are optimized for oxidative reactions. The high reductive activity of mitochondria and extrahepatic organs that is unusual for foreign compound metabolism might be explained by the involvement of enzymes that also play a major role in the metabolism of endogenous compounds.
Future work will attempt the purification of this P450 enzyme from human liver microsomes and mitochondria and will also be directed toward the elucidation of the human esterases involved in the activation of ximelagatran.
In conclusion, it can be summarized that orally available double prodrugs such as ximelagatran are metabolized by enzymes present in several organs and cell organelles. One responsible enzyme system seems to consist of cytochrome b5,b5 reductase, and a P450 enzyme and is not influenced by oxygen.
Acknowledgments
We are grateful to Astra for financial support and cooperation on this project. We also thank S. Wichmann and M. Wollny for technical assistance, W. Karhan and S. Friedrich for providing the enzymes for reconstitution experiments, and S. Deters and S. Mau for cooperation in preparing mitochondria and microsomes.
Footnotes
- Abbreviations used are::
- DLPC
- dilaurylphosphatidylcholine
- HPLC
- high-performance liquid chromatography
- Received September 11, 2002.
- Accepted February 10, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}