


Abstract
Lenvatinib, a potent inhibitor of multiple tyrosine kinases, including vascular endothelial growth factor receptors 2 and 3, generated unique metabolites after oral administration of [14C]lenvatinib (30 mg/kg) to a male cynomolgus monkey. Lenvatinib was found to be transformed to a GSH conjugate, through displacement of an O-aryl moiety, at the quinoline part of the molecule in the liver and kidneys. The GSH conjugate underwent further hydrolysis by γ-glutamyltranspeptidase and dipeptidases, followed by intramolecular rearrangement, to form N-cysteinyl quinoline derivatives, which were dimerized to form disulfide dimers and also formed an N,S-cysteinyl diquinoline derivative. In urine, a thioacetic acid conjugate of the quinoline was also observed as one of the major metabolites of lenvatinib. Lenvatinib is a 4-O-aryl quinoline derivative, and such compounds have been known to undergo conjugation with GSH, accompanied by release of the O-aryl moiety. Because of intramolecular rearrangement in the case of lenvatinib, hydrolysis of the GSH conjugate yielded N-cysteinylglycine and N-cysteine conjugates instead of the corresponding S-conjugates. Because the N-substituted derivatives possess free sulfhydryl groups, dimerization through disulfide bonds and another nucleophilic substitution reaction with lenvatinib resulted in the formation of disulfanyl dimers and an N,S-cysteinyl diquinoline derivative, respectively. Characteristic product ions at m/z 235 and m/z 244, which were associated with thioquinoline and N-ethylquinoline derivatives, respectively, were used to differentiate S- and N-derivatives in this study. On the basis of accurate mass and NMR measurements, a unique metabolic pathway for lenvatinib in monkey and the proposed formation mechanism have been elucidated.
Introduction
Lenvatinib mesylate (E7080; 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]7-methoxyquinoline-6-carboxamide mesylate) is a potent inhibitor of multiple receptor tyrosine kinases, such as vascular endothelial growth factor (VEGF) (Matsui et al., 2008b). VEGF plays an important role in tumor growth and is involved in angiogenesis, in relation to the development of metastasis. Lenvatinib showed potent antitumor efficacy with a broad spectrum of human tumor xenograft models, on the basis of inhibitory activity toward VEGF receptors 2 and 3 (Matsui et al., 2008a; Ikuta et al., 2009; Ogino et al., 2011).
Lenvatinib generated various metabolites in male cynomolgus monkey after oral administration of [14C]lenvatinib at 30 mg/kg, although it was metabolically stable in in vitro hepatic incubations. On the basis of metabolite identification with LC/MS and NMR, we found that lenvatinib was conjugated with GSH, with elimination of the chlorophenoxy group, and the GSH conjugate was further biotransformed to unique metabolites that were intramolecularly rearranged from S-conjugate to N-conjugate and dimerized. In general, GSH conjugates can be excreted directly in bile from the liver or catabolized through the mercapturic acid pathway, mainly in kidneys, for elimination (Commandeur et al., 1995). GSH conjugates can be initially metabolized by γ-glutamyltranspeptidase (γ-GTP), a specific enzyme for hydrolysis of γ-glutamyl peptide bonds, which results in the formation of S-cysteinylglycine derivatives. Then, S-cysteinylglycine derivatives can be hydrolyzed by dipeptidases (DPs) to form S-cysteine derivatives, which can yield the formation of mercapturic acid derivatives through conjugation by N-acetyltransferases.
In this study, lenvatinib was found to form a substituted S-GSH conjugate of the methoxyquinoline, with concomitant elimination of the chlorophenoxy moiety. A similar metabolic pathway was reported for a 4-O-aryl quinoline derivative, AMG 458 (1-(2-hydroxy-2-methylpropyl)-N-[5-(7-methoxyquinolin-4-yloxy)pyridin-2-yl]-5-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide), which undergoes GSH conjugation with elimination of the O-aryl group (Teffera et al., 2008). In this report, a substituted GSH conjugate, from which the O-aryl group had been displaced, was excreted in bile and was further metabolized to a mercapturic acid derivative in kidney after oral administration of AMG 458 to rats. In contrast, the substituted GSH conjugate of lenvatinib was further metabolized to an N-cysteinyl(glycinyl)quinoline derivative instead of the S-cysteinyl(glycine) and mercapturic acid derivatives observed for monkey. The N-conjugates of the quinoline indicated that an intramolecular rearrangement occurred after formation of the corresponding S-conjugates. The intramolecular rearrangement reaction has not been reported for GSH conjugates of aromatic substructures, although acyl-GSH conjugates were reported to generate N-cysteinyl(glycine) derivatives as the products of intramolecular rearrangement reactions after processing by γ-GTP and DPs (Tate, 1975; Fitch et al., 2004; Grillo et al., 2008). The N-derivatives subsequently formed disulfide-linked dimers or underwent aromatic substitution reactions with unchanged lenvatinib under concentrated conditions, such as in the urinary bladder or gall bladder. On the basis of accurate mass and NMR measurements, we demonstrate a unique metabolic pathway for lenvatinib. We also propose the formation mechanism for the metabolites observed in monkey in vivo in this study.
Materials and Methods
Materials.
Radiolabeled lenvatinib mesylate ([14C]E7080; 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]7-methoxy[2-14C]quinoline-6-carboxamide mesylate) was obtained from GE Healthcare (Chalfont St. Giles, UK), with a specific activity of 59 mCi/mmol. The radiochemical purity was more than 99% during this study. Unlabeled lenvatinib was synthesized at FUJIFILM Finechemicals (Fukushima, Japan). Reduced glutathione, S-methylcysteine, ammonium acetate, and N-acetylcysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO). l-Cysteine, acetonitrile, and methanol were obtained from Wako Pure Chemicals (Osaka, Japan). Liquid scintillation cocktail (Ultima-Flo M) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Cynomolgus monkey liver S9, Sprague-Dawley rat liver S9, and pooled human liver S9 were purchased from XenoTech, LLC (Lenexa, KS).
Dosing for Male Cynomolgus Monkey.
The animal experiments were performed in a facility accredited by the Center for Accreditation of Laboratory Animal Care and Use, Japan Health Sciences Foundation. The experimental protocol was approved by the institutional animal care and use committee, and the study was performed according to the Eisai animal experimentation regulations. An appropriate amount of lenvatinib, dissolved in 3 mM hydrochloric acid, was mixed with [14C]lenvatinib (2.143 mg/ml, 8.878 MBq/ml, dissolved in 3 mM hydrochloric acid) to yield a dosing solution of 3 mg/ml (1.27 MBq/ml). [14C]Lenvatinib was administered orally to a male cynomolgus monkey, at 30 mg/kg (12.7 MBq/kg), once per day on days 1, 2, and 4. After the first dose, urine and feces were collected for 3 days. The monkey was euthanized 4 h after the last dose, and blood, bile in the gall bladder, and the liver were obtained. Blood was collected into tubes containing sodium heparin as an anticoagulant and was centrifuged (at 3000 rpm for 10 min at 4°C) to yield plasma. A liver sample was homogenized in 2 volumes of ice-cold water by using a Physcotron homogenizer (MICROTEC CO., LTD., Chiba, Japan) at 4°C. All samples collected were stored at −80°C before metabolite analysis.
Sample Preparation Procedure for Radio-LC/MS Analysis.
Radioactivity in plasma and liver homogenate was extracted by using 2 volumes of methanol after the addition of 0.1 volume of 0.1 M phosphate buffer (pH 7.4). The extracts were dried under nitrogen, and the resulting residue was reconstituted with 50% methanol. The resulting samples were centrifuged (at 12,000 rpm for 10 min at 4°C) to yield supernatants, which were used for radio-LC/MS analysis. Urine was centrifuged (at 12,000 rpm for 10 min at 4°C) to yield a supernatant, which was used for radio-LC/MS analysis. Bile in the gall bladder was diluted 10-fold with water. The resulting sample was centrifuged (at 3000 rpm for 10 min at 4°C) to yield a supernatant, which was used for radio-LC/MS analysis.
Radio-LC/MS Analysis.
The radio-LC/MS system consisted of an accurate mass spectrometer (Synapt G2; Waters, Milford, MA), an Acquity UPLC system (Waters), and a radioisotope detector (Radiomatic 625 TR; PerkinElmer Life and Analytical Sciences). Analytical conditions for the mass spectrometer were as follows: capillary voltage, 3.2 kV; source temperature, 120°C; desolvation temperature, 400°C; cone gas flow, 50 l/h; desolvation gas flow, 800 l/h. The mass spectrometer was calibrated with cluster ions generated from sodium formate (10 mM solution dissolved in water/isopropanol, 1:9, by volume), in the mass range of 100 to 1500 Da. Mass spectral data were acquired in the positive-ion mode, with continuous correction with external standard solution (0.5 ng/ml leucine-encephalin, infused with 10-s intervals at a flow rate of 50 μl/min; m/z 556.2771), and centroids were calculated automatically. Full-scan mass spectra were acquired from m/z 100 to 1300. Aliquots of the samples (urine, bile in gall bladder, plasma, and liver homogenate) containing 40,000 to 50,000 dpm of radioactivity were injected onto an Atlantis T3 HPLC column (5 μm, 4.6 × 150 mm; Waters). Chromatography was performed with a gradient system with solvent A of water/100 mM ammonium acetate/formic acid (900:100:0.5, by volume) and solvent B of acetonitrile/100 mM ammonium acetate/formic acid (900:100:0.5, by volume), with a flow rate of 1 ml/min. The initial mobile phase was 100% solvent A. The percentage of solvent B was linearly increased to 5% over 5 min, to 15% over the next 10 min, and to 50% over the next 35 min. Solvent B was then increased to 90% over 1 min and was maintained at this level for 14 min. The eluent from the analytical column was introduced into the radioisotope detector with a split ratio of 4:1 (radioisotope detector/mass spectrometer, by volume). Radiochromatograms were obtained by mixing the eluent from the column with liquid scintillation cocktail (Ultima-Flo M), with flow rates of 0.8 and 1.6 ml/min, respectively. Mass spectral data were acquired and processed by using MassLynx 4.1 software (Waters). The radioisotope detector was controlled with ProFSA 3.3.12.79 software (PerkinElmer Life and Analytical Sciences).
Isolation of Metabolites from Monkey Urine.
The predominant metabolites in urine (C1, C3, and C4), which were observed at retention times between 12.2 and 14.1 min (see Fig. 2), were isolated with preparative HPLC. The system consisted of a LC-10AD VP pump, an SIL-HTC autosampler, an SPD-10A VP photodiode array detector, a DGU-14A degasser, and a CTO-10AC VP column oven (Shimadzu, Kyoto, Japan). Separation of metabolites was conducted by using a gradient system with solvent A of 0.1% formic acid in water and solvent B of 0.1% formic acid in acetonitrile. The initial mobile phase was 100% solvent A. The percentage of solvent B was linearly increased to 10% over 5 min, to 20% over the next 20 min, and to 100% over the next 5 min, after which conditions were maintained for the next 5 min. The column was equilibrated with the initial mobile phase before each injection. Chromatography was conducted with an Atlantis T3 preparative HPLC column (10 × 250 mm; Waters), with a flow rate of 4 ml/min and monitoring at a wavelength of 336 nm. Each fraction obtained was analyzed with the Synapt G2/UPLC system equipped with an Atlantis T3 column (4.6 × 150 mm), as described above. The fractions containing the targeted protonated molecular ions, at m/z 293.0591 (C1), 641.1483 (C3), and 698.1697 (C4), were evaporated under a nitrogen gas flow for further purification. The resulting residues were reconstituted with 30% acetonitrile. After centrifugation (at 12,000 rpm for 10 min at 4°C), three fractions, containing C1, C3, and C4, were injected onto an Atlantis dC18 column (4.6 × 150 mm; Waters), with a flow rate of 1 ml/min, and the eluent was monitored at a wavelength of 336 nm. Isocratic elution with 10% solvent B was used for separation of C1. Purification of C3 and C4 was conducted with a gradient solvent system, with an initial mobile phase of 10% solvent B followed by a linear gradient over 10 min to 100% solvent B, which was maintained for the subsequent 2 min. Before injection, the column was equilibrated with 10% solvent B for 7 min. The fractions were analyzed with the Synapt G2/UPLC system, and three fractions, containing C1, C3, and C4, were freeze-dried before NMR analysis. Isolated amounts and purities of C1, C3, and C4 were as follows: C1, 34 μg and 90%; C3, 41 μg and >95%; C4, 134 μg and 80%, respectively.
NMR Spectroscopy.
The structures of the isolated metabolites (C1, C3, and C4) were investigated by using one- and two-dimensional NMR experiments. All NMR data were recorded with a Unity INOVA, 500-MHz, NMR spectrometer (Varian, Palo Alto, CA) equipped with a 5-mm, z-gradient, indirect-detection probe. The isolated metabolites and lenvatinib were dissolved in ∼0.6 ml of methanol-d4. One-dimensional proton NMR and two-dimensional correlation spectroscopy and nuclear Overhauser effect spectroscopy (NOESY) data were acquired for each metabolite. The proton chemical shifts were referenced to an internal solvent peak for CD2HOD at 3.30 ppm. All spectra were recorded at 30°C.
In Vitro Incubation Conditions.
Lenvatinib was dissolved in dimethyl sulfoxide to yield a 10 mM solution. For investigation of the contribution of enzymatic activities to lenvatinib metabolism, incubations were conducted in potassium phosphate buffer (pH 7.4) containing 5 mM GSH and 50 μM lenvatinib, in the presence or absence of both 2 mg/ml liver S9 (Sprague-Dawley rats, cynomolgus monkeys, and humans) and NADPH, for 0, 15, 30, and 60 min at 37°C. For investigation of the reactivity of nucleophiles toward lenvatinib, incubations were conducted in potassium phosphate buffer (pH 7.4) containing 50 μM lenvatinib, in the presence or absence of 5 mM nucleophile (GSH, NAC, l- cysteine, or S-methylcysteine), for 0, 30, 60, and 120 min at 37°C. Incubations were terminated with the addition of an equal volume of 5% formic acid in acetonitrile/methanol (2:1, by volume). After centrifugation (at 12,000 rpm for 10 min at 4°C), an aliquot of supernatant was introduced into an LC/MS system (Quantum/Accela; Thermo Fisher Scientific, Waltham, MA). Chromatography was conducted with an Acquity ethylene bridged hybrid HPLC column (1.7 μm, 2.1 × 100 mm; Waters), at a flow rate of 0.5 ml/min. Mobile phases were composed of solvent A of 100 mM ammonium acetate in water and solvent B of 100 mM ammonium acetate in 90% acetonitrile. The initial mobile phase was 100% solvent A; solvent B was linearly increased to 50% over 5 min and to 100% over the next 1 min and then was maintained at 100% for an additional 2 min. Before each injection, the analytical column was equilibrated with solvent A. Lenvatinib was monitored on the basis of the fragment ions at m/z 202, 312, and 370 that were generated from a protonated molecular ion of m/z 427. The expected GSH, NAC, cysteine, and S-methylcysteine adducts of lenvatinib, with molecular ions at m/z 508, 364, 322, and 336, respectively, were monitored on the basis of the product ions at m/z 244 and 235.
Results
Monkey In Vivo Metabolites of Lenvatinib.
The structure of [14C]lenvatinib is illustrated in Fig. 1. The urine and feces samples collected over days 1 to 3 yielded 19% and 59% recovery of radioactivity, respectively (in total, 78% of the dose). A major fecal component observed with radiochromatography was unchanged lenvatinib, which accounted for 51% of the total dose (86% of fecal radioactivity). Typical radiochromatograms for the four in vivo matrices obtained after dosing of cynomolgus monkey with [14C]lenvatinib at 30 mg/kg are shown in Fig. 2. Seven major radioactive components, including unchanged lenvatinib, were observed and were defined as C1 to C7, according to the order of their retention times. The accurate mass spectra and chromatographic data are summarized in Table 1. With the exception of lenvatinib, all of these metabolites were eluted at retention times between 12 and 17 min (Table 1). Accurate mass analysis indicated that all of the metabolites contained sulfur but not chlorine, which suggests that loss of the chlorophenoxy moiety occurs during metabolism (Table 1).

Structure of lenvatinib mesylate (*, 14C-labeled position).

Representative radiochromatograms of urine (A), bile in the gall bladder (B), liver homogenate (C), and plasma (D) after oral administration of [14C]lenvatinib at 30 mg/kg (12.7 MBq/kg) per day to a male cynomolgus monkey. Urine was pooled for 24 h after the first dose. Bile in the gall bladder, liver, and plasma were obtained 4 h after the last dose. Bile in the gall bladder was diluted 10-fold with water for the analysis. Radioactivity in liver and plasma was extracted with methanol, and the extract was dried under a nitrogen gas flow. The resulting residue was reconstituted with 50% methanol. Aliquots of the samples were diluted for radio-LC/MS analysis to yield 40,000 to 50,000 dpm in each injection.
Summary of seven radioactive components of [14C]lenvatinib after oral administration to male cynomolgus monkey at 30 mg/kg
Structure Elucidation of In Vivo Metabolites of Lenvatinib.
Three metabolites found in urine, C1, C3, and C4, were isolated with preparative HPLC, and their structures were determined with NMR spectroscopy and accurate MS. The structures of other metabolites, C2, C5, and C6, were determined on the basis of accurate mass spectral data. The MS and NMR data for lenvatinib were obtained and used to aid structure elucidation of the metabolites.
Lenvatinib (C7) eluted at 46.4 min and showed a protonated molecular ion of m/z 427.1169 (C21H20ClN4O4+, +0.2 ppm) with a typical chlorine isotope pattern, as shown in Fig. 3. Lenvatinib yielded product ions at m/z 370.0562, 353.0297, and 327.0534, related to the fragmentation of the cyclopropylurea moiety (Figs. 4 and 5). The proton NMR spectral data for lenvatinib (Table 2) were used to interpret the metabolite data, as described below.
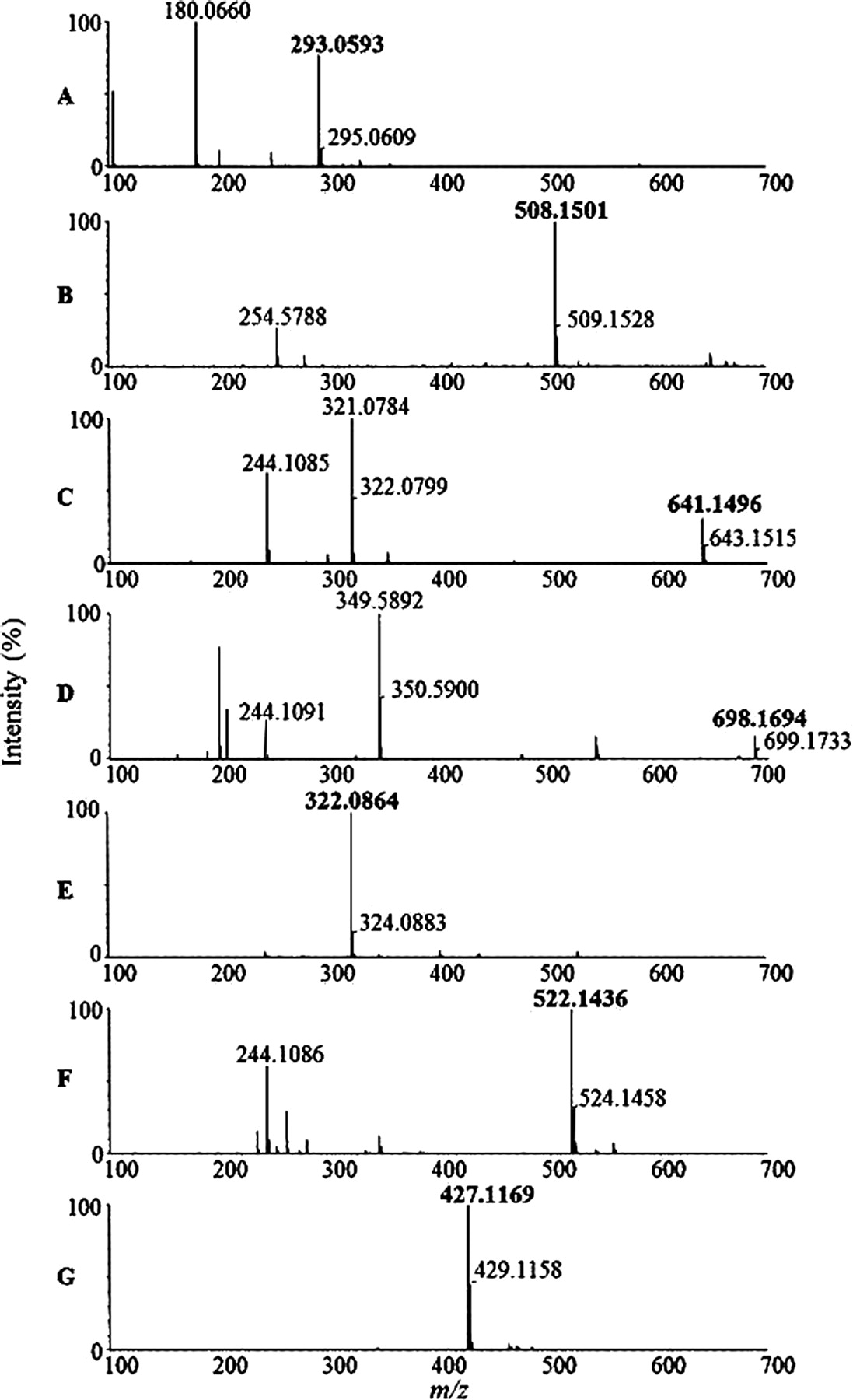
Accurate mass spectra of the predominant radioactive components, C1 (A), C2 (B), C3 (C), C4 (D), C5 (E), C6 (F), and C7 (lenvatinib) (G).

Product ion spectra of C1 at m/z 293 (A), C2 at m/z 508 (B), C3 at m/z 641 (C), C4 at m/z 698 (D), C5 at m/z 322 (E), C6 at m/z 522 (F), and C7 (lenvatinib) at m/z 427 (G).

Proposed structures and fragment patterns of the six metabolites and lenvatinib.
Summary of proton NMR chemical shifts for lenvatinib, C1, C3, and C4
C1, which was found in urine, gave a protonated molecular ion of m/z 293.0593 with a retention time of 12.2 min (Figs. 2 and 3). The chemical formula was proposed as C13H13N2O4S+ (+0.7 ppm), which corresponded to displacement of the chlorophenoxy moiety of lenvatinib with a mercaptoacetic acid (Table 1). Although a minor fragment ion at m/z 235.0490 (Figs. 4 and 5) suggested that C1 contained the thioquinoline scaffold, the other observed product ions could not be assigned. Therefore, the structure of C1 was confirmed with NMR spectroscopy. The proton NMR spectrum of C1 showed the proton signals of the quinoline moiety, whereas the signals of the chlorophenoxy and cyclopropylurea moieties present in the spectrum of lenvatinib were not observed. Instead, a singlet methylene signal was observed at δ 3.84 ppm (2H), which was assignable to the mercaptoacetic acid moiety (Table 2). In the NOESY spectrum of C1, a correlation was observed between the methylene protons of the mercaptoacetic acid moiety (H-6) and the quinoline proton (H-2), as shown in Table 2. This observation revealed the attachment site of the thioacetic acid to be the 4-position of the quinoline moiety. On the basis of the accurate mass and NMR data, the structure of C1 was proposed to be a 4-mercaptoacetic acid derivative of the methoxyquinoline.
C2 was observed in the liver sample and showed a protonated molecular ion of m/z 508.1501, which eluted at a retention time of 13.1 min (Figs. 2 and 3). The proposed chemical formula of C21H26N5O8S+ (+0.8 ppm) corresponded to a GSH conjugation of the quinoline moiety, with loss of the chlorophenoxy and cyclopropylurea moieties of lenvatinib. The product ions at m/z 379.1032, 235.0490, and 218.0290 indicated the neutral loss of pyroglutamate, γ-glutamylalanylglycine, and both γ-glutamylalanylglycine and an amino group of the amide moiety, respectively (Figs. 4 and 5). The fragment ion at m/z 235.0490 was consistent with the thioquinoline derivative, which suggests that the sulfhydryl group of GSH is bound to the quinoline moiety (Fig. 5). The retention time and LC/MS data for C2 were the same as those for the product produced from the mixture of lenvatinib and GSH in phosphate buffer (Figs. 6 and 7). Therefore, C2 was determined to be a GSH conjugate of the methoxyquinoline.

Contributions of enzymatic activities to the formation of C2. Lenvatinib (50 μM) was incubated with GSH (5 mM) with rat liver S9 fractions, cynomolgus monkey liver S9 fractions, human liver S9 fractions, or no enzymes, in the presence or absence of NADPH. A and C, the proportions of lenvatinib remaining in the presence of NADPH (A) and in the absence of NADPH (C) are shown. B and D, the formation of C2 is shown as the mass area of C2, as monitored with the product ions at m/z 202, 312, and 370 from the molecular ion at m/z 427, in the presence of NADPH (B) and in the absence of NADPH (D).

Reactions of nucleophiles with lenvatinib. Lenvatinib (50 μM) was incubated with 5 mM GSH, NAC, cysteine, or S-methylcysteine or without a nucleophile for 0, 30, 60, and 120 min. The reaction was terminated with the addition of an equal volume of 5% formic acid in acetonitrile. A, proportions of lenvatinib remaining in the presence of GSH, NAC, cysteine, and S-methylcysteine and in the absence of nucleophile. B, mass areas of conjugates generated from each nucleophile and lenvatinib. The expected substituted GSH, NAC, cysteine, and S-methylcysteine conjugates of lenvatinib, with proposed molecular ions at m/z 508, 364, 322, and 336, respectively, were monitored with product ions of m/z 244 and 235.
C3 was observed in bile in the gall bladder and in urine and eluted at a retention time of 13.5 min (Table 1; Fig. 2). The mass spectrum of C3 showed a protonated molecular ion at m/z 641.1496 (Fig. 3) with the proposed chemical formula of C28H29N6O8S2+ (+2.0 ppm), consistent with a homodimer of the cysteine conjugate of the methoxyquinoline moiety. The fragment ions at m/z 354.0502, 288.0934, and 244.1064 were proposed to be ions of the disulfanyl N-cysteine derivative of the quinoline, the N-alanine derivative of the quinoline, and the N-ethylquinoline derivative, respectively (Figs. 4 and 5). In contrast to C2, a key fragment ion at m/z 244.1064 indicated that the cysteinyl amine group was attached to the quinoline moiety. On the basis of the assignment of m/z 244, the fragment ion at m/z 354.0502 was proposed to be the disulfanyl N-cysteine derivative of the quinoline. These mass spectral data suggested that C3 is the homodimer of two molecules of N-cysteine conjugates of the methoxyquinolines. The structure of C3 was further investigated through proton NMR spectral analysis of the isolated material. The proton NMR spectrum of C3 demonstrated a set of proton signals for the quinoline moiety of lenvatinib. Instead of the chlorophenoxy moiety, signals attributable to the methine and methylene protons of the cysteine group were observed at δ 4.47 ppm (dd, J = 9.5 Hz) and at δ 3.51 (dd, J = 14.5 Hz) and 3.22 ppm (dd, J = 14.9 Hz), respectively (Table 2). These NMR data also supported the symmetric dimer structure of C3, consisting of two N-cysteinyl quinoline substructures. In the NOESY spectrum of C3, a correlation was observed between the methylene proton of the cysteine group (H-6) and the quinoline proton (H-2), as shown in Table 2. This observation indicated the N-substitution of the cysteine group at the 4-position of the quinoline moiety of lenvatinib. On the basis of the accurate mass and NMR spectral data, C3 was found to be a symmetric disulfide dimer consisting of two N-cysteine conjugates at the methoxyquinoline.
C4 was found only in urine, at a retention time of 14.1 min, and showed a protonated molecular ion at m/z 698.1694 that generated the product ions observed at m/z 411.0778 and 244.1064 (Figs. 2⇑⇑–5). The proposed chemical formula of C30H32N7O9S2+ (+0.4 ppm) was equivalent to that of C3 with the addition of one molecule of glycine, consistent with C4 being a heterodimer of cysteine and cysteinylglycine conjugates of the methoxyquinoline moiety (Table 1). Similar to C3, C4 generated the characteristic fragment ion of m/z 244.1064, which indicated that cysteine and/or cysteinylglycine was attached to the quinoline moiety with the cysteinyl amino group (Fig. 5). Furthermore, the fragment ion of m/z 411.0778, which was assigned as a disulfanyl N-cysteinylglycine conjugate at the quinoline moiety, indicated that the respective N-cysteine and N-cysteinylglycine conjugates at the quinolones were linked through a disulfide bond (Fig. 5). The structure of C4 was confirmed with NMR analysis. It showed two signal sets for the quinolines, whereas the signals of both the chlorophenoxy and cyclopropylurea moieties were not observed (Table 2). In addition to the signals of two quinoline moieties, signal sets assignable to two cysteine groups and a glycine group were observed, as shown in Table 2. In the NOESY spectrum of C4, correlations were observed between the methylene protons of two cysteine groups (H-6 and H-6′) and the quinoline protons (H-2 and H-2′) (Table 2). These data suggested that both cysteine and cysteinylglycine groups would attach to the 4-positions of the quinolines through the cysteinyl amino groups. On the basis of these spectral data, C4 was proposed to be an asymmetric disulfanyl dimer of N-cysteine and N-cysteinylglycine conjugates at the methoxyquinolines.
C5 was found in bile in gall bladder and in the liver sample, with a retention time of 14.5 min, and showed a protonated molecular ion at m/z 322.0864 (Figs. 2 and 3). The proposed chemical formula of C14H16N3O4S+ (+2.5 ppm) corresponded to a cysteine conjugate of the methoxyquinoline moiety. The product ions at m/z 276.0821, 244.1064, and 218.0937 indicated neutral loss of the carboxyl group, of both the sulfhydryl and carboxyl groups, and of the thioacetic acid moiety of cysteine, respectively (Figs. 4 and 5). The characteristic fragment ion observed at m/z 244.1064 indicated that the cysteinyl amino group was attached to the quinoline (Fig. 5). The retention time and LC/MS data for C5 were the same as those for the product produced from the mixture of lenvatinib and cysteine in phosphate buffer (Fig. 7). Therefore, C5 was determined to be an N-cysteine conjugate at the methoxyquinoline.
C6 was found in bile in the gall bladder, in urine, and in liver samples, with a retention time of 17.1 min (Table 1), and showed a protonated molecular ion at m/z 522.1436 (Figs. 2 and 3). The proposed chemical formula of C25H24N5O6S+ (+1.1 ppm) corresponds to two methoxyquinoline moieties conjugated to cysteine (Table 1). The product ions at m/z 505.1212 and 244.1064 were consistent with neutral loss of an amino group and of both the thioquinoline and carboxyl groups, respectively (Figs. 4 and 5), and the product ion at m/z 244.1064 indicated that the cysteine was attached through its amino group to one of the quinolines (Fig. 5). The chemical formula is consistent with the cysteinyl sulfhydryl group being attached to the quinoline moiety of a second lenvatinib molecule, with concomitant loss of the chlorophenoxy moiety, which means that the cysteinyl amine and sulfhydryl groups are each bound to a methoxyquinoline moiety. Therefore, C6 was proposed to be an N,S-cysteine conjugate of two methoxyquinoline moieties.
Contributions of Enzymes to Formation of C2.
To investigate the contributions of cytochromes P450 (P450s) and glutathione S-transferases to the formation of C2, lenvatinib and GSH were incubated in the presence or absence of liver S9 fractions (rat, monkey, and human), in both the presence and absence of NADPH. The proportions of lenvatinib remaining were monitored on the basis of the product ions at m/z 202, 312, and 370. Formation of C2 was monitored through detection of the characteristic fragment ion at m/z 235, which was generated from the protonated molecular ion at m/z 508. Lenvatinib concentrations decreased in a time-dependent manner in the presence of GSH, accompanied by C2 formation, in the presence and absence of both NADPH and the S9 fraction (Fig. 6).
Reactions of Nucleophiles with Lenvatinib.
To investigate the mechanism of formation of the sulfur-containing metabolites, lenvatinib was incubated in potassium phosphate buffer (pH 7.4) fortified with the nucleophiles GSH, NAC, cysteine, and S-methylcysteine, at 37°C. The proportion of lenvatinib remaining was monitored as described above. At the same time, the expected GSH, NAC, cysteine, and S-methylcysteine conjugates of the methoxyquinoline moiety were monitored on the basis of the product ions at m/z 244 and 235, which were generated from the proposed molecular ions at m/z 508 (quinoline plus GSH; C2), 364 (quinoline plus NAC), 322 (quinoline plus cysteine; C5), and 336 (quinoline plus S-methylcysteine). As shown in Fig. 7, the level of lenvatinib decreased in the presence of GSH, NAC, and cysteine, which have free sulfhydryl groups in their structures. It was also found that lenvatinib formed corresponding conjugates with GSH, NAC, and cysteine at the methoxyquinoline moiety, with concomitant elimination of the chlorophenoxy and cyclopropylurea moieties. In contrast, S-methylcysteine did not react with lenvatinib to form any conjugates.
Discussion
Metabolites found in the cynomolgus monkey after oral administration of [14C]lenvatinib are thought to be formed through a three-step reaction, as follows: 1) displacement of the chlorophenoxy moiety of lenvatinib with GSH to yield C2, 2) hydrolysis of C2 by γ-GTP and DPs to yield C5 and N-cysteinylglycine conjugates at the quinoline moiety, followed by intramolecular rearrangement from S- to N-cysteinyl quinoline derivatives, and 3) dimerization through disulfide bond formation or another nucleophilic substitution reaction with unchanged lenvatinib, to generate C3, C4, and C6.
As summarized in Table 1, the expected chemical formulas of all major metabolites (C1 to C6) (Table 1) were assigned as sulfur-containing conjugates of the methoxyquinoline moiety, with loss of the chlorophenoxy moiety of lenvatinib. On the basis of accurate mass and NMR analyses of the metabolites, it was determined that lenvatinib underwent GSH conjugation at the quinoline moiety with elimination of the chlorophenoxy moiety to generate C2, followed by sequential GSH catabolism that resulted in the formation of C1, C3, C4, C5, and C6. As shown in Figs. 4 and 5, C2 showed a characteristic product ion at m/z 235, which was interpreted as a thioquinoline derivative; this indicated that the sulfhydryl group of GSH was attached to the quinoline. Metabolites C3 to C6 showed a unique fragment ion at m/z 244, consistent with an N-ethylquinoline derivative, which indicated that the cysteinyl amino group was bound to the quinoline (Figs. 4 and 5). These key fragment ions suggested that intramolecular displacement from the S-cysteinyl(glycine) conjugates to the corresponding N-conjugates occurred after hydrolytic processing of C2. NMR analyses of purified metabolites C3 and C4 strongly supported the formation of N-conjugates at the 4-position of the quinoline, which allowed dimerization through disulfide bonds.
As shown in Fig. 6, A and B, NADPH did not affect the reaction of lenvatinib with GSH, which indicates that P450s are not involved. Figure 6, C and D, shows that formation of C2 was observed at the same levels in phosphate buffer and in S9 fractions. Figure 6 clearly indicates that C2 formation is spontaneous, with no species differences and no contributions from enzymes such as P450s and glutathione S-transferases. As shown in Fig. 7, NAC reacted with lenvatinib in a manner analogous to that of GSH, to generate an S-NAC conjugate of the methoxyquinoline, with elimination of the chlorophenoxy moiety. Cysteine also spontaneously reacted with lenvatinib, which resulted in the formation of C5. Only S-methylcysteine did not yield any conjugates with lenvatinib. Therefore, a free sulfhydryl group is necessary for the primary intermolecular displacement of the chlorophenoxy moiety of lenvatinib by nucleophiles. In a comparison of the results for cysteine with those for NAC and GSH, cysteine could yield the N-cysteinyl conjugate with the quinoline to form C5, whereas NAC and GSH generated only the S-conjugate of the quinoline, which indicates that the intramolecular rearrangement requires the cysteinyl amino group (Fig. 7).
As summarized in Fig. 8, lenvatinib primarily undergoes a nucleophilic substitution reaction with a sulfhydryl residue but not with an amino group. If an intramolecular cysteinyl amine group exists, then it can attack the 4-position carbon of the methoxyquinoline to form an N-cysteinyl quinoline derivative, through a hypothetical, five-membered, ring intermediate. Because the N-conjugates possess a free sulfhydryl group in their structures, they can form disulfide bonds to yield dimeric metabolites such as C3 and C4 or can undergo another nucleophilic substitution reaction with unchanged lenvatinib to generate an N,S-cysteinyl diquinoline derivative such as C6.

Proposed formation mechanism for the metabolites of lenvatinib (C2–C6). Cys-Gly, cysteinylglycine; Cys, cysteine; Gly, glycine.
On the basis of the formation mechanism, the metabolic pathway of lenvatinib is proposed to be as follows. In the liver, lenvatinib is conjugated with GSH or cysteine to form C2 and C5, respectively. An intermolecular nucleophilic substitution reaction between C5 and an unchanged lenvatinib molecule produces C6. Because γ-GTP and DPs are present in the gall bladder of macaques (Hinchman and Ballatori, 1990), C2 is sequentially catabolized after excretion into bile to give, after intramolecular rearrangement, the N-cysteinyl quinoline derivative C5. The dimeric metabolite C3 was not detected in the liver; therefore, the dimerization process might occur only under concentrated conditions, such as in bile in the gall bladder. The metabolism of lenvatinib after circulation into the kidneys would be similar to that in the liver. In kidneys, lenvatinib can be primarily transformed to C2, which can be subsequently hydrolyzed by γ-GTP and DPs and also can rearrange, which causes the generation of an N-cysteinylglycinyl quinoline derivative and C5. These metabolites can then be concentrated and dimerized in the urinary bladder to yield C3 and C4. C1, a mercaptoacetic acid conjugate of the quinoline moiety, was detected only in urine. According to previous reports, mercaptoacetic acid conjugates have been known to be derived from S-cysteine conjugates generated from the corresponding GSH conjugate (Lertratanangkoon et al., 1982; Stillwell et al., 1982; Richardson et al., 1998; Bloemen et al., 2001). The resulting cysteine conjugate can undergo oxidative deamination and decarboxylation reactions to yield a mercaptoacetic acid conjugate. These have mainly been observed as rat and mouse urinary metabolites. In the case of lenvatinib, it is likely that C1 is generated through a similar metabolic pathway from the GSH conjugate, C2, in kidneys.
Displacement of an O-aryl moiety by GSH, analogous to that reported here for lenvatinib, has been reported for the quinolone derivative AMG 458 (Teffera et al., 2008). In this case, the GSH conjugate was further metabolized to the corresponding mercapturic acid conjugate, which was identified as the major urinary metabolite for rats. In the same manner, because lenvatinib is also a 4-O-aryl quinoline derivative, the electron-deficient nature of the quinoline 4-position carbon enables a nucleophilic substitution reaction with GSH to generate C2 in the liver. The thioether bond of C2 is stable, because the cysteinyl amino group is masked with a γ-glutamyl peptide bond. Interestingly, once C2 is hydrolyzed by γ-GTP, and subsequently by DPs, to the corresponding S-cysteinylglycine and S-cysteine conjugates, the sulfur of the thioether bond is displaced, in an intramolecular rearrangement, by the cysteinyl amino group, which leads to the formation of N-cysteinyl(glycine) conjugates. In the transition state, the sulfhydryl group may be a better leaving group than the amine group, and thus the C-S bond is cleaved to yield the N-conjugates. The data showed that C5 can stably exist in the liver although it has a free sulfhydryl group in the structure. Once it is excreted into bile or urine, however, C5 seems to dimerize with itself or with the N-cysteinylglycinyl quinoline derivative through disulfide bonds. C5 also reacts with unchanged lenvatinib, which results in the formation of the N,S-cysteinyl diquinoline derivative, C6.
The chlorophenoxy moiety, which is released after elimination of the quinoline moiety of lenvatinib during the substitution reaction with GSH, has been detected with MS as a phenol derivative and both the sulfate and glucuronic acid conjugates of the phenol in plasma, bile, urine, and feces. The chlorophenoxy moiety of lenvatinib was not radiolabeled in this study, and its metabolic fate requires further investigation. To clarify the metabolic pathways of lenvatinib, we are planning an additional mass balance study in monkeys with lenvatinib radiolabeled at the chlorophenoxy group.
In summary, lenvatinib underwent a unique metabolic pathway involving hydrolysis of the GSH conjugate C2 by γ-GTP and DPs, followed by the formation of intramolecular rearrangement products such as C5. Furthermore, the rearranged metabolites underwent dimerization through disulfide bonds or a nucleophilic substitution reaction with another lenvatinib molecule, to form C3, C4, and C6. Finally, C1 also could be derived from C2 through a series of metabolic reactions.
Authorship Contributions
Participated in research design: Inoue and Mizuo.
Conducted experiments: Inoue, Asai, and Mizuo.
Wrote or contributed to the writing of the manuscript: Inoue, Asai, Mizuo, Fukuda, Kusano, and Yoshimura.
Acknowledgments
We thank Linda Buckley for assistance with language editing. We also appreciate Desmond O'Connor, Dr. George Lai, and Dr. Nancy Wong for scientific suggestions in this work and editorial assistance with this manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- VEGF
- vascular endothelial growth factor
- LC
- liquid chromatography
- MS
- mass spectrometry
- NAC
- N-acetylcysteine
- γ-GTP
- γ-glutamyltranspeptidase
- DP
- dipeptidase
- NOESY
- nuclear Overhauser effect spectroscopy
- HPLC
- high-performance liquid chromatography
- UPLC
- ultraperformance liquid chromatography
- AMG 458
- 1-(2-hydroxy-2-methylpropyl)-N-[5-(7-methoxyquinolin-4-yloxy)pyridin-2-yl]-5-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carboxamide
- P450
- cytochrome P450.
- Received October 18, 2011.
- Accepted December 29, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}