


Abstract
This study was carried out to determine the metabolic pathways of buspirone and cytochrome P450 (P450) isoform(s) responsible for buspirone metabolism in human liver microsomes (HLMs). Buspirone mainly underwent N-dealkylation to 1-pyrimidinylpiperazine (1-PP), N-oxidation on the piperazine ring to buspirone N-oxide (Bu N-oxide), and hydroxylation to 3′-hydroxybuspirone (3′-OH-Bu), 5-hydroxybuspirone (5-OH-Bu), and 6′-hydroxybuspirone (6′-OH-Bu) in HLMs. The apparent Km values for buspirone metabolite formation in pooled HLMs were 8.7 (1-PP), 34.0 (Bu N-oxide), 4.3 (3′-OH-Bu), 11.4/514 (5-OH-Bu), and 8.8 μM (6′-OH-Bu). CYP3A inhibitor ketoconazole (1 μM) completely inhibited the formation of all major metabolites in HLMs (0-16% of control), whereas the chemical inhibitor selective to other P450 isoforms had little or no inhibitory effect. Recombinant CYP3A4, CYP3A5, and CYP2D6 exhibited buspirone oxidation activities among nine P450 isoforms tested. The overall metabolism rate of 5 μM buspirone by CYP3A4 was 18-fold greater than that by CYP2D6 and 35-fold greater than that by CYP3A5. In a panel of HLMs from 16 donors, buspirone metabolism correlated well CYP3A activity (r2 = 0.85-0.96, ρ < 0.0005), but not the activities of other P450 isoforms. The metabolism rates of buspirone in CYP2D6 poor-metabolizer genotype HLMs were comparable to those in pooled HLMs. Taken together, these data suggest that CYP3A, mostly likely CYP3A4, is primarily responsible for the metabolism of buspirone in HLMs.
Buspirone is the first marketed anxiolytic drug from the azapirone class of compounds (Fulton and Brogden, 1997). It is as effective as the benzodiazepines for the treatment of anxiety, but buspirone produces fewer adverse side-effects such as sedation, motor impairment, and dependence liability (O'Hanlon, 1991). Unlike benzodiazepine anxiolytics, buspirone has little affinity for the γ-aminobutyric acid-benzodiazepine complex. Its primary pharmacological action is believed to be associated with the binding to 5-hydroxytryptamine subtype 1A receptor (5-HT1A) receptor, resulting in the inhibition of the activity of serotonergic neurons through down-regulation (Goa and Ward, 1986; Fulton and Brogden, 1997). Buspirone, originally approved by the Food and Drug Administration (FDA) for the treatment of generalized anxiety disorder in 1986, has been shown to be efficacious for the treatment of a variety of mental disorders, including panic disorder, major depression, obsessive-compulsive disorder, and social phobia (Fulton and Brogden, 1997; Apter and Allen, 1999; Sramek et al., 2002).
Buspirone undergoes extensive first-pass metabolism in humans, resulting in a bioavailability of less than 5%, although it is almost completely absorbed after a single oral administration (Mayol et al., 1985; Gammans et al., 1986). Urinary excretion is the major elimination pathway in humans, accounting for 60% of the total oral dose of [14C]buspirone (Mayol et al., 1985). 1-Pyrimidinylpiperazine (1-PP), 6′-hydroxybuspirone (6′-OH-Bu), and multiple secondary metabolites (Fig. 1) are the major drug-related components in the urine, whereas the parent drug only accounts for less than 1% of total urinary radioactivity (Jajoo et al., 1989b). Most of the secondary metabolites in human urine, such as 5-hydroxy-1-pyrimidinylpiperazine (5-OH-1-PP), 5,6′-dihydoxybuspirone (5,6′-di-OH-Bu), and a γ-lactone metabolite (Oxa-Bu), are derived from 1-PP, 6′-OH-Bu, and 5-OH-Bu. Unchanged buspirone accounts for less than 2% of the total radioactivity) in human plasma (Gammans et al., 1986). Buspirone metabolites, 1-PP, 5-OH-Bu, and a conjugate of 5-OH-Bu, have been found in human plasma (Gammans et al., 1986). However, human plasma metabolite profiles after oral administration of radiolabeled buspirone have not been reported. Buspirone is also extensively metabolized in rats following oral administration (Caccia et al., 1983; Jajoo et al.,1989a). 1-PP, 5-OH-Bu, 6′-OH-Bu, 3′-OH-Bu, and their further metabolites are found to be the major drug-related components in rat bile and urine. These data suggest that N-dealkylation to form 1-PP and hydroxylation to form multiple monohydroxylated metabolites are the primary metabolic pathways responsible for buspirone clearance in humans and rats. In vitro metabolism of buspirone in rats has been investigated in several systems prepared from liver tissues, including S-9 preparation (Kerns et al., 1997), liver slices (Goldthwaite et al., 1996), microsomal preparation, and hepatocytes (Jajoo et at., 1990). Major in vitro metabolic reactions in rats are the formation of 1-PP, 3′-OH-Bu, 5-OH-Bu, and 6′-OH-Bu, consistent with the in vivo metabolism pathways in rats (Jajoo et al., 1989a). However, little information on in vitro buspirone metabolism for humans is available. Gepirone, a chemical analog of buspirone, is primarily metabolized by CYP3A4 to form 1-PP and multiple monohydroxylated metabolites in HLMs (von Moltke et al., 1998; Greenblatt et al., 2003).

Proposed metabolic pathways of buspirone in human liver microsomes. The 14C-label is designated by *. The numbering system is adopted from a previously reported system (Jajoo et al., 1989a). The primary P450 enzyme responsible for major metabolic pathways in HLMs is also listed.
Although specific human P450 isoforms involved in the formation of in vitro buspirone metabolites have not been characterized, the effects of coadministration of CYP3A inhibitor or inducer on the pharmacokinetics of buspirone in humans have been extensively investigated. Results from these studies demonstrate that CYP3A inhibitors, verapamil, diltiazem, erythromycin, itraconazole, and grapefruit juice, substantially increase the area under the curve (AUC) and the maximum concentration (Cmax) of buspirone in human plasma, presumably by inhibiting CYP3A-mediated metabolic clearance (Kivisto et al., 1997; Lamberg et al., 1998a; Lilja et al., 1998). In addition, a CYP3A inducer, rifampicin, decreases the AUC and Cmax of buspirone in human plasma by 90 and 84%, respectively (Lamberg et al., 1998b; Kivisto et al., 1999). These observations strongly suggest that CYP3A isoforms play an important role in the metabolism of buspirone in humans. The main objectives of the present study were to determine buspirone metabolic pathways in HLMs, and to identify the primary P450 isoform(s) involved in these pathways.
Materials and Methods
Chemicals and Materials. [14C]Buspirone was synthesized at Bristol-Myers Squibb Company (Princeton, NJ), with a specific activity of 27 μCi/mg and a radiochemical purity >97.5%. Reference standards for buspirone metabolites, 1-PP, 5-OH-1-PP, 3′-OH-Bu, 5-OH-Bu, 6′-OH-Bu, Oxa-Bu, 5,6′-di-OH-Bu, and Bu N-oxide (Fig. 1), were also prepared at Bristol-Myers Squibb. Pooled HLMs, CYP2D6 poor-metabolizer genotype HLMs, recombinant P450 isoforms containing coexpressed human P450 reductase (Supersomes CYP1A2, CYP2A6, CYP2B6, CYP2C9-Arg144, CYP2C19, CYP2D6-Val374, CYP2E1, CYP3A4, and CYP3A5), and (+)-N-3-benzyl-nirvanol were purchased from BD Gentest (Woburn, MA). A panel of human liver microsomes from 16 donors (P450 reaction phenotyping kit) was purchased from XenoTech (Kansas City, KS). Enzyme activities of P450 isoforms in the panel of HLMs were predetermined based on the following biotransformation reactions: 7-ethoxyresorufin O-dealkylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6), S-mephenytoin N-demethylation (CYP2B6), diclofenac 4′-hydroxylation (CYP2C9), S-mephenytoin 4′-hydroxylation (CYP2C19), dextromethorphan O-demethylation (CYP2D6), chlorzoxazone 6-hydroxylation (CYP2E1), and testosterone 6β-hydroxylation (CYP3A).
Identification of Metabolites in HLMs. Incubations were carried out at 37°C in a shaking water bath for 5 to 30 min. The final incubation mixtures contained [14C]buspirone (40 μM), pooled human liver microsomes (1.0 mg/ml), and NADPH (500 μM) in 100 mM sodium phosphate buffer (pH 7.4). The reactions were started by the addition of a NADPH solution after a 3-min preincubation, and stopped by the addition of an equal volume of ice-cold methanol. The suspension was centrifuged (13,000 rpm, 10 min), and an aliquot (200 μl) of the supernatant was analyzed directly by a HPLC/radio flow detector (RFD) for metabolite quantification and by LC/MS/MS for structural elucidation, as described below.
Enzyme Kinetic Experiments in HLMs. [14C[Buspirone, at concentrations ranging from 2.5 to 150 μM, was incubated with pooled human liver microsomes (0.1 mg/ml) and NADPH (500 μM) in 100 mM sodium phosphate buffer (pH 7.4) for 5 min. Incubation reactions were started and stopped as described above. After centrifugation, the supernatant was directly analyzed by an off-line HPLC/microplate scintillation counter (MSC), as described below (Zhu et al., 2000). Under these incubation conditions, the formation of buspirone secondary metabolites was insignificant. In preliminary experiments, time courses (0-20 min) of product formation, at various microsomal protein concentrations (0.1-0.3 mg/ml), were determined to establish incubation conditions of enzymatic reaction linearity. The apparent Michaelis-Menten parameters (Km and Vmax) for the formation of 1-PP, 3-OH-Bu, 6′-OH-Bu, 5-OH-Bu, and Bu N-oxide were determined using nonlinear regression analysis (GraFit; Erithacus Software, Horley, Surrey, UK). Intrinsic clearance was determined by dividing Vmax by Km.
Chemical Inhibition Experiments. [14C[Buspirone, at a concentration of 5 μM, was incubated with pooled human liver microsomes (0.2 mg/ml) and NADPH (500 μM) in the absence (control) and presence of selective chemical inhibitors of P450 isoforms for 10 min. The following P450 inhibitors were used (Rodrigues, 1999; Tucker et al., 2001; Suzuki et al., 2002; Bjornsson et al., 2003): furafylline (20 μM) for CYP1A2, coumarin (20 μM) for CYP2A6, sulfaphenazole (10 μM) for CYP2C9, (+)-N-3-benzyl-nirvanol (3 μM) for CYP2C19, quinidine (1 μM) for CYP2D6, 4-methylpyrazole (50 μM) for CYP2E1, and ketoconazole (1 μM) for CYP3A. Stock solutions of the chemical inhibitors were prepared in acetonitrile. The final concentration of acetonitrile in incubations was 1.0%. The incubation with the mechanism-based inhibitor furafylline was started by the addition of [14C]buspirone after preincubation of liver microsomes and NADPH with or without (control) the chemical inhibitor for 20 min. The incubation reactions with other chemical inhibitors were started by the addition of NADPH. All incubations were stopped by the addition of an equal volume of chilled methanol. After centrifugation, the supernatant was directly analyzed by a HPLC/RFD. CYP3A inhibition samples were analyzed by a HPLC/MSC.
Experiments with Recombinant P450 Isoforms. In an initial screening study, [14C]buspirone (10 and 100 μM) was incubated with microsomes from baculovirus-infected insect cells containing recombinant human P450 isoforms (Supersomes) and NADPH (500 μM) in sodium phosphate (100 mM, pH 7.4) for 10 min. CYP3A4 contained coexpressed human P450 reductase and cytochrome b5, whereas other P450 isoforms contained human P450 reductase only. The Supersomes were used at P450 protein concentrations of 100 pmol/ml for CYP1A2, CYP2B6, CYP2C19, CYP2D6, and CYP3A4, and 200 pmol/ml for CYP2A6, CYP2C9, CYP2E1, and CYP3A5. Incubation reactions were started and stopped as described above. After centrifugation, the supernatant was directly analyzed by a HPLC/RFD as described below. In a follow-up study, the catalytic activities of buspirone-metabolizing P450 isoforms were quantitatively determined in the incubations of [14C]buspirone (5 μM) with CYP2D6 (100 pmol/ml, 10 min), CYP3A4 (50 pmol/ml, 1 min) and CYP3A5 (200 pmol/ml, 10 min). Buspirone metabolites in these incubations were analyzed by a HPLC/MSC as described below.
Correlation Experiments. [14C[Buspirone, at a concentration of 5 μM, was incubated separately with a panel of human liver microsomes (0.1 mg/ml) from 16 individual donors in the presence of NADPH (500 μM) for 10 min. Incubation reactions were started by the addition of NADPH and stopped by the addition of 2 volumes of chilled methanol. Metabolites formed in the incubations were quantitatively determined by a HPLC/MSC as described below. The correlations of the formation rates of major metabolites with enzymatic activities of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A were calculated using Microsoft Excel (Microsoft, Redmond, WA).
Metabolism in CYP2D6 Poor-Metabolizer (PM) Genotype HLMs. [14C[Buspirone, at concentrations of 5 μM (0.1 mg of microsomal protein/ml, 5 min) and 0.5 μM (0.2 mg of microsomal protein/ml, 10 min), was incubated separately with three individual CYP2D6 PM HLMs. [14C]Buspirone was also incubated with three pooled HLM samples (control) under the same conditions. The incubations were started by the addition of NADPH (500 μM) and stopped by the addition of 2 volumes of chilled methanol. After centrifugation, the supernatant was analyzed by a HPLC/MSC as described below.
HPLC/Radio Flow Detection. The HPLC method used a Zorbax RX-C8 column (4.6 × 250 mm) and a linear stepwise gradient with solvent A (0.01% trifluoroacetic acid in water) and solvent B (acetonitrile). Solvent B in the gradient started at 8% and then changed as follows: 8% (8 min), 40% (30 min), 90% (35 min), and 8% (40 min). HPLC analyses were performed on a Shimadzu Class VP System (Shimadzu, Columbia, MD) equipped with two pumps (model LC-10AD), an autoinjector (model SIL 10 AD), and a diode array detector (model SPD-MA10A) at a flow rate of 1 ml/min. Radioactivity in HPLC eluent was determined using a RFD (β-RAM; IN/US Systems, Tampa, FL). The HPLC eluent was mixed with ULTIMA-FLO M cocktail (PerkinElmer Life and Analytical Sciences, Boston, MA) at a ratio of 1:3, and then the mixture passed through a 400-μl liquid detection cell in the RFD. Concentrations of major metabolites were determined based on the following equation: Concentration of metabolite = Relative radioactivity concentration of the metabolite peak (%) × Initial concentration of buspirone.
Accordingly, the rates of formation of the major metabolites were calculated. The rate of overall buspirone metabolism was estimated based on the conversion of buspirone to the total radioactive metabolites.
HPLC/Microplate Scintillation Counting. An off-line HPLC/MSC was used for the quantification of radioactive metabolites when a high sensitivity for radiodetection was required (Zhu et al., 2000). The same HPLC method was used as described above. HPLC eluent was collected into 96-well plates coated with solid scintillators at a rate of 15 s per well (Deep-Well LumaPlate; PerkinElmer Life and Analytical Sciences) and then dried using a speed vacuum system (SpeedVac AES-2010; Savant Instruments, Holbrook, NY). Radioactivity of the residues in 96 wells was determined with a TopCount microplate scintillation counter (PerkinElmer Life and Analytical Sciences). Up to 12 wells were simultaneously analyzed with a 10-min counting time. Metabolite concentrations in incubations were calculated based on the same equation described above.
LC/MS/MS. LC/MS/MS analysis was carried out for metabolite identification by using the Shimadzu HPLC system (described above) coupled with a Finnigan TSQ quantum triple quadrupole mass spectrometer (Thermo Finnigan, San Jose, CA). HPLC eluent was introduced to the mass spectrometer at a flow rate of 200 μl/ml after splitting. The mass spectrometer was operated in the positive electrospray mode at a capillary temperature of 300°C. MS/MS analysis was performed using nitrogen as the collision gas, and the collision energy was set at 28 to 35 eV.
Results
Metabolite Profiling and Identification. A typical HPLC radiochromatogram of buspirone metabolites in HLMs is presented in Fig. 2A. In addition to the parent drug, five major radioactivity peaks corresponding to metabolites M2, M3, M6, M7, and M8 were observed. Three minor metabolites, namely, M1, M4, and M5, were also detected in HLMs. M4 and M5 were overlapped in the radiochromatogram (Fig. 2A). M1 (5.5 min) was detected by LC/MS/MS (data not shown), but not by radioactivity analysis due to its low concentration in HLMs. These metabolites were not observed in control incubation samples, in which NADPH was not added. The structures (Fig. 1) of buspirone metabolites were determined by LC/MS/MS, and chromatographic and mass spectral comparisons with reference standards. The protonated molecular ions (MH+) and product ion spectral data of buspirone metabolites, along with fragmentation interpretation, are listed in Table 1.

Representative HPLC-radiochromatograms of incubation mixtures of [14C]buspirone with human liver microsomes (A) and recombinant CYP3A4 (B). [14C]Buspirone was incubated with human liver microsomes (A; 40 μM buspirone, 1 mg of microsomal proteins/ml, 20 min) and recombinant CYP3A4 (B; 32 μM buspirone, 3 pmol of P450 protein/ml, 10 min). Metabolites were identified by LC/MS/MS, and by chromatographic and spectral comparison with authentic reference standards: M2 (1-PP), M3 (3′-OH-Bu), M4 (Oxa-Bu), M5 (5,6′-di-OH-Bu), M6 (6′-OH-Bu), M7 (5-OH-Bu), and M8 (Bu N-oxide). M1 (5-OH-1-PP, retention time 5.5 min) was detected by LC/MS (data not shown), but not by a HPLC/RFD.
MS fragments and their interpretation of buspirone metabolites in human liver microsomes A similar MS/MS fragmentation pattern of buspirone under ionspray ionization conditions was previously reported (Kerns et al., 1997).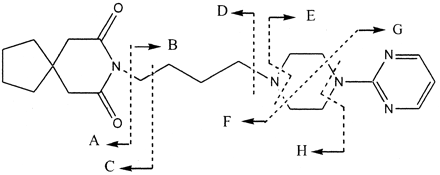
M2 was identified as 1-PP. It displayed an MH+ ion at m/z 165 and two prominent product ions at m/z 148 and 122, corresponding to the fragment ions E and G, respectively. M1 was identified as 5-OH-1-PP. It had an MH+ ion (m/z 181) 16 mass units greater than that for 1-PP, and displayed the same fragmentation pattern as that for 1-PP. M3, M4, M6, M7, and M8 had the same MH+ ion (m/z 402) 16 mass units greater than that for the parent drug (m/z 386), indicating that an oxygen atom was incorporated into these metabolites. M3 was identified as 3′-OH-Bu. The product ion spectrum of M3 (Fig. 3A) displayed multiple fragment ions, generated from the major fragmentation pathways, as those of buspirone (Table 1). The product ion at m/z 184 (fragment A) indicates that the hydroxylation occurred on the moiety of azaspironedecanedione. M4 was identified as Oxa-Bu, and M6 was identified as 6′-OH-Bu. The major product ions of 3′-OH-Bu, 6′-OH-Bu, and Oxa-Bu were identical. M5 was identified as 5,6′-di-OH-Bu. Like 5-OH-1-PP, it showed a diagnostic ion at m/z 138 (fragment ion G), consistent with the hydroxylation on the pyrimidine ring. M7 was identified as 5-OH-Bu. The product ion spectrum of M8 (Bu N-oxide) is presented in Fig. 3B, which is consistent with the structure of the N-oxide on the piperazine ring (Fig. 3B). The loss of an oxygen (-16) from the N-oxide group led to the formation of unique fragments at m/z 177 and 277. The retention times and product ion spectra of all the metabolites (M1-M8) were the same as those of the reference standards (data not shown). Therefore, the structures of these buspirone metabolites formed in HLMs were confirmed.

Product ion spectra of buspirone metabolites 3′-OH-Bu (A) and Bu N-oxide (B). The spectra were obtained by collision-induced dissociation of the MH+ ions at m/z 402. The interpretation of key fragment ions is also presented in Table 1.
Enzyme Kinetics of Buspirone Metabolism in HLMs. The apparent enzyme kinetic parameters for the formation of the major buspirone metabolites in pooled human liver microsomes are summarized in Table 2. Under the incubation conditions, the conversion of the primary metabolites to secondary metabolites such as 5,6-di-OH-Bu and Oxa-Bu were negligible. The formation of 1-PP, 3′-OH-Bu, 6′-OH-Bu, and Bu N-oxide by HLMs conformed to monophasic Michaelis-Menten kinetics, suggesting these metabolic reactions are catalyzed predominantly by a single P450 isoform or by more than one isoform with similar Km values. For example, an Eadie-Hofstee plot (Fig. 4A, inset) of the 6′-OH-Bu formation appeared linear over the substrate concentrations ranging from 2.5 to 150 μM, with an apparent Km of 8.8 μM. The formation of 5-OH-Bu by HLMs seems to follow biphasic Michaelis-Menten kinetics, with one low apparent Km of 11.4 μM and one high apparent Km of 514 μM (Fig. 4B; Table 2), indicating that two P450 isoforms may play a role in the 5-OH-Bu formation. As indicated by the apparent CLint values, the in vitro intrinsic clearance corresponding to each metabolic pathway was 6′-OH-Bu > 1-PP > 5-OH-Bu ≈ 3′-OH-Bu > Bu N-oxide. The formation of 6′-OH-Bu was the most predominant metabolic pathway in HLMs (Fig. 2), accounting for more than one-third of the total in vitro intrinsic clearance (Table 2).
Enzyme kinetic parameters of buspirone metabolism in HLM [14C]Buspirone, at concentrations ranging from 2.5 to 150 μM, was incubated with pooled human liver microsomes (0.1 mg/ml) and NADPH (500 μM) in 100 mM sodium phosphate buffer (pH 7.4) for 5 min. Data presented in this table were averaged from analyses of duplicate incubations.
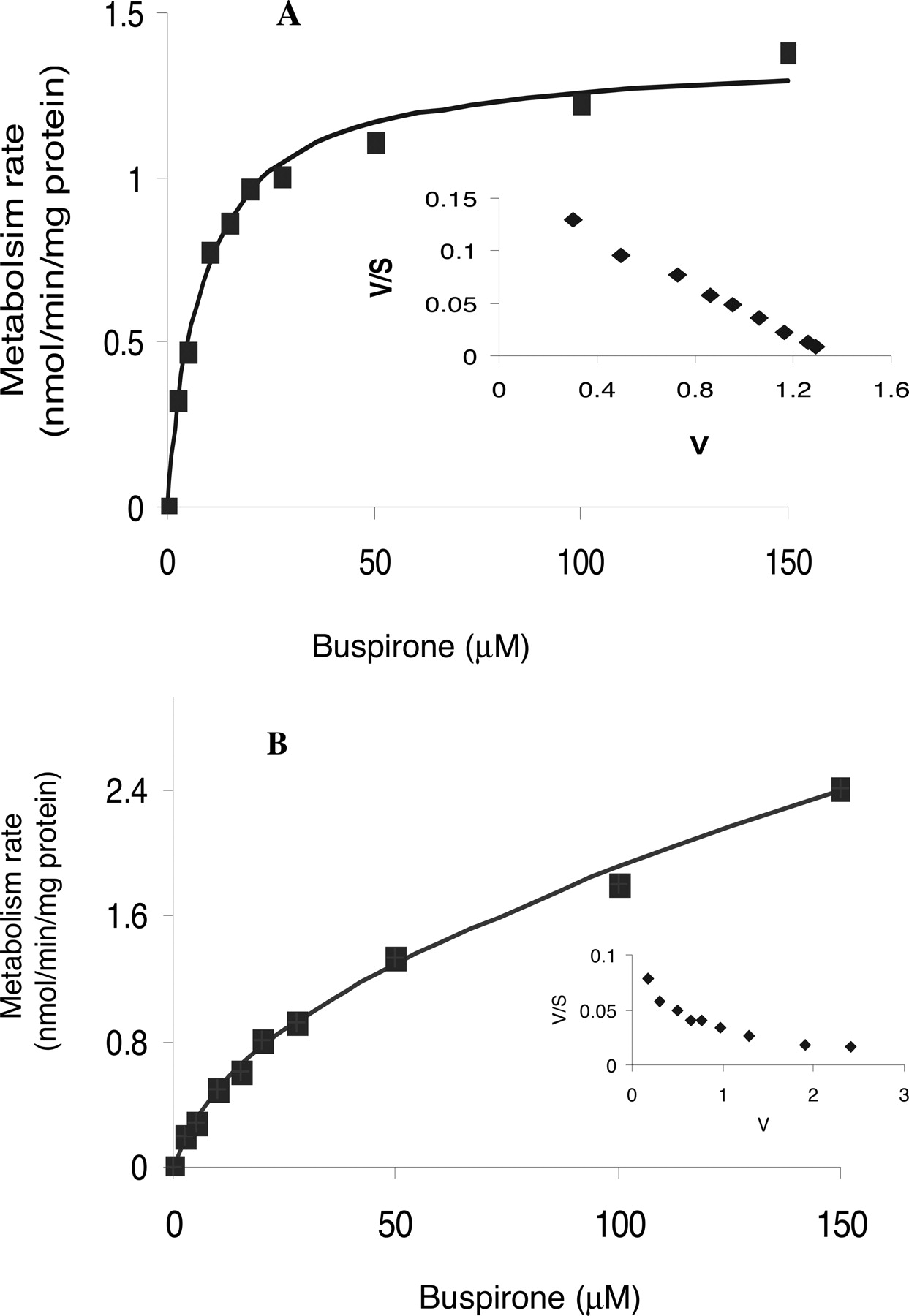
Kinetic plots of 6′-OH-Bu and 5-OH-Bu formation in HLMs. Substrate saturation plots of the formation of 6′-OH-Bu (A) and 5-OH-Bu (B) in HLMs are shown. Inset, Eadie-Hofstee plots. Data presented in this figure were averaged from analyses of duplicate incubations. Enzyme kinetic parameters obtained from the plots are shown in Table 2.
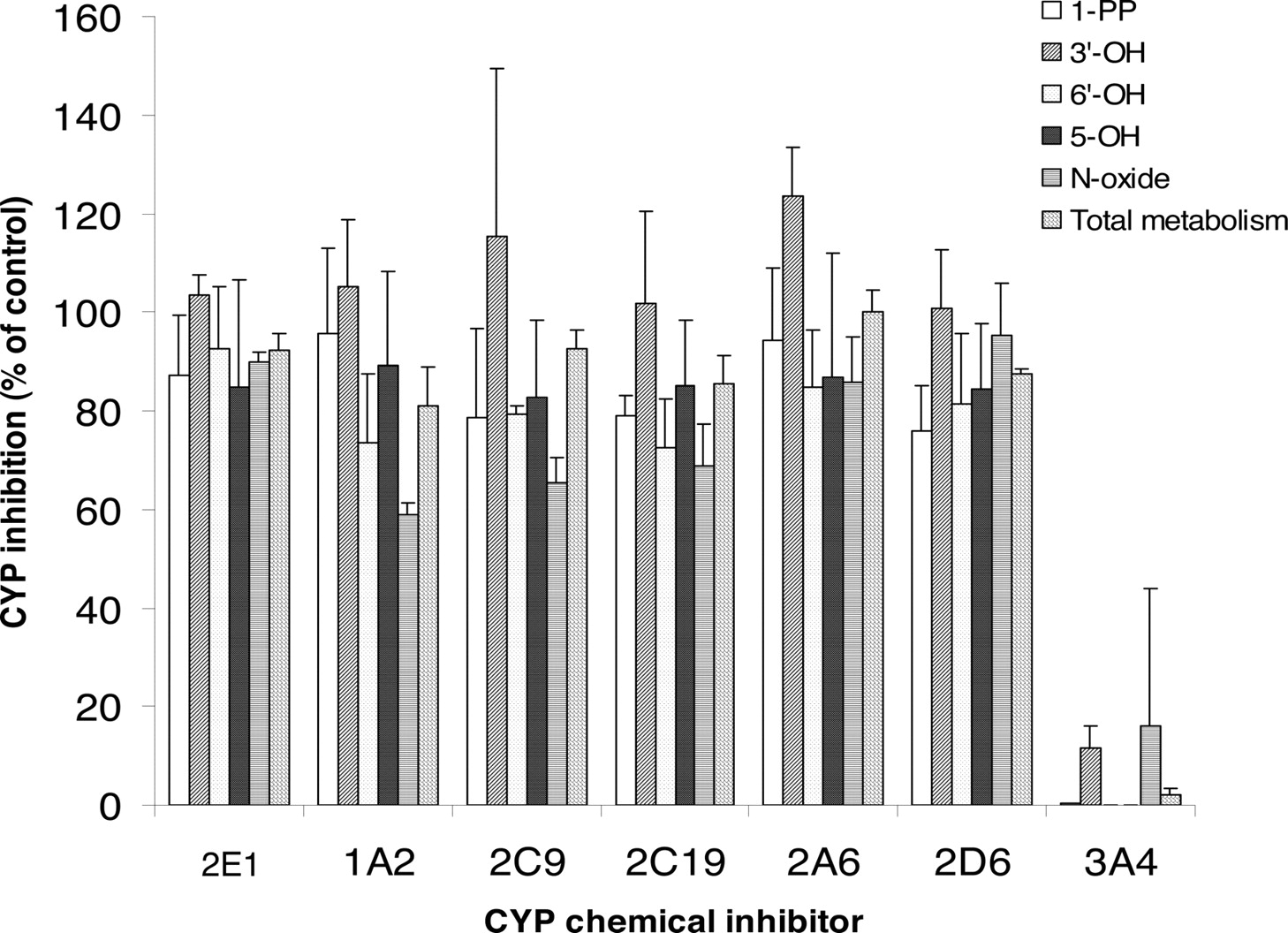
Metabolism Inhibition by Chemical Inhibitors of P450 Isoforms. The effects of various P450 isoform-specific chemical inhibitors on buspirone metabolism were determined at the 5 μM substrate concentration in pooled HLMs (Fig. 5). Ketoconazole (1 μM), a potent CYP3A4/5 inhibitor, completely or very significantly inhibited the formation of major metabolites, 1-PP, 3′-OH-Bu, 5-OH-Bu, 6′-OH-Bu, and Bu N-oxide (0-16% of control). The chemical inhibitors selective to CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP2E1 had little or no inhibitory effect on the metabolism of buspirone in HLMs (Fig. 5).
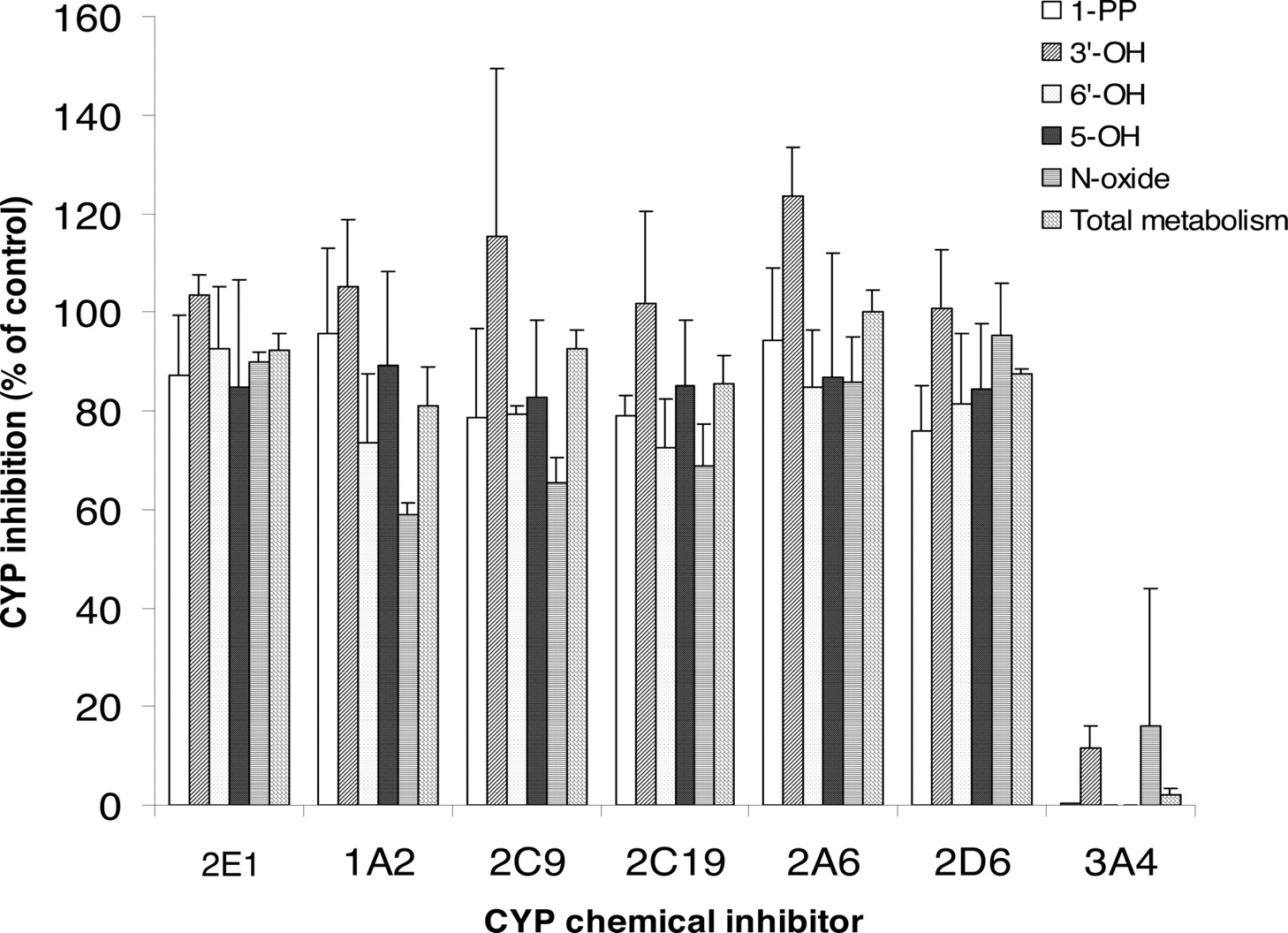
Inhibition of buspirone metabolism in human liver microsomes by chemical inhibitors selective to P450 isoforms. [14C]Buspirone (5 μM) was incubated with pooled HLMs (0.2 mg of protein/ml) for 10 min in the presence of the chemical inhibitors selective to CYP1A2 (20 μM furafylline), CYP2A6 (20 μM coumarin), CYP2C9 (10 μM sulfaphenazole), CYP2C19 [3 μM (+)-N-3-benzyl-nirvanol], CYP2D6 (1 μM quinidine), CYP2E1 (50 μM 4-methylpyrazole), and CYP3A (1 μM ketoconazole) (Rodrigues, 1999; Tucker et al., 2001; Suzuki et al., 2002; Bjornsson et al., 2003). The data presented are the average from the analysis of triplet incubations. The rate of the overall metabolism was calculated based on the conversion of buspirone to the total metabolite peaks.
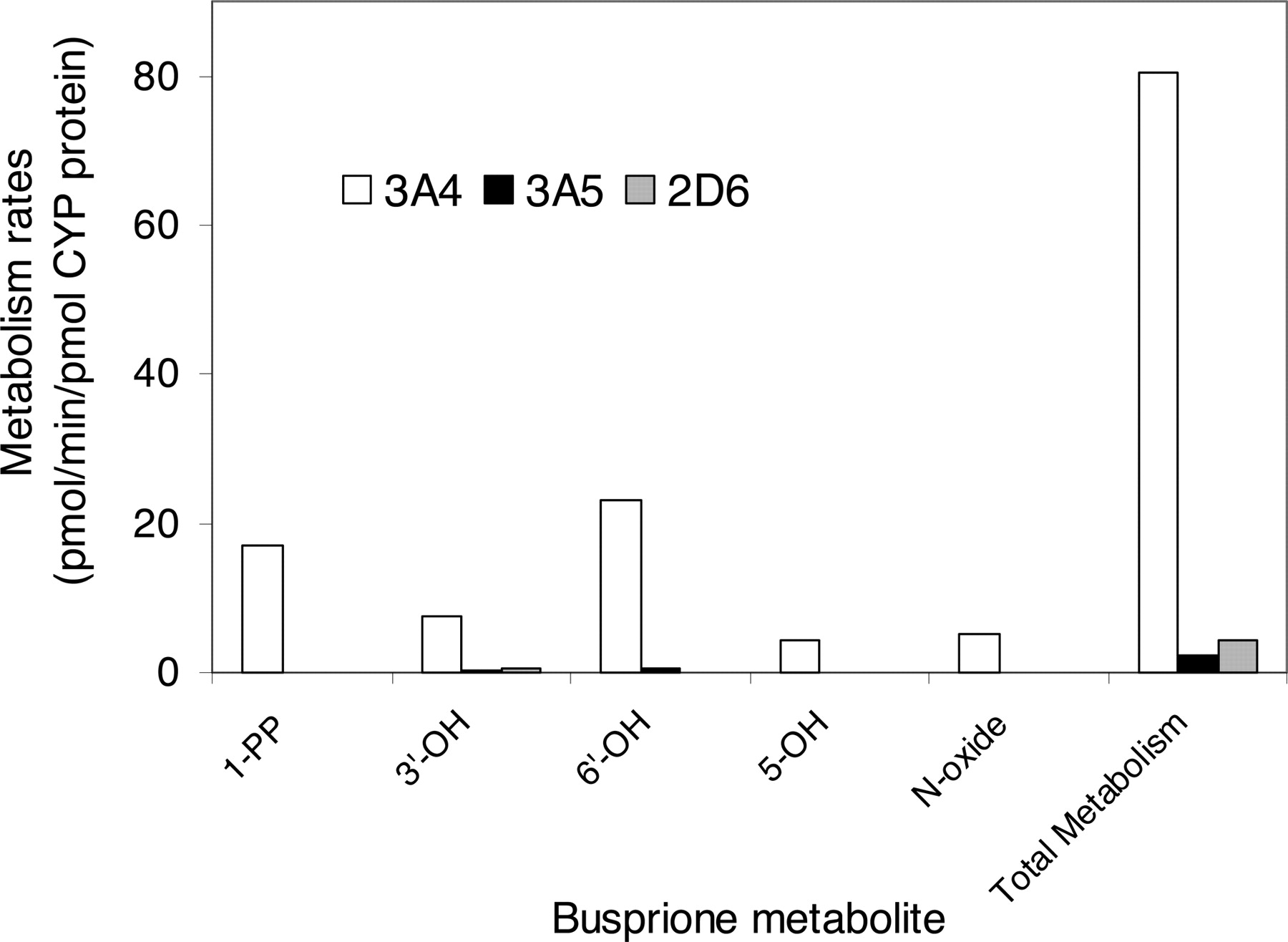
Metabolism in Recombinant P450 Isoforms. In the initial screening study with recombinant P450 isoforms, CYP3A4, CYP3A5, and CYP2D6 exhibited catalytic activity for the oxidation of buspirone (10 and 100 μM), whereas no significant metabolites were detected in the incubations with CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, and CYP2E1. CYP3A4 (Fig. 2B) and CYP3A5 (data not shown) catalyzed the formation of all major metabolites, 1-PP, 3′-OH-Bu, 6′-OH-Bu, 5-OH-Bu, and Bu N-oxide, similar to those catalyzed by HLMs (Fig. 2A). The buspirone metabolism rates in recombinant CYP2D6, CYP3A4, and CYP3A5 were further determined at the 5 μM substrate concentration (Fig. 6). The formation rates of the five major metabolites catalyzed by CYP3A4 were 13- to 44-fold greater than those by CYP2D6, and 37- to 170-fold greater than those by CYP3A5. The overall metabolism rate by CYP3A4 was 18-fold greater than that by CYP2D6 and 35-fold greater than that by CYP3A5.
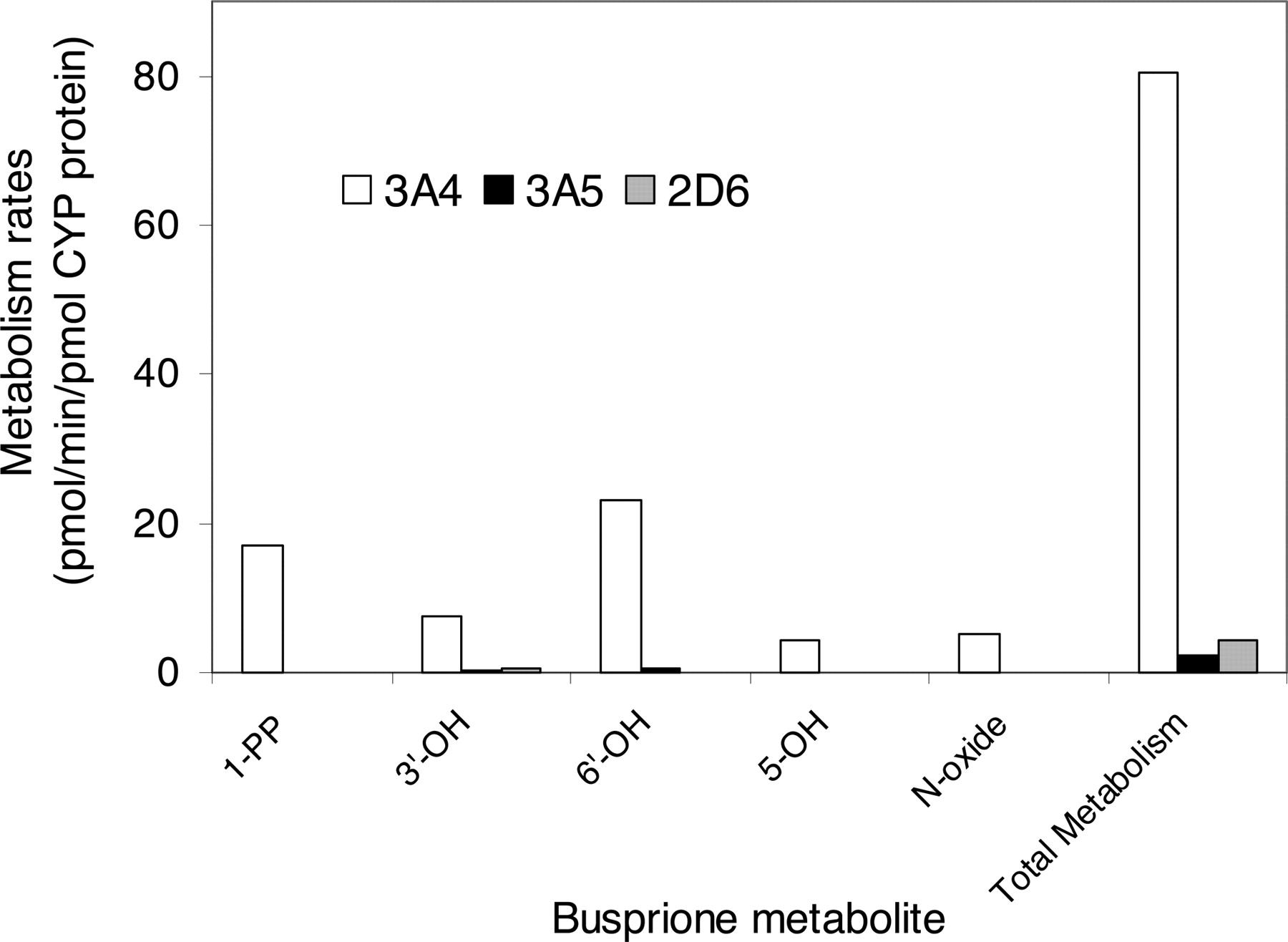
Metabolism rates of 5 μM buspirone by recombinant CYP2D6, CYP3A4, and CYP3A4. The data presented are an average from the analysis of duplicate incubations. The rate of the overall metabolism was calculated based on the conversion of buspirone to the total metabolite peaks.
Correlation of Metabolism with P450 Isoform Activities. The rates for the major metabolite formation in a panel of liver microsomes from 16 individual subjects were determined at a 5 μM substrate concentration. The rates of the overall buspirone metabolism varied approximately 9.3-fold among the liver microsomal preparations evaluated. The significant individual variability of buspirone metabolism in HLMs is in agreement with the variation (25-fold) of the plasma exposures of buspirone in 26 human subjects after oral administration of 40 mg of buspirone (Gammans et al., 1985). The correlation between the rates of metabolite formation and P450 marker activities is shown in Table 3. Testosterone 6β-hydroxylation, a marker for CYP3A activity, strongly correlated with the formation rates of all major metabolites (r2 = 0.85 to 0.96, ρ < 0.0005) and the overall metabolism rates (r = 0.92, ρ < 0.0005). The enzyme activities of P450 isoforms other than CYP3A did not correlate with buspirone metabolism rates in the panel of human liver microsomes (Table 3).
Correlation of P450 activities with the metabolism rates of buspirone in a panel of human liver microsomes Buspirone (5 μM) was incubated separately with 16 individual HLM.
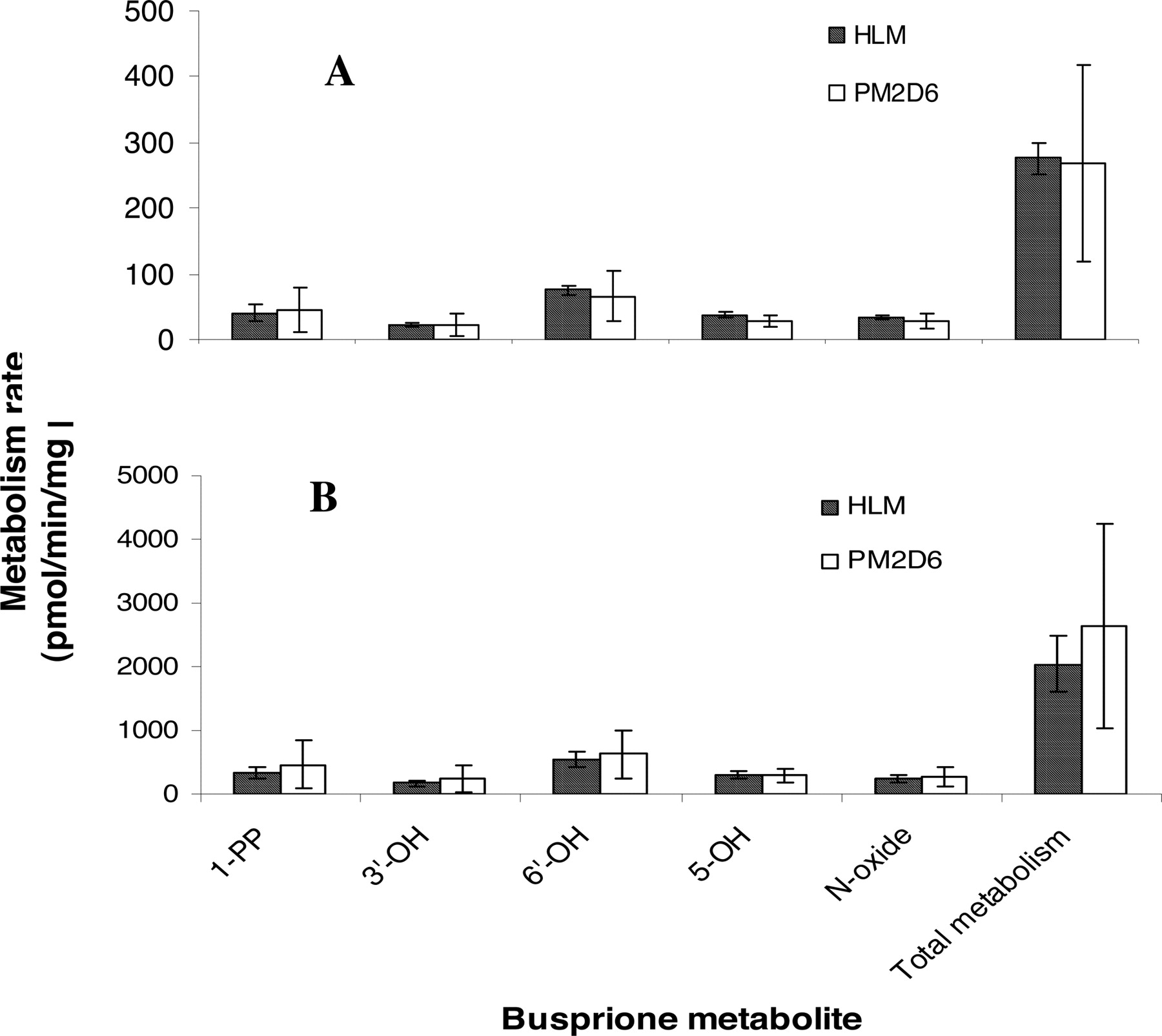
Metabolism by CYP2D6 PM HLMs. To further evaluate the role of CYP2D6 in buspirone metabolism, metabolism rates of buspirone (5.0 and 0.5 μM) were determined in the incubations with three individual CYP2D6 PM HLMs and three pooled HLMs. The mean values of CYP3A activity in the two HLM groups were the same (Table 4). As shown in Fig. 7, the formation rates of the major buspirone metabolites in CYP2D6 PM HLMs were similar to those in pooled HLMs. The individual variability of metabolite formation rates by CYP2D6 MP HLMs was severalfold greater than those by pooled HLMs (Fig. 7), consistent with the variation of CYP3A4 activity in the CYP2D6 MP HLM samples (Table 4). The relative rates of the overall buspirone metabolism in CYP2D6 PM HLMs were at the same level as those in pooled HLMs (Table 4). The relative rates were calculated by dividing the buspirone metabolism rates by the predetermined CYP3A enzyme activity so that the difference of the CYP3A4 contribution to the metabolism in these two HLM groups was eliminated. In addition, the relative rates of the overall buspirone metabolism (5 μM) in the three HLM samples, which had the lowest CYP2D6 activity in the panel of human liver microsomes tested in the correlation experiment, were comparable to those in the three pooled HLMs (Table 4).
Comparison of the relative metabolism rates of buspirone in CYP2D6 PM HLM, HLM with low CYP2D6 activity, and pooled HLM
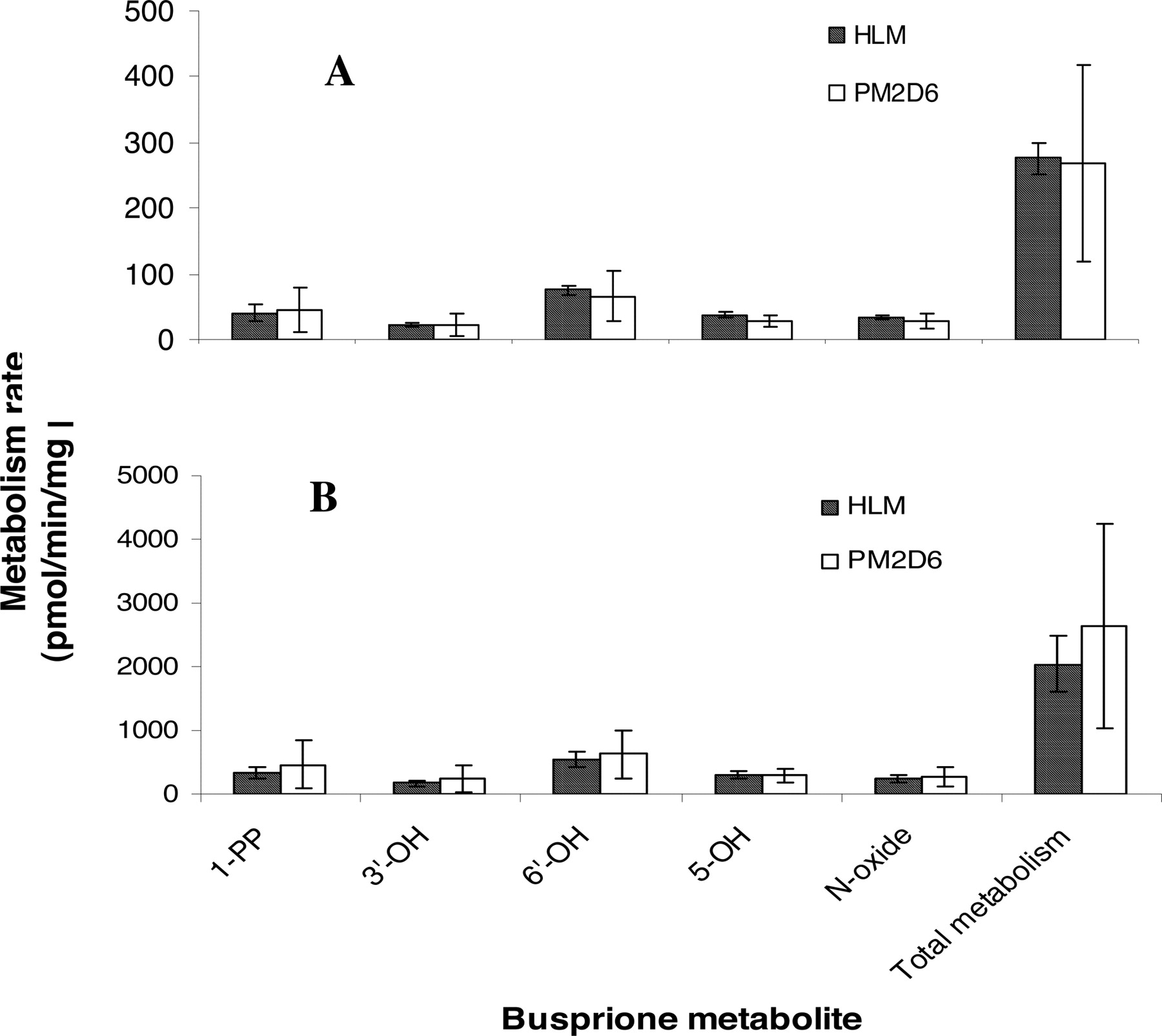
Comparison of buspirone metabolism in CYP2D6 PM HLMs (PM2D6) and pooled HLMs. Incubations were carried out at the 0.5 μM (A) and 5 μM (B) concentrations of buspirone in three individual CYP2D6 PM HLMs and three pooled HLMs. The mean values of CYP3A activity in the two groups of HLMs were at the same level (Table 4). The data presented are an average from the analysis of triplicate incubations.
Discussion
Although buspirone has been used for the treatment of generalized anxiety disorder for 18 years, little information on in vitro metabolism of buspirone in humans is available. This study was attempted to determine the metabolic pathways of buspirone in HLMs and P450 isoform(s) responsible for the formation of these metabolites. Results from the metabolite profiling and identification experiments demonstrate that buspirone is primarily metabolized to 1-PP, 3′-OH-Bu, 6′-OH-Bu, 5-OH-Bu, and Bu N-oxide in HLMs (Fig. 2A). A similar metabolite profile was observed in the buspirone incubations with recombinant CYP3A4 (Fig. 2B) and CYP3A5 (data not shown). The structures of buspirone metabolites (M1-M8) were unambiguously identified by LC/MS/MS analyses (Table 1) and comparisons with authentic reference standards. Bu N-oxide was not seen in previous in vitro and in vivo metabolism studies (Jajoo et al., 1989a,b, 1990). Based on these findings, the major biotransformation pathways of buspirone in HLMs are proposed (Fig. 1.) These include hydroxylation on the azaspironedecanedione moiety to form 3′-OH-Bu and 6′-OH-Bu, hydroxylation on the pyrimidine ring to form 5-OH-Bu, N-dealkylation of the substituted butyl side chain to give 1-PP, and N-oxidation on the piperazine ring to give Bu N-oxide. Some of these primary metabolites undergo further enzymatic reactions to form secondary metabolites, including 5-OH-1-PP and 5,6′-di-OH-Bu. In addition, base-catalyzed rearrangement of 6′-OH-Bu results in the formation of Oxa-Bu (Jajoo et al., 1989b).
The buspirone metabolism to 1-PP, 6′-OH-Bu, and 5-OH-Bu in HLMs is consistent with in vivo metabolism in humans. As reported previously, oxidation of buspirone, followed by urinary excretion, was the major elimination route of buspirone in humans (Jajoo et al., 1989b). 1-PP, 5-OH-1-PP, and 6′-OH-Bu were the most abundant metabolites in the human urine. 5-OH-Bu was found in plasma at a level comparable to that of buspirone (Gammans et al., 1985), but not in the urine after oral administration (Jajoo et al., 1989b). Most likely, 5-OH-Bu was extensively converted to secondary metabolites, since substantial amounts of 5-OH-1-PP, 5,6′-di-OH-Bu, and 5-OH-Oxa-Bu were found in human urine (Jajoo et al., 1989b). However, buspirone metabolism to 3′-OH-Bu and Bu N-oxide, although clearly existing in HLMs, has not been observed in humans. Like 1-PP, 5-OH-Bu, and 6′-OH-Bu (Jajoo et al., 1989b), both 3′-OH-Bu and Bu N-oxide undergo further biotransformation reactions to form multiple di-hydroxyl metabolites in HLMs (Zhu et al., 2002). Those secondary metabolites could be excreted into urine, since at least two unknown di-hydroxyl metabolites were observed in human urine (Jajoo et al., 1989b). Furthermore, 3′-OH-Bu, Bu N-oxide, and their further metabolites could be eliminated via biliary excretion. A significant amount of radioactivity (approximately 29% of the total dose) was found in feces after the parent drug was almost completely absorbed following oral administration of [14C]buspirone in humans (Mayol et al., 1985). Bu N-oxide showed a unique product ion spectrum under electrospray ionization (Fig. 3; Table 1), which can be utilized for its detection in humans by LC/MS/MS.
Several lines of evidence from the present P450 reaction phenotyping study strongly suggest that buspirone is primarily metabolized by CYP3A, mostly likely CYP3A4, in HLMs. First, the formation of all major metabolites and the overall metabolism of buspirone were completely or very significantly inhibited by 1 μM ketoconazole, a CYP3A-specific inhibitor (Fig. 5). The chemical inhibitors selective to CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, and CYP2E1 had little or no inhibitory effect on buspirone metabolism (Fig. 5). Second, recombinant CYP3A4 exhibited very high buspirone oxidation activity, whereas other P450 isoforms had no or minimal buspirone oxidation activity (Fig. 6). Furthermore, the metabolism of buspirone in a panel of human liver microsomes correlated well with enzyme activity only for CYP3A, among eight P450 isoforms tested (Table 3). Finally, the in vitro P450 reaction phenotyping data are in agreement with the in vivo observations that CYP3A4 inhibitors verapamil, diltiazem, (Lamberg et al., 1998a), erythromycin, itraconazole (Kivisto et al., 1997), and grapefruit juice (Lilja et al.,1998) substantially increase the AUC and Cmax of buspirone. In contrast, the CYP3A4 inducer rifampicin significantly decreases the AUC and Cmax of buspirone (Lamberg et al., 1998b; Kivisto et al., 1999).
Although the initial screening experiment with recombinant P450 isoforms found that CYP2D6 had buspirone oxidation activity, the results from the studies with P450 chemical inhibitors and CYP2D6 PM HLMs strongly suggest that the contribution of CYP2D6 to buspirone metabolism in HLMs is minimal, compared with that of CYP3A. As indicated in Fig. 5, quinidine (1 μM) had little or no inhibitory effect on the formation of major buspirone metabolites (76-100% of control). In addition, the metabolism of buspirone (5 μM) in the CYP2D6 PM HLMs and the HLMs with very low CYP2D6 activity was as fast as that in pooled HLMs (Fig. 7; Table 4). The buspirone metabolism in the CYP2D6 PM HLMs and pooled HLMs was also determined at the 0.5 μM substrate concentration, which is more relevant to the maximal buspirone concentration in humans (Mahmood and Sahajwalla 1999). Metabolite formation rates at the 0.5 μM buspirone concentration were not significantly different in the two HLM groups (Table 4; Fig. 7). Furthermore, the role of CYP2D6 in buspirone metabolism was evaluated by the quantitative analyses of CYP3A4-, CYP3A5-, and CYP2D6-mediated metabolism under more suitable incubation conditions. The rate of the overall buspirone metabolism by CYP2D6 was about 18-fold less than that by CYP3A4 (Fig. 6). These observations, together with the consideration that the CYP3A content is more than 10-fold greater than that of CYP2D6 in human livers (Rodrigues 1999), provide additional evidence for the insignificant role of CYP2D6 in buspirone metabolism. These in vitro data are consistent with the results from a recent clinical drug-drug interaction study, which showed that deramciclane, an in vitro CYP2D6 inhibitor, did not affect buspirone pharmacokinetics (Laine et al., 2003).
Rapid metabolism of buspirone by HLMs (Table 2) and CYP3A4 (Fig. 6) predicts extensive first-pass metabolism of buspirone in humans, which is consistent with previous observations in humans (Mayol et al., 1985). The in vitro metabolism data also support the observation that buspirone was rapidly eliminated from humans, mainly via metabolism, rather than direct excretion of the parent drug (Jajoo et al., 1989b). Several clinical drug interaction studies showed that the increase of buspirone AUC by CYP3A4 inhibitors varied from 3.4- to 19-fold, which appeared to correlate with the potencies of the CYP3A inhibitors (Mahmood and Sahajwalla, 1999). For example, coadministered itraconazole, a potent CYP3A4 inhibitor, produced 15.8- to 19.2-fold elevation of the buspirone AUC, whereas erythromycin, a moderate CYP3A4 inhibitor, increased the buspirone AUC by 5.2- to 5.9-fold (Kivisto et al., 1997). The elevation of buspirone AUC was approximately 1.5-fold greater than that of midazolam AUC by the same CYP3A4 inhibitors (Bjornsson at al., 2003). Buspirone has been recommended as one of the preferred in vivo probe substrates for the evaluation of CYP3A inhibition by the FDA (FDA/CDER, 1999). The results from the present study provide in vitro metabolism evidence for the use of buspirone as the CYP3A probe substrate in clinical drug interaction studies.
In summary, we have demonstrated that the major metabolic pathways (Fig. 1) of buspirone in HLMs are N-dealkylation to 1-PP, N-oxidation to Bu N-oxide, and hydroxylation to 3′-OH-Bu, 5-OH-Bu, and 6′-OH-Bu. Bu N-oxide is a newly identified buspirone metabolite. Among the P450 isoforms evaluated, CYP3A, most likely CYP3A4, is the primary P450 enzyme responsible for the formation of these metabolites in HLMs (Fig. 1). Although CYP2D6 has buspirone oxidation activity, its contribution to the overall metabolism of buspirone in HLMs appears to be very minimal.
Acknowledgments
We thank Dr. Robert F. Mayol for intellectual input during the course of the study. We also thank Dr. Kent Rinehart and colleagues in the Radiosynthesis Group at Bristol-Myers Squibb Company for the synthesis of [14C]buspirone and metabolite reference standards.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.000836.
-
ABBREVIATIONS: FDA, Food and Drug Administration; P450, cytochrome P450; HLM, human liver microsome; 1-PP, 1-pyrimidinylpiperazine; 3′-OH-Bu, 3′-hydroxybuspirone; 5-OH-Bu, 5-hydroxybuspirone; 6′-OH-Bu, 6′-hydroxybuspirone; Bu N-oxide, buspirone N-oxide; 5-OH-1-PP, 5-hydroxy-1-pyrimidinylpiperazine; 5,6′-di-OH-Bu, 5,6′-dihydroxybuspirone; Oxa-Bu, 3-oxo-N-[4-[-(2-pyrimidinyl)-1-piperazinyl]butyl]-2-oxaspiro-[4,4]nonane-1-carboxamide; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; LC/MS/MS, liquid chromatography/tanden mass spectrometry; MH+, protonated molecular ion; MSC, microplate scintillation counter; RFD, radio flow detector; Km, Michaelis content; Vmax, maximal initial reaction velocity; CLint, intrinsic clearance (Km/Vmax); AUC, area under the plasma concentration-time curve; Cmax, maximum plasma concentration.
- Received June 3, 2004.
- Accepted January 3, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}