


Abstract
Verapamil inhibition of CYP3A activity results in many drug-drug interactions with CYP3A substrates, but the mechanism of inhibition is unclear. The present study showed that verapamil enantiomers and their major metabolites [norverapamil and N-desalkylverapamil (D617)] inhibited CYP3A in a time- and concentration-dependent manner by using pooled human liver microsomes and the cDNA-expressed CYP3A4 (+b5). The values of the inactivation kinetic parameters kinact and KI obtained with the cDNA-expressed CYP3A4 (+b5) were 0.39 min-1 and 6.46 μM for R-verapamil, 0.64 min-1 and 2.97 μM for S-verapamil, 1.12 min-1 and 5.89 μM for (±)-norverapamil, and 0.07 min-1 and 7.93 μM for D617. Based on the ratio of kinact and KI, the inactivation potency of verapamil enantiomers and their metabolites was in the following order: S-norverapamil > S-verapamil > R-norverapamil > R-verapamil > D617. Using dual beam spectrophotometry, we confirmed that metabolic intermediate complex formation with CYP3A was the mechanism of inactivation for all compounds. The in vitro unbound fraction was 0.84 for S-verapamil, 0.68 for R-verapamil, and 0.84 for (±)-norverapamil. A mechanism-based pharmacokinetic model predicted that the oral area under the curve (AUC) of a CYP3A substrate that is eliminated completely (fm = 1) by the hepatic CYP3A increased 1.6- to 2.2-fold after repeated oral administration of verapamil. For midazolam (fm = 0.9), a drug that undergoes extensive intestinal wall metabolism, the predicted increase in oral AUC was 3.2- to 4.5-fold. The predicted results correlate well with the in vivo drug interaction data, suggesting that the model is suitable for predicting drug interactions by mechanism-based inhibitors.
Verapamil is a calcium channel blocker widely used in the treatment of angina pectoris, coronary artery disease, cardiac arrhythmias, and hypertension (McTavish and Sorkin, 1989). Clinically available formulations of verapamil are racemic mixtures of S- and R-enantiomers, which have different pharmacokinetic and pharmacological properties. S-verapamil has been shown to have a negative dromotropic effect on atrioventricular conduction that is 10 to 18 times greater than R-verapamil in man (Echizen et al., 1985). S-verapamil is preferentially eliminated during first-pass metabolism, and as a consequence, the plasma concentration ratio of R- to S-verapamil is around 5:1 after oral administration and is approximately 2:1 after intravenous administration (Vogelgesang et al., 1984). O-Demethylation and N-dealkylation are the two major metabolic pathways of verapamil. CYP3As are the primary enzymes involved in N-dealkylation, which produces two major metabolites, norverapamil and N-desalkylverapamil (D6171) (Kroemer et al., 1992).
Verapamil inhibits the metabolism of several coadministered CYP3A substrates (Robson et al., 1988; Backman et al.,1994; Kantola et al., 1998; Lamberg et al., 1998). For example, in healthy volunteers, the oral AUCs of midazolam, cyclosporine A, simvastatin, and buspirone increase 2- to 6-fold after multiple doses of verapamil treatment. These in vivo drug interactions are not anticipated, however, because steady-state plasma concentrations of verapamil are far below the competitive inhibition constant (Ki) estimated in vitro. These data suggest that the inhibition may involve the metabolites of verapamil and/or verapamil may not act as a reversible inhibitor of CYP3A enzymes in vivo.
Verapamil and its metabolites contain amine functional groups (Fig. 1), which are common features for mechanism-based inactivators that form a metabolic intermediate complex (MIC). A mechanism-based inactivator is catalytically activated by a target enzyme to a reactive intermediate, that then inactivates the enzyme before its release from the active site (Silverman, 1995). The inhibitory effects of the inactivation remain after elimination of the inhibitors from the body and can lead to serious side effects (Kanamitsu et al., 2000). In the case of mechanism-based inactivation through MIC formation, inactivators are catalytically oxidized to intermediates or products that coordinate tightly to the prosthetic heme of the P450, leading to irreversible inactivation of the enzyme under physiological conditions. Primary amines are required for complex formation with amine-containing compounds, but secondary and tertiary amines are suitable precursors of the P450 complexes if they are N-dealkylated in situ to primary amines (Ortiz de Montellano and Correia, 1995). Previous studies showed that verapamil forms a MIC with CYP3A (Ma et al., 2000), but the capability of verapamil enantiomers and their metabolites to form a MIC with CYP3A is not defined. The aims of the present study are to test whether verapamil enantiomers are mechanism-based inhibitors, to investigate the potential role of verapamil metabolites in drug interactions, and to quantitatively predict the extent of inhibition of CYP3A-mediated metabolism after repeated oral administration of verapamil using a mechanism-based inhibition model.
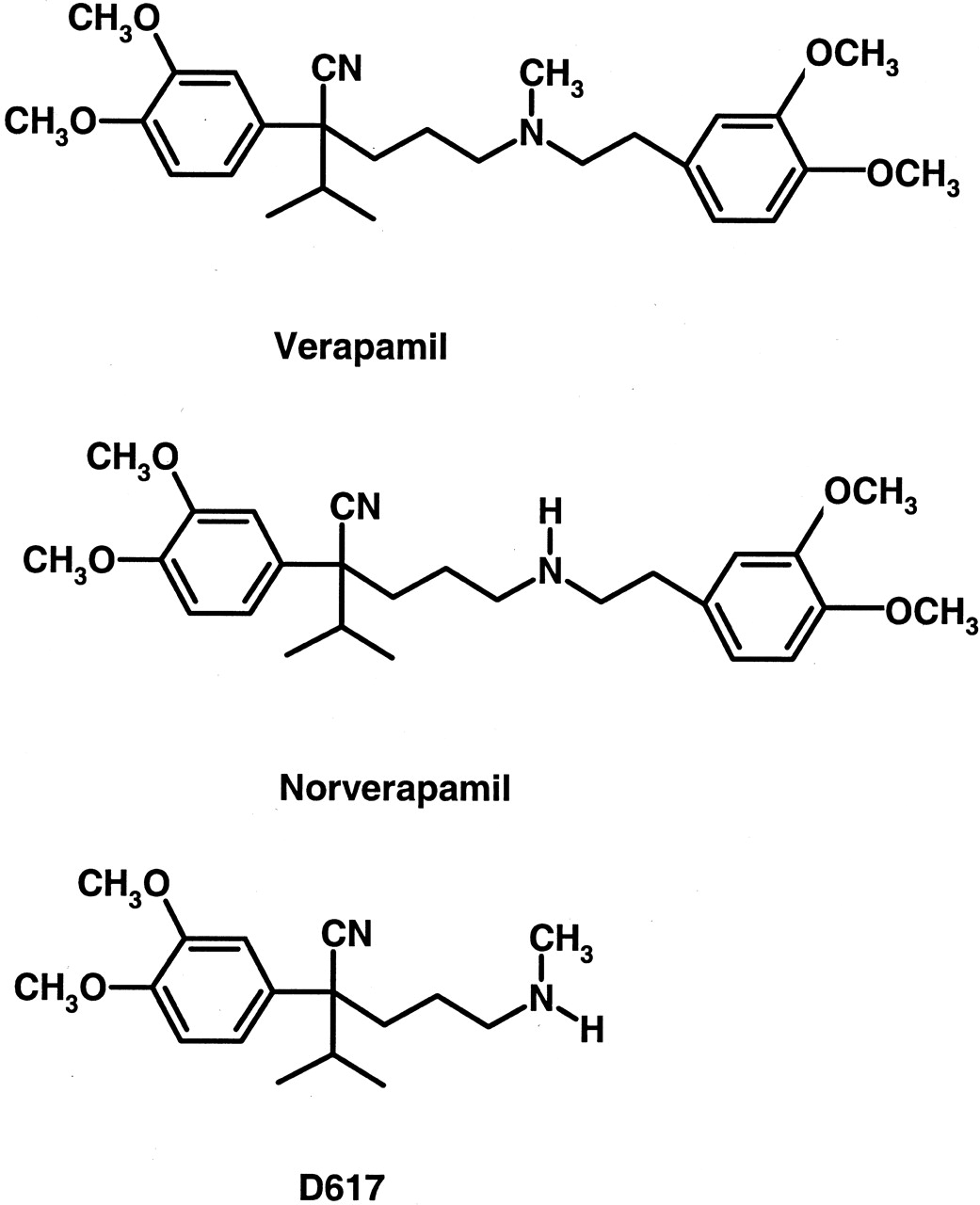
Chemical structures of verapamil, norverapamil, and D617.
All three compounds contain amine functional groups and have the potential to form metabolic intermediate complexes with CYP3A.
Materials and Methods
Chemicals. Dextromethorphan, (±)-verapamil, R-verapamil, S-verapamil, and (±)-norverapamil were purchased from Sigma/RBI (Natick, MA). D617 was generously supplied by Abbott GmbH & Co. (Ludwigshafen, Germany). Testosterone, 6β-hydoxytestosterone, and desmethyldiazepam were purchased from Sigma-Aldrich (St. Louis, MO). NADPH (98%) was purchased from Roche Diagnostics (Indianapolis, IN). All other reagents were of high-performance liquid chromatography (HPLC) grade.
Human Liver Microsomes (HLMs) and cDNA-Expressed Human P450s. The HLMs were prepared from five human liver tissues obtained at surgery in accordance with protocols approved by the Institutional Review Board of Indiana University (Indianapolis, IN) (Gorski et al., 1994). Microsomal fractions were prepared and pellets were suspended in a buffer to a protein concentration of 20 mg/ml and were kept at -80°C (Gorski et al., 1994). Cytochrome P450 was quantified by the method of Omura and Sato (1964). Protein concentration of the pooled HLMs was assayed using the Lowry method (Lowry et al., 1951). The cytochrome P450 concentration of the pooled HLMs was 0.3 nmol/mg of protein. Microsomes containing the cDNA-expressed CYP3A4 (+b5) (Supersomes) were purchased from BD Gentest (Woburn, MA), and the microsomal protein concentration and P450 content were provided by the manufacturer.
Free Fractions of Verapamil and Norverapamil in HLMs and Supersomes. The binding of verapamil and norverapamil to microsomes was measured by the ultrafiltration method using an MPS ultrafiltration device (Millipore Corporation, Bedford, MA). Verapamil or norverapamil was mixed with pooled HLMs (1.6 mg/ml) or cDNA-expressed CYP3A4 (+b5) and the final concentration of verapamil or norverapamil was 10 μg/ml. The mixture (0.5 ml) was placed in the sample reservoir of the MPS device in triplicate. After incubation for 30 min (37°C), each sample was centrifuged at 1200g for 15 min, yielding 70 to 100 μl of filtrate. A 70-μl aliquot of each filtrate was used to determine the unbound concentration of verapamil or norverapamil. The same volume was taken from the sample reservoir just before centrifugation to determine the total concentrations. Dextromethorphan (internal standard, 500 ng) was added to 70 μl of ultrafiltrate or microsomal suspension. Sixty microliters of the filtrate or 20 μl of the microsomal suspension was injected directly onto the HPLC column without extraction. The free fraction of verapamil or norverapamil was calculated as the ratio of the filtrate concentration to the total concentration. Preliminary experiments indicated that binding to the ultrafiltration device was not significant.
Quantitation of CYP3A Inactivation. Testosterone 6β-hydroxylation was used as a marker to quantify CYP3A activity. HLMs (0.8 mg/ml) or the cDNA-expressed CYP3A4 (+b5) were preincubated with various concentrations of verapamil enantiomers or their metabolites, respectively, in the presence of NADPH at 37°C for various time durations. Fifty microliters of preincubation mixture was transferred to a tube containing 250 μM testosterone and 1 mM NADPH in 0.1 M sodium phosphate buffer (950 μl). A saturating concentration of testosterone was chosen to measure the activity of all remaining free CYP3A. The testosterone 6β-hydroxylation reaction was determined at 37°C for 10 min, and the reaction was terminated by adding 1 ml of ice-cold acetonitrile.
HPLC Assay of 6β-Hydoxytestosterone, Verapamil, and Norverapamil. 6β-Hydroxytestosterone concentration was determined as previously described (Zhao et al., 2002). The HPLC system used a 5-μm C18 column (Luna; Phenomenex, Torrance, CA) and a mobile phase of methanol/20 mM sodium phosphate buffer (60:40, v/v), containing 0.1% (v/v) triethylamine adjusted to pH 6.3 with orthophosphoric acid, pumped at a flow rate of 1 ml/min and ultraviolet detection at a wavelength of 254 nm. Standard curves ranged from 25 ng/ml (limit of quantification) to 4000 ng/ml. Quality control samples (n = 2; concentrations of 150 and 1500 ng/ml) were run with each set of unknowns. Standard curves were deemed acceptable if the quality controls concentrations estimated from the standard curve were within 10% of their prepared concentrations. If the estimates were >10%, then the standard curve and unknown samples were discarded and repeated.
Verapamil and norverapamil were determined by HPLC. The HPLC consisted of a 5-μm C18 column (Luna) and a mobile phase of methanol/20 mM, pH 6.3, sodium phosphate buffer (70:30, v/v containing 0.1% (v/v) triethylamine) pumped at 1 ml/min with fluorescence detection at an excitation wavelength of 280 nm without emission filter.
Normal-phase HPLC was used for the chiral separation of norverapamil (Brandsteterova et al., 2001) to obtain the individual norverapamil enantiomers for use in the inactivation assay. Fifty micrograms of racemic norverapamil was injected onto a Chiralpak AD column (0.46 cm × 25 cm; Chiral Technologies Inc., Daicel Chemical Industries Ltd., Düsseldorf, Germany). The mobile phase consisted of a mixture (90:10, v/v) of hexane and ethanol containing 0.1% (v/v) triethylamine and pumped at 1 ml/min. Detection was by fluorescence at an excitation wavelength of 210 nm with a 300-nm emission filter.
Spectrophotometric Detection of Metabolic Intermediate Complex. cDNA-expressed CYP3A4 (+b5) was used to characterize MIC formation associated with the metabolism of verapamil. MIC was measured as a function of time in samples containing R-verapamil, S-verapamil, norverapamil, or D617 at 10 μM in a microsomal buffer (100 mM sodium phosphate buffer, 5 mM magnesium chloride, pH 7.4). Spectral differences between the reference and sample cuvettes were obtained with a dual-beam spectrophotometer (Uvikon 933 double-beam UV-visible spectrophotometer; Research Instruments International, San Diego, CA) by scanning from 400 to 500 nm and monitoring every 5 min for up to 60 min. In each case, the sample cuvette contained 200 pmol of CYP3A4 (+b5), inactivator, and 1 mM NADPH. The reference cuvette contained everything in the sample cuvette, but no inactivator was added. All MIC formation experiments were maintained at 37°C and initiated by the addition of NADPH in a final incubation volume of 1 ml. The extent of MIC formation formed was quantified based on previous reported extinction coefficient (455–490 nm) value of 64 M-1/cm-1 (Pershing and Franklin, 1982).
Estimation of Inactivation Constants. For incubations with HLMs or cDNA-expressed CYP3A4 (+b5), the natural logarithm of the percentage of the remaining CYP3A activity was plotted against the preincubation time. The observed inactivation rate constants (kobs) were determined from the slopes of the initial linear decline in activity (Silverman, 1995). The parameters kinact and KI were obtained from plotting kobs against the inhibitor concentration by using the nonlinear regression program (WinNonlin 1.1; Pharsight, Mountain View, CA) according to eq. 1 (Mayhew et al., 2000): 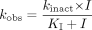 where kinact is the rate constant that defines the maximal rate of inactive enzyme formation, I is the initial concentration of the inhibitor, and KI is the inhibitor concentration when kobs = kinact/2. Equation 1 assumes that there is negligible change of I in the incubation period and that loss of enzyme is solely due to inactivation by the inhibitor. The unbound KI (KI,u) was calculated from KI and fu,m, where fu,m is the free fraction of the drugs in the microsomes.
where kinact is the rate constant that defines the maximal rate of inactive enzyme formation, I is the initial concentration of the inhibitor, and KI is the inhibitor concentration when kobs = kinact/2. Equation 1 assumes that there is negligible change of I in the incubation period and that loss of enzyme is solely due to inactivation by the inhibitor. The unbound KI (KI,u) was calculated from KI and fu,m, where fu,m is the free fraction of the drugs in the microsomes.
Prediction of the Extent of Inhibition in Vivo. When an enzyme is inactivated by one drug, the intrinsic clearance of a second drug that primarily relies on that enzyme is reduced. The theoretical background on the prediction of in vivo CYP3A4 inhibition using a mechanism-based model has been described in detail elsewhere (Mayhew et al., 2000). Briefly, under the baseline condition, the rate of change of active enzyme concentration (dEt/dt) is determined by the balance between the rate of de novo synthesis (R0) and the rate of degradation (eq. 2). 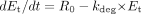 kdeg is the endogenous degradation rate constant of the enzyme, and Et is the total active enzyme concentration.
kdeg is the endogenous degradation rate constant of the enzyme, and Et is the total active enzyme concentration.
At steady state (dEt/dt = 0), the concentration of the enzyme (Ess) is defined by: 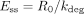
In the presence of a mechanism-based inhibitor (assuming the unbound plasma concentration of the inhibitor is far below the Ki), the steady-state enzyme concentration (Ess) is given by eq. 4: 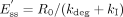 where kI is the in vivo inactivation rate constant of the enzyme. In turn, the steady-state enzyme concentration determines the intrinsic clearance of a substrate (Wilkinson and Shand, 1975),
where kI is the in vivo inactivation rate constant of the enzyme. In turn, the steady-state enzyme concentration determines the intrinsic clearance of a substrate (Wilkinson and Shand, 1975), 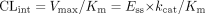 Equation 5 assumes that at the enzyme active site [S] << Km. Vmax and Km are the maximal rate and the Michaelis constant of the affected pathway of metabolism, and kcat is the first-order rate constant that relates Vmax to Ess.
Equation 5 assumes that at the enzyme active site [S] << Km. Vmax and Km are the maximal rate and the Michaelis constant of the affected pathway of metabolism, and kcat is the first-order rate constant that relates Vmax to Ess.
If the affected drug is completely absorbed into the portal vein and the liver is the only route for its elimination, the oral clearance of the affected drug can be expressed as (Wilkinson and Shand, 1975): 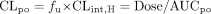 AUCpo is obtained from time 0 to infinity after a single oral dose or over a dosing interval when the affected drug is administered orally to steady state. fu is unbound fraction of the affected drug in the plasma, and CLint,H is the hepatic intrinsic clearance.
AUCpo is obtained from time 0 to infinity after a single oral dose or over a dosing interval when the affected drug is administered orally to steady state. fu is unbound fraction of the affected drug in the plasma, and CLint,H is the hepatic intrinsic clearance.
The extent of a drug-drug interaction can be described by the ratio of the AUCpo in the inactivated state over AUCpo in the baseline state. Assuming that fu, Km, and kcat are not affected by inhibitor coadministration, the oral AUC ratio of the affected drug in the presence and in the absence of the inhibitor can be predicted by combining eqs. 3, 4, 5, and 6: 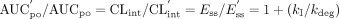 The in vivo inactivation rate constant kI can be substituted with the predicted inactivation rate constant kobs (eq. 1) under the assumption that the in vitro inhibitory constants are directly applicable to the in vivo interaction (eq. 8):
The in vivo inactivation rate constant kI can be substituted with the predicted inactivation rate constant kobs (eq. 1) under the assumption that the in vitro inhibitory constants are directly applicable to the in vivo interaction (eq. 8):  where Iu, the unbound inhibitor concentration in hepatocytes, is assumed to be equal to the unbound average plasma concentration of the inhibitor at the steady state. When KI,u >> Iu, eq. 8 can be simplified to:
where Iu, the unbound inhibitor concentration in hepatocytes, is assumed to be equal to the unbound average plasma concentration of the inhibitor at the steady state. When KI,u >> Iu, eq. 8 can be simplified to: 
When multiple inactivators are present in the systemic circulation simultaneously, the extent of interaction is given by: 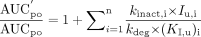 where n is the number of inhibitors present. When the CYP3A pathway is only one of multiple elimination pathways in the liver:
where n is the number of inhibitors present. When the CYP3A pathway is only one of multiple elimination pathways in the liver:  where fm represents the fraction of the total hepatic elimination that is due to the CYP3A pathway in the absence of the inhibitor (Rowland and Matin, 1973). For drugs that are CYP3A substrates, significant first-pass metabolism may occur in the intestinal wall. Therefore, the ratio of AUCpo is determined by both hepatic intrinsic clearance and intestinal wall availability.
where fm represents the fraction of the total hepatic elimination that is due to the CYP3A pathway in the absence of the inhibitor (Rowland and Matin, 1973). For drugs that are CYP3A substrates, significant first-pass metabolism may occur in the intestinal wall. Therefore, the ratio of AUCpo is determined by both hepatic intrinsic clearance and intestinal wall availability. 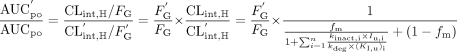 where FG and F′G are the intestinal wall availabilities in the absence and presence of inhibitor, in this case, verapamil. FG is a function of the intrinsic clearance at the intestinal wall (CLint,G),
where FG and F′G are the intestinal wall availabilities in the absence and presence of inhibitor, in this case, verapamil. FG is a function of the intrinsic clearance at the intestinal wall (CLint,G),  where A is the absorption constant that may be a function of epithelial permeability and intestinal blood flow. Assuming that A is unaffected by the presence of the inhibitor, the ratio of F′G to FG can be estimated from the relative change in CLint,G caused by the inhibitor (eq. 14):
where A is the absorption constant that may be a function of epithelial permeability and intestinal blood flow. Assuming that A is unaffected by the presence of the inhibitor, the ratio of F′G to FG can be estimated from the relative change in CLint,G caused by the inhibitor (eq. 14):  The ratio of intestinal intrinsic clearances in the absence and presence of the inhibitors can be estimated from the in vitro data using eq. 15:
The ratio of intestinal intrinsic clearances in the absence and presence of the inhibitors can be estimated from the in vitro data using eq. 15:  The approach presented in eq. 15 assumes that the kinetic parameters obtained using human liver microsomes are applicable for the enterocytes and that the CYP3A pathway is the only elimination pathway present in the intestinal wall. IG is the unbound inhibitor concentration in the enterocytes.
The approach presented in eq. 15 assumes that the kinetic parameters obtained using human liver microsomes are applicable for the enterocytes and that the CYP3A pathway is the only elimination pathway present in the intestinal wall. IG is the unbound inhibitor concentration in the enterocytes.
The time-dependent changes in the concentration of the active enzyme in the liver in the presence of the inhibitor can be expressed as:  where t is the time after the inhibitor is given. Substituting eqs. 3 and 4 into eq. 16, the fraction of initial CYP3A content will be expressed as:
where t is the time after the inhibitor is given. Substituting eqs. 3 and 4 into eq. 16, the fraction of initial CYP3A content will be expressed as: 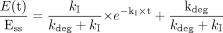
As stated above, the in vivo inactivation rate constant kI can be substituted with the in vitro predicted inactivation rate constant kobs (eq. 1).
Pharmacokinetic parameters of verapamil and norverapamil from the literature (Table 2) and the inactivation parameters estimated in vitro were combined to predict the extent of the in vivo drug interactions. The degradation rate constant, kdeg, was assumed to equal that estimated for the CYP3A degradation half-life in the rat (Correia, 1991) and in CYP3A4-expressing Caco-2 cells (Malhotra et al., 2001), because the corresponding value for CYP3A in human is not available.
Pharmacokinetic parameters used in the prediction
Results
Inhibition of CYP3A4 Activity in Vitro by Verapamil and Metabolites. Coincubation of testosterone and S-verapamil (up to 40 μM) for 10 min in the presence of NADPH and pooled human liver microsomes failed to reveal any significant reversible inhibition of CYP3A activity (Fig. 2A). Time-dependent inhibition of an enzyme is one of the characteristics of mechanism-based inhibition, and therefore S-verapamil was preincubated with the pooled human liver microsomes and cDNA-expressed human CYP3A4 (+b5), respectively. After preincubation (up to 20 min), the reaction mixture of S-verapamil and the enzyme was diluted 20-fold to ensure that reversible inhibition was minimal. The extent of CYP3A inhibition increased as preincubation times and S-verapamil concentrations increased (Fig. 2B). After a 20-min preincubation with 10 μM S-verapamil, the testosterone 6β-hydroxylation activity of the pooled human liver microsomes was reduced by approximately 90% of the no-preincubation control (Fig. 2B). Similar results were also obtained for R-verapamil and verapamil enantiomers when the cDNA-expressed CYP3A4 (+b5) was used (data not shown). Figure 3, A and B, shows the plots of inactivation rate constants against inhibitor concentrations for R- or S-verapamil. The estimated values of the maximum inactivation rates and the dissociation constants of verapamil enantiomers for CYP3A were obtained by fitting the inactivation profiles (Figs. 2B, 3, A and B) into eq. 1 and are presented in Table 1. The values of kinact obtained by using the cDNA-expressed human CYP3A4 (+b5) were approximately 5-fold higher than those obtained by using the pooled human liver microsomes. The estimated values of KI were similar for R-verapamil using either the cDNA-expressed human CYP3A4 (+b5) or the pooled human liver microsomes. However, for S-verapamil, the value of KI obtained by using the pooled human liver microsomes was approximately 2-fold less than that obtained by using the cDNA-expressed human CYP3A4 (+b5).
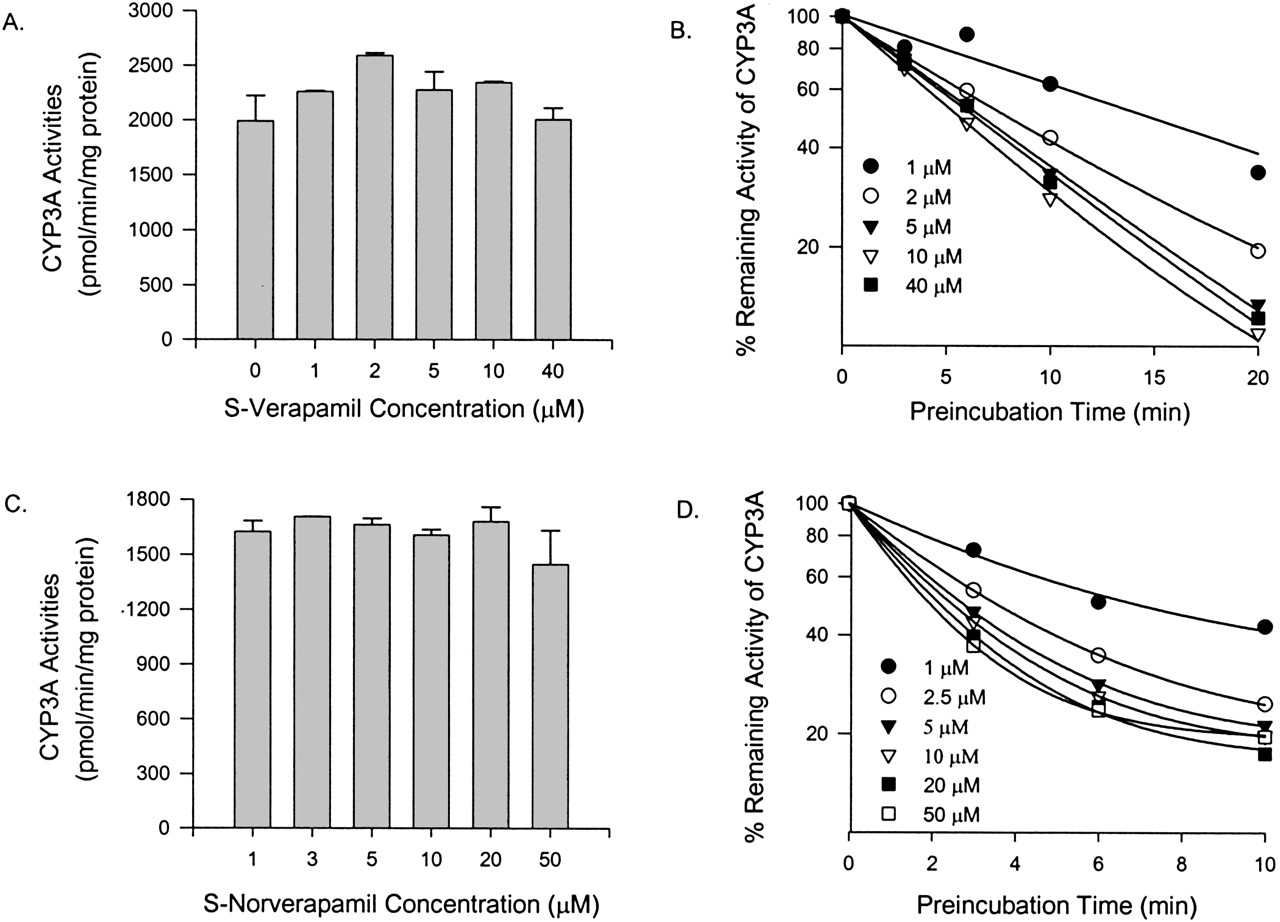
Effect of S-verapamil and S-norverapamil on testosterone 6β-hydroxylation activity of CYP3A in pooled human liver microsomes.
CYP3A activities at 0-min preincubation time when incubated with various concentrations of S-verapamil (A) and S-norverapamil (C). Time- and concentration-dependent inhibition of CYP3A activity by S-verapamil (B) and S-norverapamil (D). Individual data points represent the mean result from duplicate incubations. The line represents the line of best fit for the data.
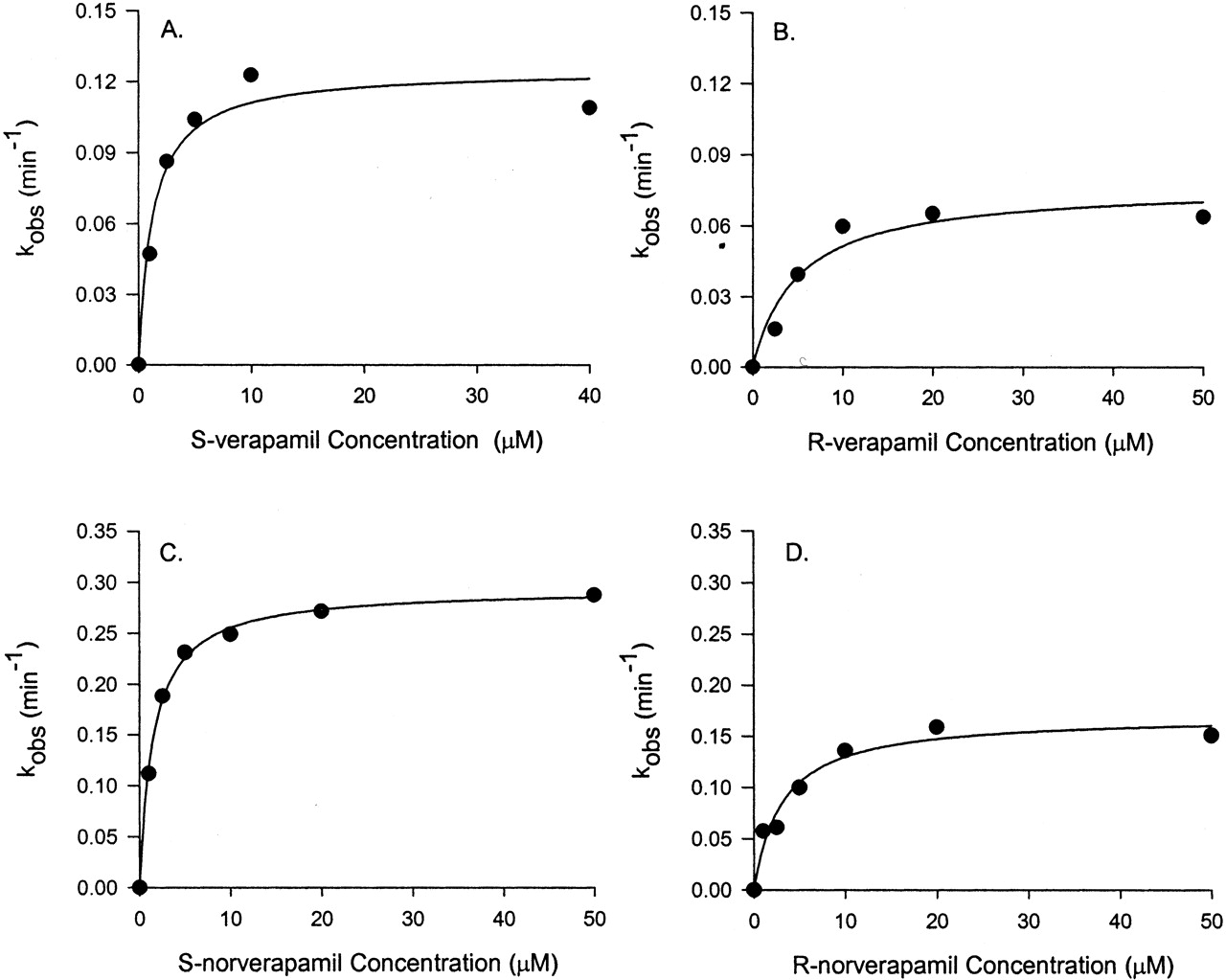
Plots of kinact against inhibitor concentration of verapamil or norverapamil from which estimations of kinetic parameters in vitro for mechanism-based inhibition were made.
Pooled human liver microsomes were incubated with verapamil and norverapamil (0–50 μM) and NADPH, and then CYP3A enzyme activity was monitored by testosterone 6β-hydroxylation. The estimates of kobs from initial rates of enzyme inactivation were plotted against inhibitor concentrations. The line represents the line of best fit with eq. 1. The corresponding kinact and KI values are presented in Table 1.
Estimated inactivation parameters of CYP3A4 by verapamil and its metabolites
Steady-state plasma concentrations of norverapamil approximate those of verapamil (Abernethy et al., 2000), and D617 is the major metabolite of verapamil found in the urine (Piotrovskii et al., 1986). Both metabolites could contribute to CYP3A inhibition in vivo. There was no apparent inhibition of CYP3A activities without preincubation with norverapamil (Fig. 2C) or D617 (data not shown). To evaluate the possibility that R-norverapamil, S-norverapamil, and D617 could be irreversible inhibitors of CYP3A, preincubations were performed with the pooled human liver microsomes or the cDNA-expressed CYP3A4 (+b5), respectively, in the presence of NADPH. S-norverapamil (Fig. 2D), R-norverapamil, and D617 (data not shown) showed time- and concentration-dependent inhibition of testosterone 6β-hydroxylase activity. For human liver microsomes, the plots of inactivation rate constants against inhibitor concentrations for R-and S-norverapamil are shown in Fig. 3, C and D. The estimated KI and kinact values are presented in Table 1. Compared with verapamil and norverapamil, D617 was a very weak inhibitor. Based on the ratio of kinact and KI obtained using the human liver microsomes and the cDNA-expressed human CYP3A4 (+b5), the potencies of verapamil enantiomers and their metabolites as irreversible inhibitors were in this order: S-norverapamil > S-verapamil > R-norverapamil > R-verapamil > D617. S-enantiomers are more potent than the R-enantiomers, and norverapamil enantiomers are more potent than their respective parent drugs.
The unbound inhibition constant value KI,u (KI,u = KI · fu,m) provides an estimate of inactivation potency that is independent of drug binding to the in vitro system. The fraction unbound in human liver microsomes and the cDNA-expressed human CYP3A4 (+b5) was determined by ultrafiltration, and the results are shown in Table 1. When microsomal binding was accounted for, the relative potency of the enantiomers of verapamil and norverapamil were unaffected.
Formation of P450 MIC. MIC formation with CYP3A4 was detected spectrophotometrically. An example of time-dependent MIC formation using S-verapamil is presented in Fig. 4. A peak absorbance difference was observed at 454 nm (Fig. 4A). The magnitude of MIC formation increased with time and was affected by the concentrations of S-verapamil R-verapamil, and norverapamil (Fig. 4B). Similar results were obtained for D617 (Fig. 4B). Only the data for 10 μM D617 are shown because of the availability problem. The observed maximal MIC formation at 10 μM of inhibitors was close to 100% of the total CYP3A in the sample for R-, S-verapamil, and (±)-norverapamil and was close to 67% for D617.
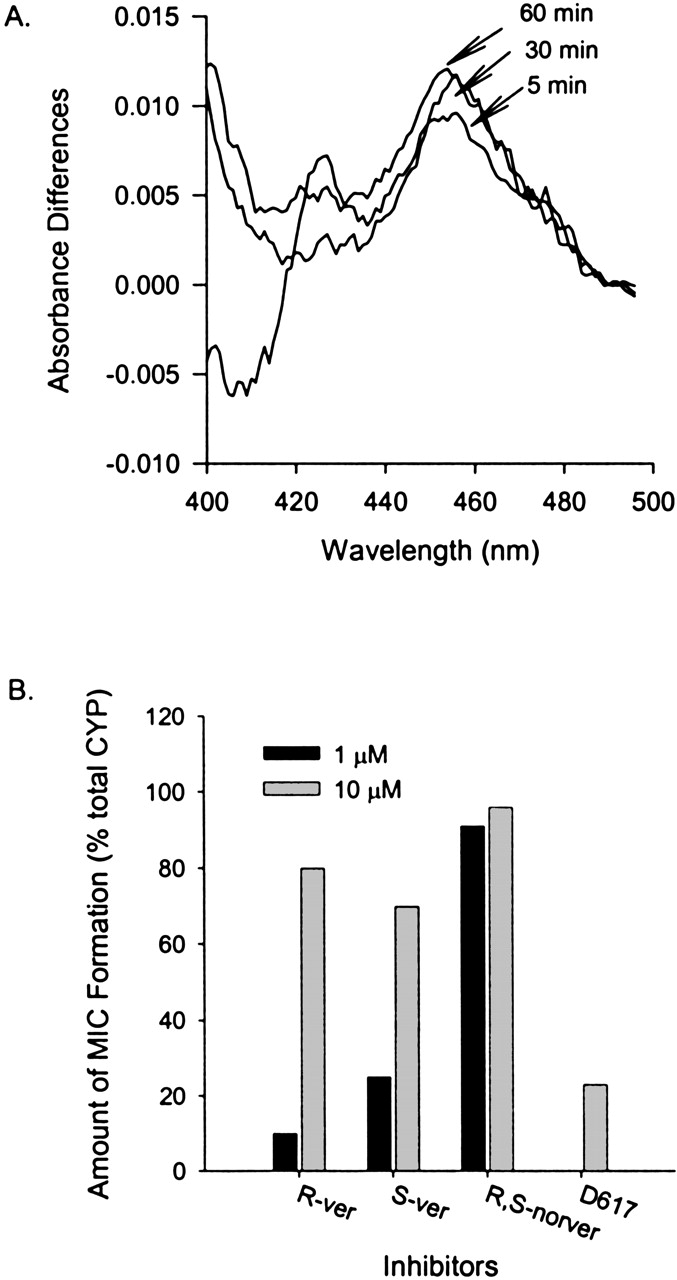
Metabolic intermediate complex formation by S-verapamil with cDNA-expressed human CYP3A4 (+b5).
A, sample cuvette contained cDNA-expressed CYP3A4 (+b5), S-verapamil (10 μM), buffer, and NADPH, whereas reference cuvette contained everything in the sample cuvette except S-verapamil. MIC formation with CYP3A4 was detected spectrophotometrically from the absorbance difference for the incubates with and without an inhibitor at wavelengths between 400 and 500 nm. Absorbance was monitored every 5 min from 0 to 60 min. Note, for the purpose of clarity, only lines representing change in absorbance difference for scans at 5, 30, and 60 min were shown in this Figure. B, the percentage of MIC formed with different concentrations of S-verapamil, R-verapamil, and norverapamil at 454 nm at 10 min. The open bar represents 1 μM, and the filled bar represents 10 μM of S-verapamil.
Quantitative Prediction of Drug Interactions in Vivo. In our initial analysis, the extent of drug interactions by verapamil was calculated as the sum of the effect of both R- and S-verapamil. Equation 10 was applied to this prediction because the unbound plasma concentrations of R-and S-verapamil are much smaller than the unbound KI. Based on pharmacokinetic parameters published in the literature and kinetics parameters estimated from the pooled human liver microsomes (Table 2), after chronic verapamil dosing, the predicted oral AUC of a coadministered CYP3A substrate that is eliminated completely by the hepatic CYP3A would increase 1.6- to 2.4-fold. However, our data show that norverapamil enantiomers also irreversibly inhibited CYP3A, and their inactivation potencies were comparable with that of S-verapamil. Therefore, we added R- and S-norverapamil to the R- and S-verapamil effect by assuming that norverapamil enantiomers have equivalent nonspecific binding in microsomes and equivalent fractions unbound in plasma (Mikus et al., 1990). When norverapamil was accounted for, the predicted increase in the oral AUC of a completely hepatically eliminated CYP3A substrate was 1.7- to 2.6-fold. When the inhibition data from cDNA-expressed human CYP3A4 (+b5) were used to make this prediction, the increase in the oral AUC of a coadministered CYP3A substrate was about 2-fold higher than that obtained by using the pooled human liver microsomes.
The first-pass metabolism in the intestinal wall is as important as that in the liver for presystemic elimination of some CYP3A substrates. Therefore, we used midazolam, a well characterized CYP3A substrate that is not transported by P-glycoprotein (P-gp), to predict a drug interaction with verapamil that accounts for CYP3A inhibition in intestinal wall and liver. We assumed that 90% of midazolam is eliminated via CYP3A-mediated oxidation in humans (Palkama et al., 1999), and the corresponding predicted increase in oral AUC was 1.6- to 2.2-fold using eq. 11. The intestinal wall bioavailability (FG) of midazolam is approximately 0.5 without coadministration of any CYP3A inhibitors (Olkkola et al., 1993; Gorski et al., 1998; Palkama et al., 1999). After multiple doses of verapamil treatment, if we assume maximal intestinal wall inhibition (F′G = 1), the predicted oral AUC increase was 3.2- to 4.5-fold using eq. 12. For drugs that have different FG and fm values than those of midazolam, a different prediction will be obtained as illustrated in Fig. 5. In general, the lower the baseline intestinal availability and the greater the fraction metabolized by CYP3A, the greater the predicted change in the oral AUC in the presence of verapamil.

Simulation of the extent of inhibition of a coadministered CYP3A substrate by verapamil.
A, the relationship between the extent of inhibition by verapamil and the fraction of the dose (fm) that is systemically eliminated through the CYP3A pathway in the absence of verapamil. Simulations were obtained from eq. 11. B, simulation of changes in the initial FG on the ratio of FG in the presence (F′G) and absence (FG) of verapamil. Simulations were obtained from eq. 14 and eq. 15. CLint,G/CLint,G in the eq. 14 can be expressed as:  Thus, (ΔCLint,G/CLint,G) × 100% can be expressed as the percentage decrease in CLint,G. Similarly, CLint,G/CL′int,G in the eq. 15 can be expressed as the reciprocal of 1 - (ΔCLint,G/CLint,G). When verapamil is not present (IG = 0), the percentage decrease in CLint,G is equal to 0. When IG >> KI, the percentage decrease in CLint,G is close to 100% (eq. 15).
Thus, (ΔCLint,G/CLint,G) × 100% can be expressed as the percentage decrease in CLint,G. Similarly, CLint,G/CL′int,G in the eq. 15 can be expressed as the reciprocal of 1 - (ΔCLint,G/CLint,G). When verapamil is not present (IG = 0), the percentage decrease in CLint,G is equal to 0. When IG >> KI, the percentage decrease in CLint,G is close to 100% (eq. 15).
Discussion
Clinical studies have shown that verapamil inhibits the metabolism of CYP3A substrates, such as midazolam, cyclosporin A, simvastatin, buspirone, and almotriptan (Robson et al., 1988; Backman et al., 1994; Kantola et al., 1998; Lamberg et al., 1998; Fleishaker et al., 2000). However, the mechanism of verapamil-mediated drug interactions has been unclear. When competitive inhibition alone is considered, verapamil-induced drug interactions are not expected because the in vitro Ki value (24 to 80 μM) (Lampen et al., 1995) is about 100-fold greater than the mean steady-state plasma concentration of verapamil after oral administration. Our data confirmed that verapamil enantiomers, D617, and norverapamil enantiomers are weak, reversible inhibitors of CYP3A that would not be expected to cause significant inhibition of CYP3A in vivo. The clinically important drug interactions by verapamil may lie in the capability of verapamil to inhibit CYP3A by forming an MIC (Ma et al., 2000).
The present study demonstrates that both R- and S-verapamil and R-and S-norverapamil are effective, irreversible inhibitors of the cDNA-expressed CYP3A4 and CYP3A activity in human liver microsomes. Spectrophotometric studies confirmed that verapamil, norverapamil, and D617 caused this essentially irreversible inhibition by forming an MIC. The steady-state plasma concentration of norverapamil is comparable with its parent drug. Therefore, the inhibition of CYP3A by norverapamil may play a significant role in CYP3A-mediated drug interactions in vivo. In contrast to the predictions from reversible inhibition, the irreversible inhibition model predicted a 2- to 4-fold increase in the AUC of a coadministered CYP3A substrate that is completely metabolized by hepatic CYP3A. The prediction arising from the irreversible inhibition model is in good agreement with the in vivo data (Backman et al., 1994), suggesting that our experimental and theoretical approach is appropriate for predicting drug interactions by mechanism-based inhibitors.
An important aspect of our predictive approach was to consider the enantiomers of verapamil and norverapamil as independent entities because they showed different inactivation potencies toward CYP3A (Fig. 3 and Table 1). The relative plasma concentrations of R- and S-verapamil and R- and S-norverapamil in humans are dependent on the design of the oral formulation and vary with the input rate into the systemic circulation (Karim and Piergies, 1995). Our model allows the flexibility to generate formulation-specific predictions for verapamil-drug interactions. The concentrations of inhibitors at the active site of the enzyme were assumed to be equivalent to the unbound concentrations in plasma that also display enantioselectivity. To complement this choice of inhibitor concentrations, we estimated the unbound KI values in vitro after the estimation of the fraction of R-verapamil, S-verapamil, and (±)-norverapamil unbound in pooled human liver microsomes. This approach has been recommended because nonspecific binding in enzyme preparations can have a significant impact on the result of in vitro-in vivo correlation studies (Obach, 1997). The inhibitory effect of D617 was not considered, although its plasma concentration is comparable with that of verapamil and norverapamil (Abernethy et al., 2000), because the inactivation potency of D617 is much less than that of verapamil and norverapamil, and D617 is not highly bound (Mikus et al., 1990). It is important to note that using cDNA-expressed CYP3A4 (+b5) as an enzyme source resulted in a marked overestimation of the extent of interaction because this enzyme preparation was not simply more active than human liver microsomes but also displayed significantly lower KI values (Table 1). Therefore, we recommend the use of pooled human liver microsomes to estimate the inactivation kinetics parameters and predict the in vivo drug interactions.
Our mechanism-based inhibition model is also suitable for predicting the extent of verapamil interactions with drugs that are extensively and exclusively metabolized in the intestinal wall by CYP3A. Previous knowledge of the initial FG and the fraction of the dose (fm) that is eliminated systemically through the CYP3A pathways is needed for the prediction of any CYP3A-mediated drug interaction. The estimates of FG and fm are often available from the literature or from the clinical pharmacological studies performed during the early phase of drug development. Alternatively, the extent of drug interactions by verapamil can be roughly estimated by combining the simulated results obtained by using eq. 11 (Fig. 5A) or eqs. 14 and 15 (Fig. 5B). As shown in eq. 12, the extent of inhibition by verapamil depends on fm and the ratio of F′G and FG for a given CYP3A substrate. The ratio of F′G and FG is influenced by two variables, the initial value of FG, which depends on the CLint,G, and the unbound inhibitor concentration in the enterocytes IG, which determine the extent of reduction in CLint,G of the affected drug. Figure 5B illustrates how changes in the two variables (FG and IG) affect the ratio of F′G and FG. The intestinal wall metabolism plays an important part in the first-pass process (Olkkola et al., 1993; Gorski et al., 1998), and if ignored, the extent drug interactions involving CYP3A will often be significantly underestimated. Using midazolam as an example, we predict that oral midazolam AUC increased 3.2- to 4.4-fold, which is slightly greater than the reported mean AUC increase of 3-fold in a single study (Backman et al., 1994). Differences between our predictions and the observed extent of interaction (Backman et al., 1994) may be related to: (1) the plasma verapamil concentration in the previous studies did not reach steady-state as the mean Cmax was reported around 0.09 μM, which is lower than the mean average steady-state verapamil concentration (0.14–0.18 μM) used in our prediction (Moreland et al., 1989; Dilger et al., 1999); and (2) the pharmacokinetics of midazolam were studied only 26 h after the onset of multiple verapamil dosing, and therefore, as predicted by eq. 17, only 30% of hepatic CYP3A would be inactivated compared with the maximal effect of 83% that would be expected under the steady-state conditions of our prediction.
Although the predicted effect of verapamil on other CYP3A substrates is close to the observed effect, it is important to acknowledge several uncertainties associated with our model that would affect the outcome of the prediction. First, steady-state unbound plasma concentrations are used in the prediction, based on the assumption that the steady-state unbound concentration of the inhibitor in the liver is equal to the unbound concentration in the hepatic capillary, but this may not be true when active transport is present. Verapamil was shown to be a substrate of P-gp (Pauli-Magnus et al., 2000) and to induce protein expression of P-gp, which functions as an efflux pump that lowers intracellular drug concentrations up to 10-fold in human colon carcinoma cells LS180 (Schuetz et al., 1996). Norverapamil was found in considerably higher concentrations in the intestinal lumen and biliary secretion compared with those in the plasma (von Richter et al., 2001), suggesting the existence of active transport probably via P-gp, which is expressed at a high level on the apical surfaces of epithelial cells in the liver (bile canaliculi) and small intestine (Cordon-Cardo et al., 1990). Therefore, in hepatocytes the intracellular concentrations of verapamil and norverapamil may be lower than the unbound plasma concentrations. On the other hand, norverapamil may passively diffuse from the systemic circulation and lumen into the enterocyte where it may be metabolized by CYP3A or actively transported back into the lumen. This recycling process could increase CYP3A exposure to norverapamil, and consequently, using unbound plasma concentration of norverapamil could underestimate the inhibition at the intestinal wall. Second, inhibitor concentrations in the portal vein during the first-pass were not considered when predicting the magnitude of drug interactions. If we consider the portal vein concentrations (0.07 μM), assuming that the average rate of presentation of 240 mg of S- or R-verapamil over a 24-h period is 10 mg/h, the blood flow of the portal vein is 63 liter/h, and the bioavailability of verapamil is around 0.2 (McTavish and Sorkin, 1989), the total verapamil concentration in the liver will be approximately 0.21 to 0.25 μM. Therefore, the predicted increase in oral AUC of midazolam will be 3.8- to 5.2-fold, which is greater than the value predicted using the systemic concentrations alone. Third, the model assumes that the amount of active CYP3A is decreased by mechanism-based inhibition and the inhibitor has no effect on the rate of CYP3A synthesis. However, verapamil is a weak inducer of CYP3A, which increased mRNA expression by 1.7-fold in LS180 cells (Schuetz et al., 1996). If this is the case for verapamil in vivo, the increased enzyme concentration might antagonize the inhibitory effect, and the predicted AUC increase will be greater than the observed one. Finally, the prediction is also affected by the enzyme degradation rate. The value of kdeg used in our prediction is the degradation rate of CYP3A in the rat or the human CYP3A4 in Caco-2 cells (Correia, 1991; Malhotra et al., 2001), because the corresponding value for CYP3A4 in human is not available.
In conclusion, our studies showed that in vivo drug interactions with verapamil are most likely due to mechanism-based inhibition of CYP3A by the enantiomers of verapamil and norverapamil via MIC formation. This inactivation of CYP3A is also consistent with the nonlinear accumulation of verapamil and its metabolites during chronic dosing (Schwartz et al., 1985). A mechanism-based inactivation model successfully predicted the extent of drug interactions from the in vitro data. Due to a combination of environmental and genetic factors substantial inter- and intraindividual variations in R- and S-verapamil and norverapamil plasma concentrations have been observed (Eichelbaum and Somogyi, 1984; Abernethy et al., 2000). Using our model, this variability could be incorporated into an individual, focused prediction scheme to identify those at greatest risk from verapamil-induced drug interactions.
Footnotes
-
↵1 Abbreviations used are: D617, N-desalkylverapamil; AUC, area under the concentration-time curve; MIC, metabolic intermediate complex; P450, cytochrome P450; HPLC, high-performance liquid chromatography; HLMs, human liver microsome; P-gp, P-glycoprotein; FG, intestinal wall bioavailability.
-
This research is supported by National Institutes of Health Grant GM67308.
- Received June 26, 2003.
- Accepted October 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}