


Abstract
Repaglinide is mainly metabolized by cytochrome P450 enzymes CYP2C8 and CYP3A4, and it is also a substrate to a hepatic uptake transporter, organic anion transporting polypeptide (OATP)1B1. The purpose of this study is to predict the dosing time–dependent pharmacokinetic interactions of repaglinide with rifampicin, using mechanistic models. In vitro hepatic transport of repaglinide, characterized using sandwich-cultured human hepatocytes, and intrinsic metabolic parameters were used to build a dynamic whole-body physiologically-based pharmacokinetic (PBPK) model. The PBPK model adequately described repaglinide plasma concentration-time profiles and successfully predicted area under the plasma concentration-time curve ratios of repaglinide (within ± 25% error), dosed (staggered 0–24 hours) after rifampicin treatment when primarily considering induction of CYP3A4 and reversible inhibition of OATP1B1 by rifampicin. Further, a static mechanistic “extended net-effect” model incorporating transport and metabolic disposition parameters of repaglinide and interaction potency of rifampicin was devised. Predictions based on the static model are similar to those observed in the clinic (average error ∼19%) and to those based on the PBPK model. Both the models suggested that the combined effect of increased gut extraction and decreased hepatic uptake caused minimal repaglinide systemic exposure change when repaglinide is dosed simultaneously or 1 hour after the rifampicin dose. On the other hand, isolated induction effect as a result of temporal separation of the two drugs translated to an approximate 5-fold reduction in repaglinide systemic exposure. In conclusion, both dynamic and static mechanistic models are instrumental in delineating the quantitative contribution of transport and metabolism in the dosing time–dependent repaglinide-rifampicin interactions.
Introduction
Drug-drug interactions (DDIs) associated with membrane transporters and metabolizing enzymes can lead to severe adverse reactions and/or reduced pharmacologic effects. In vitro tools are valuable in assessing the involvement of such individual processes in the drug disposition. For example, human liver microsomes are extensively used in predicting the metabolic clearance of xenobiotics in human (Obach et al., 2006). Recently, sandwich-cultured human hepatocytes were demonstrated as a useful in vitro tool to characterize the hepatobiliary transport mechanism (Abe et al., 2009; Jones et al., 2012; Kimoto et al., 2012). However, integrating these in vitro data to quantitatively project the interplay between metabolizing enzymes and transporters and their impact on drug disposition continues to be a challenge. Recently, physiologically-based pharmacokinetic (PBPK) modeling has demonstrated utility in predicting drug pharmacokinetics and evaluating the DDI potential (Huang and Rowland, 2012; Rostami-Hodjegan, 2012). The implementation of PBPK is being increasingly considered in drug discovery and development, due to its versatility and the availability of commercial packages [e.g., GastroPlus (Simulations Plus, Inc., Lancaster, CA), PK-Sim (Bayer Technology Services GmbH, Leverkusen, Germany), and Simcyp (SimCYP Ltd., Sheffield, UK)]. Furthermore, the latest US Food and Drug Administration and European Medicines Agency guidelines on drug interactions suggested the use of mechanistic modeling to quantitatively predict the magnitude of DDIs in various clinical situations (European Medicines Agency, 2012; US Food and Drug Administration, 2012).
Repaglinide is an antidiabetic drug used to treat type 2 or non–insulin-dependent diabetes mellitus (Scott, 2012). It lowers the blood glucose levels by stimulating the release of insulin from the pancreas while interfering with the ATP-dependent potassium channels in the β-cell membrane. Repaglinide is majorly metabolized by the cytochrome P450 (P450) isoenzymes CYP2C8 and CYP3A4, and it is also a substrate to a hepatic uptake transporter, organic anion transporter polypeptide (OATP)1B1 (Bidstrup et al., 2003; Niemi et al., 2003c; Kajosaari et al., 2005a; Niemi et al., 2005; Menochet et al., 2012; Sall et al., 2012). The plasma exposure of repaglinide is altered by several drugs that inhibit CYP2C8, CYP3A4, and OATP1B1 (Tornio et al., 2012). Notably, gemfibrozil caused an increase in area under the plasma concentration-time curve (AUC) of repaglinide up to 8.3-fold (Honkalammi et al., 2011a), mainly due to mechanism-based inactivation of CYP2C8 by its major circulating metabolite, gemfibrozil 1-O-β-glucuronide, and competitive inhibition of OATP1B1 by both gemfibrozil and the metabolite (Fujino et al., 2003; Shitara et al., 2004). Oxidative biotransformation of repaglinide by both CYP2C8 and CYP3A4, with CYP3A4 fraction metabolism (fm)CYP3A4 of 0.29–0.45, has been reported in vitro (Bidstrup et al., 2003; Kajosaari et al., 2005a). However, an apparent in vitro–in vivo disconnect in fm of the two CYPs has been suggested, based on the recent clinical repaglinide-gemfibrozil DDIs studies (Honkalammi et al., 2011a, 2012). In these studies, using a static modeling approach, repaglinide fmCYP2C8 was estimated to be >0.85 in vivo. Previously, we developed a whole-body PBPK model for repaglinide, which suggested that repaglinide systemic disposition is dependent on the hepatic uptake clearance, and that the change in its AUC is influenced by hepatic uptake clearance, intrinsic metabolic clearances, and the fm (Varma et al., 2013). Our mechanistic evaluation demonstrated that repaglinide-gemfibrozil DDIs can be quantitatively described with the in vitro fm values.
The antituberculosis agent rifampicin is a typical inducer of drug transporters and metabolizing enzymes, particularly CYP3A4 (Niemi et al., 2003a). DDIs were reported when repaglinide was administered with rifampicin. Notably, repaglinide dosing time relative to rifampicin treatment significantly influences the magnitude of repaglinide systemic exposure change. For example, only a 31% decrease in repaglinide AUC was observed when repaglinide was ingested 1 hour after the last dose of rifampicin treatment (Hatorp et al., 2003). In a separate study, repaglinide AUC decreased 57% when administered 12.5 hours following the last oral dose of rifampicin (Niemi et al., 2000). Yet in another study, repaglinide AUC was decreased by ∼50 and 80% when administered concomitantly and 24 hours after the last rifampicin dose, respectively (Bidstrup et al., 2004). We hypothesized that the effects of rifampicin on the repaglinide disposition could be complex, involving induction of CYP3A4 and inhibition of hepatic uptake or metabolism; these multiple mechanisms need to be considered to quantitatively rationalize the observed dosing time–dependent interactions. In this study, we used a dynamic mechanistic (whole-body PBPK) model of repaglinide to simulate the plasma concentration-time profiles and further assess the repaglinide dosing-time dependent interactions with rifampicin. In addition, a static mechanistic “extended net-effect” model considering enzyme- and transporter-mediated disposition of repaglinide was developed to assess the interactions with rifampicin.
Materials and Methods
PBPK Modeling and Simulations.
Whole-body PBPK modeling and simulations of clinical pharmacokinetics and DDIs were performed using the population-based absorption, distribution, metabolism, and excretion simulator, Simcyp (version 11.0). The repaglinide model was built using the physicochemical properties and in vitro preclinical data, such as human plasma unbound fraction, blood-to-plasma ratio, and metabolic intrinsic clearance values (Table 1). Complete details of the repaglinide PBPK model have been described elsewhere (Varma et al., 2013). In brief, to obtain the distribution of repaglinide into all organs except the liver, we adopted a full-PBPK model using Rodgers and Rowland’s method (Rodgers and Rowland, 2006) considering rapid equilibrium between blood and tissues. Permeability-limiting hepatic disposition of repaglinide was considered, for which sinusoidal active uptake intrinsic clearance and passive diffusion obtained from sandwich-cultured human hepatocyte studies were incorporated (Varma et al., 2013). Hepatic microsomal CLint,met (intrinsic clearance) and fm contribution of CYP3A4 to the total metabolic clearance (fmCYP3A4) used in the current modeling represent 131 µl/min per mg and 0.29, respectively (Kajosaari et al., 2005a; Varma et al., 2013). The model with these initial (transport and metabolism) input parameters resulted in underprediction of hepatic clearance. Therefore, an empirical scaling factor (SFactive) for the hepatic sinusoidal active uptake (SFactive = 16.9), estimated by top-down model fitting to the intravenous data were applied, while keeping the rest of the input parameters same as that of the initial model (Jones et al., 2012; Varma et al., 2012b). The advanced dissolution, absorption, and metabolism model, or ADAM, was adopted to capture intestinal absorption and predict oral pharmacokinetics of repaglinide. The rifampicin model (input parameters, Table 1) was directly adopted from the Simcyp compound library (Varma et al., 2012b). Rifampicin interaction parameters against CYP3A4, CYP2C8, and OATP1B1 were generated in-house or extracted from literature. Each simulation was performed for 50 subjects (5 trials × 10 subjects). The virtual populations of healthy subjects had a body weight of 70 kg, ranged in age from 18 to 65 years, and included both sexes. Dose, dosing interval, and dosing duration of repaglinide and rifampicin were identical to that reported in individual clinical studies.
Summary of input parameters for repaglinide dynamic and static mechanistic models
Static Mechanistic “Extended Net-Effect” Model.
The area under the plasma concentration-time curve ratio (AUCR) of repaglinide in the presence (AUC′po) and absence (AUCpo) of rifampicin treatment can be described as in eq. 1 (Fahmi et al., 2008).
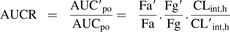 (1)
(1)Fa′ and Fa represent the fraction of drug absorbed from the intestine, and Fg′ and Fg represent the fraction of drug escaping gut-wall metabolism in the presence and the absence of rifampicin, respectively. Repaglinide is a highly permeable drug with almost complete absorption (>95%) (Hatorp et al., 1998; Varma et al., 2010), therefore the Fa′/Fa term was assumed to be 1 (see Discussion). CLint,h and CL′int,h represent the intrinsic hepatic clearance in the absence and presence of the rifampicin, respectively. Due to the primary involvement of active uptake and P450-mediated metabolism in the hepatic disposition of repaglinide (Varma et al., 2013), its overall intrinsic hepatic clearance can be mathematically defined by extended clearance concept, as in eq. 2 (Liu and Pang, 2005; Shitara et al., 2006; Shitara and Sugiyama, 2006; Camenisch and Umehara, 2012; Barton et al., 2013). (2) where PSuptake and PSefflux are the uptake and efflux intrinsic clearances across the sinusoidal membrane. PSinflux,active, PSefflux,active and PSpd are sinusoidal active uptake, active efflux, and passive diffusion intrinsic clearances, respectively. CLint,CYP3A4 and CLint,CYP2C8 are metabolic intrinsic clearances.
(2) where PSuptake and PSefflux are the uptake and efflux intrinsic clearances across the sinusoidal membrane. PSinflux,active, PSefflux,active and PSpd are sinusoidal active uptake, active efflux, and passive diffusion intrinsic clearances, respectively. CLint,CYP3A4 and CLint,CYP2C8 are metabolic intrinsic clearances.
Assuming active efflux across sinusoidal membrane (PSefflux,active) is negligible, eq. 2 can be rewritten as:
 (3)
(3)Similar to that used in the PBPK model, SFactive represents empirical scaling factor for active uptake estimated by matching the in vitro CLint,h to the in vivo CLint,h obtained from intravenous pharmacokinetics of repaglinide (eq. 9) (Table 1). The in vitro intrinsic values were scaled assuming the following: 118 × 106 hepatocytes g−1 liver, 39.8 mg microsomal protein g−1 liver, 24.5 g liver kg−1 body weight (mean values used in the healthy volunteer population file of Simcyp version 11.0).
In the presence of rifampicin, the expected net effect of competitive inhibition of CYP3A4 and CYP2C8, induction of CYP3A4 and competitive inhibition of active uptake (OATP1B1) can be illustrated by eq. 4.
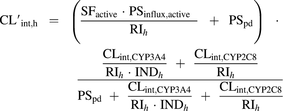 (4)
(4)RIh is the competitive inhibition term, and INDh is the hepatic CYP3A4 induction term (eq. 5) (Fahmi et al., 2008; Giacomini et al., 2010; Barton et al., 2013).
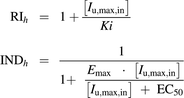 (5)
(5)Ki is the inhibition constant, and Iu,max,in is the maximum unbound rifampicin concentration at the inlet to liver after repaglinide dosing. Rifampicin fraction unbound in the in vitro incubations was assumed to be one. The values were taken from portal vein concentration-time profile predicted using PBPK (Simcyp) model (Varma et al., 2012b). Emax represents the maximum-fold induction, and EC50 is the concentration of inducer associated with half-maximum induction.
Assuming the gut metabolism of repaglinide is determined by only CYP3A4 (CYP2C8 expression in the gut is negligible) (Paine et al., 2006), the change of the fraction of drug escaping intestinal extraction in the presence of perpetrator can be defined by eq. 6 (Fahmi et al., 2008).
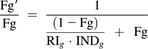 (6)
(6)RIg and INDg are the reversible inhibition and induction terms for CYP3A4-mediated gut metabolism (eq. 7).
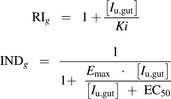 (7)
(7)Iu,gut, the free intestinal rifampicin concentration, was estimated by eq. 8.
 (8)
(8)Dose, Ka, fa, fu,gut and Qgut [248 ml/min (Fahmi et al., 2008)] represent total dose given orally, absorption rate constant, fraction absorbed, fraction unbound in the gut, and enterocytic blood flow, respectively. Fg of repaglinide was estimated to be 0.94 based on the current PBPK (Simcyp) model. Also, to maintain consistency between the model predictions, all the parameters used for static modeling are the same as used for PBPK modeling (Table 1).
In vivo CLint,h was calculated using the well-stirred liver model (Pang and Rowland, 1977).
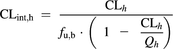 (9)
(9)CLh is the hepatic blood clearance obtained from intravenous total plasma clearance corrected for Rb. The fu,b represents the fraction unbound in blood, and Qh represents the average hepatic blood flow of 20.7 ml/min/kg (Kato et al., 2003).
Model Predictability.
The model-predicted AUC ratios were compared with the observed values using percentage prediction error (PPE), shown in eq. 10. Prediction bias and precision were also assessed with root mean square error (RMSE), shown in eq. 11, and average-fold error (AFE), shown in eq. 12.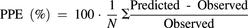 (10)
(10) (11)
(11) (12)N is the number of observations.
(12)N is the number of observations.
Results
Dynamic (PBPK) Model Predictions.
A whole-body PBPK model, assuming permeability-limited hepatic disposition, was used to assess repaglinide DDIs with rifampicin. The PBPK model adequately described repaglinide plasma concentration-time profile after a single intravenous and an oral dose (Fig. 1A). The simulated mean plasma concentration–time profile of repaglinide following rifampicin treatment is in good agreement with the observed data, where repaglinide was dosed 12.5 hours after the last dose of rifampicin (Fig. 1B). Furthermore, the magnitude of repaglinide-rifampicin interactions with concomitant or time-separated dosing are well predicted; the model predicted least and largest changes in exposure when repaglinide was administered 1 hour and 24 hours after the last dose of rifampicin, respectively (Fig. 2). For all the DDI predictions, rifampicin was considered to induce CYP3A4 activity (EC50 – 0.228 µM; Emax – 49.2), as well as reversibly inhibit OATP1B1 (Ki – 0.93 µM), CYP3A4 (Ki – 18.5 µM) and CYP2C8 (Ki – 30.2 µM) (Table 1).
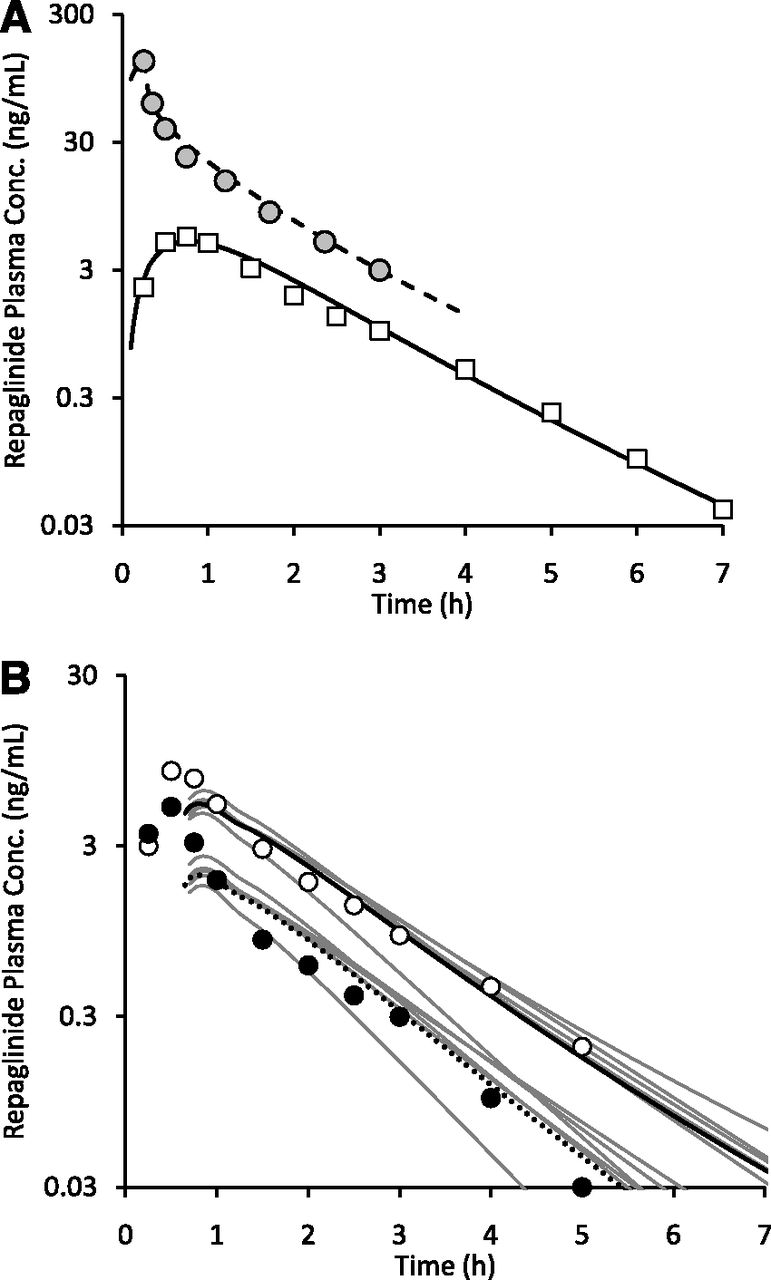
PBPK model predictions of repaglinide pharmacokinetics and drug–drug interactions with rifampicin. (A) Observed and simulated mean plasma concentration (conc.)-time profiles of repaglinide following single 2-mg intravenous infusion (circles and dashed line) and 0.5-mg oral dose (squares and solid line) (Hatorp et al., 1998; Skerjanec et al., 2010). (B) Mean plasma concentration-time profiles of single 0.5-mg oral repaglinide dosed 12.5 hours after the last dose of once-daily 600 mg rifampicin is shown. Open (solid line) and closed (dotted line) data points represent mean observed (simulated) values in the control and rifampicin treatment groups, respectively (Niemi et al., 2000). Gray lines represent plasma concentration-time profiles in individual trials (5 trials × 10 subjects).
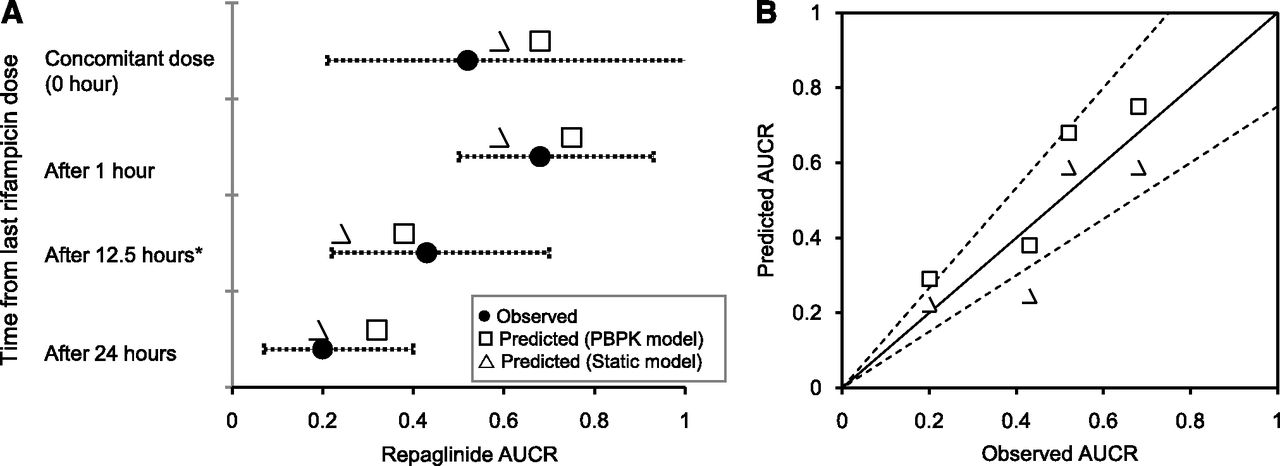
(A) Dynamic and static model predicted AUCRs of repaglinide dosed at different time after the last dose of rifampicin. (B) Observed verses predicted AUCRs. Observed mean AUCRs were taken from separate clinical studies (Niemi et al., 2000; Hatorp et al., 2003; Bidstrup et al., 2004). For dynamic model simulations, dosage regimen of repaglinide and rifampicin is similar to the original reported study design: *5-day rifampicin treatment of these data points – all other points involves 7-day rifampicin treatment. Error bars represent range or 90% confidence interval of the observed AUCRs. Dashed lines represent ± 25% deviation from unity (solid line).
Static Mechanistic Model Predictions.
A static mechanistic model was devised based on the extended clearance concept to predict the change in overall hepatic clearance. The predictions based on static model are in good agreement with the observed values, as well as with the AUCRs predicted by the PBPK model (Fig. 2). In general, the static model predicted AUCR within 25% of the observed values (Table 2). Interestingly, sensitivity analysis of fmCYP3A4 input with a fixed CLint,met (131 µl/min per mg) showed no significant effect on the AUCRs (Fig. 3).
Summary of the dynamic and the static mechanistic model–based predictions of repaglinide-rifampicin interactions

Effect of hepatic fraction metabolism (fmCYP3A4) on the static model-based predictions of AUCRs of repaglinide dosed at different time after the last dose of rifampicin.
Individual Components of the Interaction.
Figure 4 shows the predicted changes in the individual components of CYP3A4 induction, OATP1B1 inhibition, and P450 inhibition, as well as the net effect of rifampicin on repaglinide pharmacokinetics. Both the dynamic and static models suggested that only OATP1B1 inhibition leads to increase in repaglinide plasma exposure by ∼2- to 3-fold, whereas the CYP3A4 induction effect resulted in an AUCR of ∼0.2- to 0.3-fold. Models predict that rifampicin show competitive inhibitory effect on OATP1B1 up to ∼12 hours post dose, resulting in partial masking of CYP3A4 induction effect when both drugs are administered in temporal proximity. In contrary, rifampicin has a negligible inhibitory effect on P450-mediated metabolism, even with concomitant dosing. In general, the CYP3A4 induction and OATP1B1 inhibition effects of rifampicin predicted by the static mechanistic model are larger than that noted with dynamic model, presumably due to the use of maximum inlet concentration. Nevertheless, the net effect of the multiple interaction components resulted in prediction of DDIs similar to those observed in the clinic.

Simulated contribution of individual components of CYP3A4 induction, OATP1B1 inhibition and P450s inhibition to the net-effect of the predicted repaglinide-rifampicin DDIs by (A) dynamic or (B) static models. Data points represent observed mean AUC ratios.
Based on PBPK modeling, rifampicin treatment resulted in a gradual increase in CYP3A4 activity in the intestine and liver by ∼25-fold and ∼15-fold, respectively (Fig. 5A). The induction effect of rifampicin was higher in the intestine, presumably due to higher exposure in the gut. Following rifampicin treatment, the fraction extracted in the gut increased from ∼6% of the dose to ∼55%, implicating that the major site of repaglinide elimination shifts from liver in the control group to the gut in the rifampicin treatment group (Fig. 5B). Interestingly, increased CYP3A4 activity in liver has a minimal effect on the hepatic clearance of repaglinide. For example, CLint,h was reduced by only ∼27%, whereas Fg was reduced by ∼74% compared with the control when repaglinide was dosed 24 hours after rifampicin treatment (Table 2). On the other hand, rifampicin showed up to a maximum of ∼60% OATP1B1 inhibition and showed no accumulation following multiple rifampicin once-daily doses (Fig. 5C). Finally, the dynamic modeling suggested that the induction effect returned to the baseline only after 4 days following the last dose of 7-day rifampicin treatment (Fig. 5A).

Effect of rifampicin treatment on the intrinsic metabolic and transport clearances of repaglinide. PBPK model simulations of the effect of rifampicin treatment on the (A) change in CYP3A4 metabolic activity in the intestine and liver, (B) fraction of repaglinide dose metabolized in intestine and liver, and (C) change in OATP1B1 activity. Arrows indicate time of single repaglinide dose following rifampicin treatment.
Discussion
This mechanistic evaluation demonstrated that differential effects of rifampicin on CYP3A4 and OATP1B1 causes varied degree of change in repaglinide systemic exposure, depending on the temporal separation of administration of two drugs. The pharmacokinetics of repaglinide was highly affected when repaglinide was administered 24 hours after the last dose of rifampicin pretreatment (Bidstrup et al., 2004). However, repaglinide ingested concomitantly or 1 hour after the last dose of rifampicin showed only low-to-moderate reduction in repaglinide systemic exposure (AUCR ∼0.52–0.68) (Hatorp et al., 2003; Bidstrup et al., 2004). Clearly, these different clinical observations can be accurately predicted using the PBPK model and the proposed static mechanistic “extended net-effect” model.
The systemic clearance of repaglinide estimated using in vitro enzyme kinetics considerably underpredicted in vivo clearance, suggesting a key role of hepatic uptake in its disposition (Hatorp et al., 1998; Kajosaari et al., 2005a). Our previous in vitro transport studies and mechanistic modeling indicated that the systemic clearance of repaglinide is determined mainly by the hepatic uptake process (Varma et al., 2013). Here, we developed PBPK and static mechanistic models for repaglinide incorporating the hepatic transport- and enzyme-mediated disposition processes, based on the in vitro input parameters. Both the models directly using the in vitro transport parameters; however, underpredicted repaglinide systemic clearance, presumably due to discrepancy in the in vitro–in vivo extrapolation of transporter-mediated uptake activity. There are many possible reasons for the potential in vitro–in vivo discrepancy in the hepatic transporter activity, including down-regulation of transporter protein and partial loss of functional activity in the in vitro system (Jones et al., 2012; Varma et al., 2012b, 2013). Therefore, an empirical scaling factor for PSactive, estimated based on “top-down” fitting to human intravenous plasma concentration-time profiles, was incorporated in the PBPK model (Fig. 1) (Watanabe et al., 2009; Jones et al., 2012; Varma et al., 2012b, 2013). Similarly, scaling factor for active uptake was applied to the static mechanistic model to match the in vitro hepatic clearance to that observed in vivo. The scaling factors estimated for both the models were similar (Table 1). Overall, the whole-body PBPK and the static mechanistic models, assuming permeability-limited hepatic disposition, adequately described the pharmacokinetics of repaglinide.
Rifampicin is a potent inducer of P450 and also shows in vitro inhibition of CYP3A4 and CYP2C8. The net effect of significant inductive as well as inhibitory effects of rifampicin on P450-mediated metabolism was thought to be responsible for the observed dosing-time dependent repaglinide-rifampicin DDIs (Niemi et al., 2000; Bidstrup et al., 2004). However, the current assessment using in vitro Ki of CYP3A4 (18.5 µM) and CYP2C8 (30.2 µM) demonstrated that the acute inhibition of metabolism had only a minimal role (Fig. 4). In contrary, reduction in hepatic uptake via inhibition of OATP1B1 caused increase in AUC, which, when combined with the induction potential of rifampicin, yielded a good prediction of AUCRs. Interestingly, change in the overall hepatic CLint due to only CYP3A4 induction following rifampicin treatment was minimal compared with the reduction noted with Fg (Table 2). Presumably, hepatic uptake being the rate-determining step in the systemic clearance of repaglinide, changes in only metabolic activity (induction or inhibition) do not significantly alter the overall hepatic clearance (Maeda et al., 2011; Varma et al., 2013). Moreover, a sensitivity analysis suggested no significant effect of fmCYP3A4 on the predicted AUCR (Fig. 3). Collectively, the mechanistic modeling suggests that the net-effect of increased CYP3A4 activity (mainly in gut) and decreased hepatic uptake determine the magnitude of repaglinide-rifampicin interactions.
Additional rifampicin-mediated induction of efflux transporters, such as P-glycoprotein (P-gp), might also play a role in decreasing the repaglinide exposure (Westphal et al., 2000). Furthermore, differential acute inhibition of intestinal efflux and chronic induction of P450s or P-gp by rifampicin could explain dosing-time dependent differences in repaglinide exposure. Rifampicin is a moderate P-gp inhibitor (IC50 – 169 μM) in vitro (Reitman et al., 2011). Also, oral exposure of digoxin was shown to increase by rifampicin co-dosing, presumably due to intestinal P-gp inhibition (Reitman et al., 2011). On the basis of our assessment, repaglinide is categorized as a biopharmaceutics class I drug (Wu and Benet, 2005; Varma et al., 2012a) exhibiting high permeability (Caco-2 permeability ∼26 × 10−6 cm/s), moderate solubility (68 µg/ml) (Mandic and Gabelica, 2006), low dosing (<4 mg), and complete absorption (>95%). Thus, while repaglinide has been shown to be a P-gp substrate with high asymmetric transport across MDCK-MDR1 cells (Korzekwa et al., 2012), P-gp is expected to have a limited role in determining the extent of repaglinide absorption (Varma et al., 2005). On the other hand, the full inductive effect is believed to be attained in about 7 days after once-daily rifampicin treatment (Niemi et al., 2003a). The PBPK model simulation of CYP3A4 activity is consistent with this, further suggesting that the average metabolic activity starts declining after 24 hours of the last dose of rifampicin (Fig. 5A). Therefore, it is unlikely that the dosing time–dependent interactions observed (low when concomitant dosing versus high interaction when dosed 24 hours after rifampicin) are due to further enhanced induction.
Although to a lesser extent relative to CYP3A4 induction, rifampicin also induces CYP2C8, which plays a key role in repaglinide elimination (Niemi et al., 2003a). It is unclear from the existing data if CYP2C8 was induced by rifampicin and to what extent. However, such information can be derived if the plasma profiles of repaglinide–circulating metabolites are available. Formation of metabolites M1 and M4 is dependent on the CYP3A4 and CYP2C8, respectively (Sall et al., 2012). Gemfibrozil, a potent mechanism-based inactivator of CYP2C8, drastically decreased plasma circulating M4 levels while significantly increasing M1 concentrations (Kajosaari et al., 2005b; Tornio et al., 2008; Honkalammi et al., 2011b,a, 2012). Similar differential metabolite pharmacokinetics may delineate the CYP3A4 versus CYP2C8 role in the observed induction effect. Nevertheless, we postulate that CYP2C8 induction only has a minimal effect on repaglinide pharmacokinetics, because (i) repaglinide systemic clearance is majorly determined by the hepatic uptake clearance and an increase in hepatic metabolic activity is less likely to affect the systemic clearance (Table 2), and (ii) as noted with the simulations here, increase in the gut metabolism is the major driver for observed decrease in repaglinide exposure (particularly when rifampicin and repaglinide doses were separated by >12 hours), and CYP2C8 contribution to the gut metabolism is believed to be relatively low (Paine et al., 2006). Nevertheless, although no clinical evidence exist, expression and in vitro data suggest that rifampicin significantly induces the intestinal CYP2C8 and CYP2C9 isoforms and may partially contribute to such interactions (Lapple et al., 2003; Glaeser et al., 2005).
Repaglinide lowers blood glucose levels by stimulating the release of insulin from the pancreas while interfering with the ATP-dependent potassium channels in the β-cell membrane. Clinical DDI studies often demonstrated relationship between plasma exposure and pharmacodynamic activity of repaglinide, with increased glucose-lowering activity by cyclosporine and gemfibrozil and decreased activity following rifampicin treatment (Niemi et al., 2003b; Honkalammi et al., 2012; Tornio et al., 2012). Therefore, an increase in repaglinide dose based on clinical response to therapy should be considered when it is coadministered with strong P450 inducers, such as rifampicin, and other therapeutic agents, such as carbamazepine, phenytoin, efavirenz, and St. John’s wort (Luo et al., 2002; Niemi et al., 2003a). Unlike rifampicin, many of these P450 inducers do not inhibit OATPs at the clinically relevant concentrations; therefore, the interactions are expected to be independent of dosing time in relative to repaglinide dose.
Involvement of similar dual effects of rifampicin has been hypothesized to explain DDIs with other P450 substrates. For example, a single intravenous dose of rifampicin increased the AUC of glyburide by ∼120%, presumably due to acute inhibition of hepatic uptake (Zheng et al., 2009). However, glyburide AUC reduced when concomitantly dosed with rifampicin following a chronic oral treatment; and a further reduction in AUC was noted when glyburide was dosed 2 days after the last dose of rifampicin. Atorvastatin-rifampicin interactions serve as another example, wherein a single intravenous dose of rifampicin increased atorvastatin AUC by ∼7-fold (Lau et al., 2007), whereas atorvastatin dosed 17 hours after a 5-day oral rifampicin treatment led to significant decrease in its systemic exposure (Backman et al., 2005). In a microdosing study, atorvastatin AUC was shown to be markedly affected by a single rifampicin dose administered simultaneously, but not by an intravenous dose of itraconazole, a potent CYP3A4 inhibitor; this finding suggests that the hepatic uptake is the rate-determining process in the hepatic clearance of atorvastatin (Maeda et al., 2011). Therefore, as demonstrated in the current study, the marked reduction in atorvastatin AUC by chronic rifampicin treatment noted in the clinic (Backman et al., 2005) could be explained by the CYP3A4 induction at the gut, with a minimal contribution of increased hepatic metabolic activity. As shown in the current study, similar dynamic and static mechanistic modeling utilizing in vitro transport and metabolic kinetics can be useful in delineating the quantitative function of individual components of these interactions.
For OATPs substrate drugs like repaglinide, where systemic clearance is determined by the hepatic uptake as well as by metabolism, mechanistic considerations assuming permeability-limited disposition are needed to accurately predict complex DDIs (Varma et al., 2013). As demonstrated in this study, the mechanistic modeling approaches using both the dynamic and static models are useful for assessing the DDI potential. Notably, when the input parameters remain the same, both models yielded similar results. The proposed static model has the advantage of being simple and more transparent and can be valuable for quantitative predictions of DDI scenarios in the drug development. Nevertheless, dynamic models can help consider extremes in population variability by incorporating variability in the in vivo drug disposition and polymorphic clearance pathways or by simulating drug disposition in disease states or special populations (Rowland et al., 2011).
In conclusion, the current PBPK and static mechanistic model-based analysis suggest that the dual effects of CYP3A4 induction and competitive inhibition of OATP1B1-mediated hepatic uptake are apparent when both drugs are administered in temporal proximity, whereas the CYP3A4 induction effect can be isolated if repaglinide and rifampicin doses are sufficiently separated in time (>12 hours after rifampicin dose). Finally, since uptake is rate-determining step in the hepatic disposition, increased CYP3A4 activity in the gut, but not the liver, majorly contributes to the increased metabolic clearance of repaglinide following rifampicin treatment.
Authorship Contributions
Participated in research design: Varma, Lin.
Conducted experiments: Varma, Lin, Bi, Rotter.
Contributed new reagents or analytic tools: Varma.
Performed data analysis: Varma, Fahmi, Lin, Lam, El-Kattan, Goosen, Lai.
Wrote or contributed to the writing of the manuscript: Varma, Lin, Bi, Rotter, Fahmi, Lam, El-Kattan, Goosen, Lai.
Footnotes
- Received December 7, 2012.
- Accepted February 7, 2013.
Abbreviations
- AUC
- area under the plasma concentration–time curve
- AUCR
- area under the plasma concentration–time curve ratio
- CLint
- intrinsic clearance
- DDI
- drug-drug interaction
- fm
- fraction metabolism
- OATP
- organic anion transporting polypeptide
- P450
- cytochrome P450
- PBPK
- physiologically-based phamacokinetic
- P-gp
- P-glycoprotein
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}