


Abstract
Pemetrexed, an anionic anticancer drug with a narrow therapeutic index, is eliminated mainly by active renal tubular secretion. The in vitro to in vivo extrapolation approach used in this work was developed to predict possible drug-drug interactions (DDIs) that may occur after coadministration of pemetrexed and nonsteroidal anti-inflammatory drugs (NSAIDs), and it included in vitro assays, risk assessment models, and physiologically based pharmacokinetic (PBPK) models. The pemetrexed transport and its inhibition parameters by several NSAIDs were quantified using HEK-PEAK cells expressing organic anion transporter (OAT) 3 or OAT4. The NSAIDs were ranked according to their DDI index, calculated as the ratio of their maximum unbound concentration in plasma over the concentration inhibiting 50% (IC50) of active pemetrexed transport. A PBPK model for ibuprofen, the NSAID with the highest DDI index, was built incorporating active renal secretion in Simcyp Simulator. The bottom-up model for pemetrexed underpredicted the clearance by 2-fold. The model we built using a scaling factor of 5.3 for the maximal uptake rate (Vmax) of OAT3, which estimated using plasma concentration profiles from patients given a 10-minute infusion of 500 mg/m2 of pemetrexed supplemented with folic acid and vitamin B12, recovered the clinical data adequately. The observed/predicted increases in Cmax and the area under the plasma-concentration time curve (AUC0–inf) of pemetrexed when ibuprofen was coadministered were 1.1 and 1.0, respectively. The coadministration of all other NSAIDs was predicted to have no significant impact on the AUC0–inf based on their DDI indexes. The PBPK model reasonably reproduced pemetrexed concentration time profiles in cancer patients and its interaction with ibuprofen.
Introduction
Pemetrexed supplemented with folic acid and vitamin B12 is used to treat non–small cell lung cancer and malignant mesothelioma, alone or in combination with cisplatin (Shih et al., 1997; Bajetta et al., 2003; Scagliotti et al., 2003; Latz et al., 2006; Takimoto et al., 2007). Pemetrexed is a hydrophilic anionic compound eliminated unchanged in the urine (70–90%) primarily by active tubular secretion, presumably by organic anion transporters (OATs) (Rinaldi, 1999; Kurata et al., 2014). The structurally related drug methotrexate, one of the first antifolates used for the treatment of cancer, is predominantly eliminated unchanged in urine, with 80% of the dose being secreted actively by the kidney. The administration of inhibitors of the OATs, such as probenecid and nonsteroidal anti-inflammatory drugs (NSAIDs), reduces the clearance of methotrexate. For example, Tracy et al. showed that naproxen, trisalicylate, and ibuprofen decreased the plasma clearance by 1.28-, 1.31-, and 1.66-fold, respectively (Cassano, 1989; Frenia and Long, 1992; Tracy et al., 1992). Additionally, the coadministration of high doses of methotrexate and ketoprofen was shown cause severe toxicities and even deaths (Thyss et al., 1986). Consequently the use of NSAIDs with high doses of methotrexate is not recommended.
Because patients undergoing chemotherapy often take nonsteroidal anti-inflammatory drugs (NSAIDS) for pain control and given that methotrexate and pemetrexed rely on renal transporters for their elimination, it was hypothesized that pemetrexed would experience the same drug interaction as methotrexate. Clinical studies demonstrated no interaction with aspirin, but a 20% increase in the area under the plasma-concentration time curve (AUC0–inf) of pemetrexed was observed when patients with normal renal function (creatinine clearance >80 ml/min) were cotreated with 400 mg of ibuprofen 4 times a day (Sweeney et al., 2006). Although it is statistically significant, this increase does not require a dose adjustment in patients with normal renal function (creatinine clearance >80 ml/min). However, because renal insufficiency increases pemetrexed exposure, caution should be used when administering NSAIDs concurrently with pemetrexed to patients with mild and moderate renal insufficiency (creatinine clearance from 45 to 79 ml/min) (Mita et al., 2006). Although not studied clinically, it is recommended that NSAIDs with short elimination half-life should be avoided for the period of 2 days before, the day of, and 2 days after administration of pemetrexed in patients with mild to moderate renal insufficiency. For NSAIDs with longer elimination half-lives, patient taking these NSAIDs should interrupt dosing for at least 5 days before, the day of, and 2 days after pemetrexed administration [Alimta (pemetrexed), Eli Lilly and Company, Indianapolis, IN; package insert available at http://labels.fda.gov/].
OATs are a group of membrane transporter proteins expressed in several epithelial tissues of the body, including the proximal tubular cells of the kidney (Erdman et al., 2006; Burckhardt, 2012). These transporters play an important role in renal elimination, by actively secreting a wide variety of endogenous and exogenous hydrophilic organic anions, including p-aminohippuric acid, prostaglandins, cephalosporins, antivirals, and anticancer drugs (Burckhardt, 2012). In the kidney, OAT1 (SLC22A6) and OAT3 (SLC22A8) are expressed on the basolateral membrane of the proximal tubular cells, where they import organic anions in an exchange of dicarboxylates such as succinate and α-ketoglutarate (Motohashi et al., 2002; Bakhiya et al., 2003; Sweet et al., 2003). OAT4 (SLC22A11) is expressed on the apical membrane of the apical tubular cells and functions as a bidirectional transporter, moving solutes from the cell into the tubular fluid and vice versa in an exchange of dicarboxylates, urate, and other organic anions (Motohashi et al., 2002; Hagos et al., 2007a; Hagos et al., 2007b). NSAIDs are a well characterized group of OAT inhibitors that are generally better inhibitors of OAT1 and OAT3 than OAT4. Although ibuprofen and diclofenac are effective inhibitors of OAT1 and OAT3, with concentration inhibiting 50% of the active transport (IC50) values in the lower micromolar range, acetaminophen, aspirin, and salicylate are low-affinity inhibitors (Khamdang et al., 2002).
Our objectives were to 1) identify the transporters responsible for the cellular transport of pemetrexed, 2) determine the kinetic parameters of pemetrexed transport, 3) quantify the inhibitory potency of a subset of NSAIDs toward these transporters, and finally 4) establish a mechanistic physiologically based pharmacokinetic (PBPK) model of pemetrexed and its interaction with ibuprofen that also could be applied to other potential inhibitors of pemetrexed renal secretion.
Materials and Methods
Chemicals.
Pemetrexed was synthesized at Eli Lilly and Company. Radiolabeled [14C]ibuprofen was purchased from American Radiochemicals (St. Louis, MO). Radiolabeled [3H]estrone-3-sulfate (E3S) was purchased from Perkin Elmer (Boston, MA). The internal standard ([13C515N]pemetrexed) and celecoxib were synthesized at Eli Lilly and Company. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Preliminary In Vitro Substrate Transporter Screen.
The preliminary uptake screen was performed according to a transient transporter expression method previously described elsewhere using HeLa cells transfected with cDNAs of human transporters OAT1 (SLC22A6), OAT2 (SLC22A7), OAT3 (SCL22A8), OAT4 (SLC22A9), OCT1 (SLC22A1), OCT2 (SLC22A2), OCTN1 (SLC22A4), OATP1A2 (SLCO1A2), OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), OATP2B1 (SLCO2B1), OATP4C1 (SLCO4C1), ASBT (SLC10A2), PEPT1 (SLC15A1), and PEPT2 (SLC15A2) (Cvetkovic et al., 1999). Drug uptake was evaluated using [14C]pemetrexed (60 μM) in medium for 10 minutes at 37°C. The reaction was terminated using ice-cold buffer, and the cells were lysed with 1% SDS. The samples were added to a vial containing Ultima Gold scintillation cocktail (Perkin Elmer), and the amount of pemetrexed in the cells was measured using a scintillation counter (Tri-Carb 2900TR; Perkin Elmer).
In Vitro Transporter Kinetic Studies.
HEK-PEAK cells were transfected with plasmids containing human OAT3 and OAT4 following previously published protocols (Godinot et al., 2003). In short, human OAT3 (NM_004254) and OAT4 (NM_080866) genes, purchased from GeneArt (Regensburg, Germany), were inserted into EW1969 plasmid before transfection of HEK-PEAK cells using an Effectene Transfection Reagent kit (Qiagen, Valencia, CA).
In vitro uptake experiments were performed based on reports published by Nozaki et al. (2004) with some modifications. HEK-PEAK cells from passages 36–75 were used 72 hours after seeding on CellBIND 12-well plates (Corning Life Sciences, Tewksbury, MA) at a density of 150,000 cells/well. The reactions were performed in uptake buffer consisting of Krebs‑Henseleit buffer at pH 7.4 supplemented with 2.5 mM CaCl2 and 10 mM HEPES. Protein quantification was performed using a BCA protein assay (Sigma-Aldrich) where bovine serum albumin was used as the standard.
OAT3 and OAT4 qualification studies were performed to assess the functionality of the HEK-PEAK transfected cells. The cells were treated with 0.5 ml of buffer containing 1 μM E3S for 2 minutes in the presence or absence of 200 μM probenecid, a known inhibitor of the OATs, at room temperature. The incubations were stopped by the addition of ice-cold buffer, followed by two washes with buffer. Cells were lysed in 0.3 ml of 0.2 N NaOH overnight at 4°C. A 0.2-ml aliquot was added to a vial containing scintillation cocktail (ScintSafe 30%; Thermo Fisher Scientific, Waltham, MA), and the cellular uptake of E3S was measured using a scintillation counter. Protein quantification was performed as mentioned previously with the remaining sample.
All pemetrexed uptake and inhibition studies were performed at room temperature with 0.5 ml of pemetrexed in Krebs‑Henseleit buffer. Before the beginning of the experiment, the vector control, OAT3 cells, and OAT4 cells were washed twice with buffer and allowed to equilibrate for 5 minutes. Time-dependent uptake studies were performed with 39 μM pemetrexed and stopped at the specified times up to 15 minutes. Concentration-dependent uptake studies were performed for 5 minutes with pemetrexed concentrations ranging from 0.01 to 117 μM. All reactions were stopped with the addition of cold buffer, and the cells were lysed in a 1:1 methanol/water (v/v) for 3 minutes. The concentration of pemetrexed was quantified using liquid chromatography with tandem mass spectrometry (LC-MS/MS).
In Vitro Transporter Inhibition Studies.
The inhibitory effects of individual NSAIDs and probenecid on pemetrexed uptake were evaluated using vector control and OAT3 and OAT4 transfected cells incubated at room temperature with 0.5 ml of pemetrexed (19.5 μM) in uptake buffer. Ibuprofen, diclofenac, naproxen, and celecoxib were added to the cells in concentrations ranging from 0.1 to 200 μM. The incubations were stopped at 5 minutes; the cells were rinsed twice with ice-cold buffer, and they were treated with 0.3 ml of 1:1 methanol/water (volume/volume) to extract the pemetrexed for LC-MS/MS quantification.
Fraction Unbound in Incubation.
The fraction unbound of ibuprofen in the incubations (fu,inc) was measured in vector control cells using [14C]ibuprofen. The conditions for this experiment were the same as those described earlier for the transport experiments. Briefly, the HEK-PEAK vector control cells were washed twice with buffer and incubated at room temperature for 5 minutes with 0.5 ml of 10 μM ibuprofen in buffer. Samples of the incubation media (200 μl) were collected at 0 and 5 minutes, placed in a vial containing 10 ml of scintillation cocktail, and quantified using a scintillation counter. The fraction of pemetrexed unbound in the incubation was assumed to be 1.
Analytic Method.
The concentration of pemetrexed in the cells was quantified by LC-MS/MS using [13C515N]pemetrexed as an internal standard. Reverse-phase chromatography was used with a flow rate of 0.6 ml/min and an injection volume of 20 μl. The analytes were chromatographically separated using a Synergi MAX-RP 80A 5μ 100 × 2.0 mm high-pressure liquid chromatography column (Phenomenex, Torrance, CA), with a gradient liquid chromatography system composed of 0.2% formic acid in water (mobile phase A) and 0.2% formic acid in methanol (mobile phase B). The gradient profile was 10% B from 0 to 0.30 minutes, 70% B at 0.70 to 2.00 minutes, and returned to 10% B at 2.20 minutes. The flow rate was 0.6 ml/min, and the analysis was performed at ambient laboratory conditions with the effluent directed to the mass spectrometer between 0.10 and 2.40 minutes.
Pemetrexed was detected in positive ion mode using a Sciex API 4000 triple quadruple mass spectrometer equipped with a Turbo Ion Spray interface and Analyst 1.4 software (Applied Biosystems/MDS, Foster City, CA). The mass/charge ratio for pemetrexed and [13C515N]pemetrexed was 426 → 297 m/z and 432 → 297 m/z, respectively. The collision energies were -30 and -36 for pemetrexed and [13C515N]pemetrexed, respectively. The dynamic range of the assay was between 0.77 and 395 ng/ml, and the samples with pemetrexed concentrations above the upper limit of quantification were diluted with buffer to concentrations within the assay range.
Three individual batches of quality control samples at four separate concentrations (0.98, 3.9, 125, and 500 ng/ml) were examined with four to six samples tested at each concentration. The results for 0.98, 3.9, 125, and 500 ng/ml samples showed an interbatch accuracy of 4.3, 3.3, 4.7, and 7.7%, respectively, and an interbatch precision of 11.1, 8.3, 7.9, and 8.4%, respectively.
Data Analysis.
The Michaelis-Menten kinetic parameters for pemetrexed transport were estimated by fitting the pemetrexed uptake data using nonlinear regression (WinNonlin v5.3; Pharsight, Sunnyvale, CA), according to the following formula: (1)where, v is the initial uptake (pmol/min per mg of protein), Vmax is the maximal uptake rate (pmol/min per mg of protein), S is the concentration of substrate (μM), Km (μM) is the Michaelis-Menten kinetic constant, and Kd (μl/min per mg of protein) is the passive permeability constant. The Kd for pemetrexed was calculated experimentally by fitting the uptake velocity of pemetrexed in the HEK-PEAK vector control cells. The passive intrinsic clearance (CLint,passive) of pemetrexed used in the model was calculated by multiplying the Kd by the number of cells in 1 milligram of protein.
(1)where, v is the initial uptake (pmol/min per mg of protein), Vmax is the maximal uptake rate (pmol/min per mg of protein), S is the concentration of substrate (μM), Km (μM) is the Michaelis-Menten kinetic constant, and Kd (μl/min per mg of protein) is the passive permeability constant. The Kd for pemetrexed was calculated experimentally by fitting the uptake velocity of pemetrexed in the HEK-PEAK vector control cells. The passive intrinsic clearance (CLint,passive) of pemetrexed used in the model was calculated by multiplying the Kd by the number of cells in 1 milligram of protein.
The inhibition constant (Ki) of the NSAIDs was calculated according to the following formula, assuming competitive inhibition: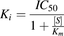 (2)where IC50 is the concentration of NSAIDs necessary to inhibit 50% of the uptake of pemetrexed, [S] is the concentration of pemetrexed in the medium, and Km is the affinity of pemetrexed for the particular organic anion transporter. The IC50 for pemetrexed was calculated with the following formula:
(2)where IC50 is the concentration of NSAIDs necessary to inhibit 50% of the uptake of pemetrexed, [S] is the concentration of pemetrexed in the medium, and Km is the affinity of pemetrexed for the particular organic anion transporter. The IC50 for pemetrexed was calculated with the following formula: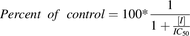 (3)where [I] is the concentration of the NSAID, and percent of control is the percentage of pemetrexed taken up by the cells in the presence of the inhibitor compared with the cells treated with only pemetrexed.
(3)where [I] is the concentration of the NSAID, and percent of control is the percentage of pemetrexed taken up by the cells in the presence of the inhibitor compared with the cells treated with only pemetrexed.
Statistical Analysis.
The in vitro experimental data are presented as mean ± S.E. or mean (% coefficient of variation). To test for significant differences between multiple groups, a one-way analysis of variance was used followed by either Dunnett’s or Bonferroni’s test. P ≤ 0.05 was considered statistically significant. The software used was GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Modeling Strategy.
The modeling strategy employed in this project was the following. 1) The active secretion of pemetrexed was defined to be via OAT3 and OAT4 on the basolateral and apical membrane of the proximal tubular cells, respectively. 2) A pemetrexed PBPK model was developed using in vitro and in silico data. 3) The model was optimized by using pemetrexed plasma concentration-time profiles from cancer patients. 4) The model was tested by changing the pemetrexed dose from 50 to 700 mg/m2, and the results were compared with other clinical studies. 5) A PBPK model for ibuprofen was developed using literature data and measured Ki and fu,inc. The model was verified using the results of the clinical interaction studies (Sweeney et al., 2006). This modeling strategy is described in detail herein.
Pemetrexed Clinical Data.
Human plasma concentration-time profiles after the administration of a 10-minute infusion of 500 mg/m2 of pemetrexed with vitamin B12 and folic acid from a published study (Sweeney et al., 2006) were used to build the model. In this study, serial blood samples were obtained predose, immediately before the end of infusion, and at 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 hours after termination of the infusion. Results from published studies were used to test and verify the model (Latz et al., 2006; Mita et al., 2006; Nakagawa et al., 2006; Sweeney et al., 2006).
Pemetrexed PBPK Model.
All simulations were performed using PKPD Profiles in the Simcyp Simulator (version 12.1; Simcyp Limited, Sheffield, United Kingdom). A full PBPK model for pemetrexed was built using measured and predicted physicochemical and biologic data (Table 1) based on the Rodgers et al. method with rapid equilibrium between blood and tissues except for the kidney, which was set up as a permeability-limited tissue (Rodgers et al., 2005; Rodgers and Rowland, 2006). The multicompartment kidney model built into Simcyp was used to incorporate the glomerular filtration and active renal secretion of pemetrexed. This mechanistic kidney model consists of three major compartments: blood, renal cells, and tubular fluid, all subdivided into several regions to approximate the anatomy and physiology of the kidney.
Physicochemical and biochemical parameters used in the pemetrexed PBPK model
Two active transport processes were enabled in the mechanistic kidney model, one on the basolateral membrane of the proximal tubular cells to simulate OAT3-mediated uptake of pemetrexed, and a second on the apical membrane of the proximal tubular cells to simulate OAT4-mediated efflux of pemetrexed into the renal tubules. The values for the passive and active membrane transport of pemetrexed were assumed to be the equal in all three compartments of the proximal tubular cells and were taken from the in vitro experiments previously described.
All other parameters used in the mechanistic kidney model were default Simcyp Simulator values with no modification unless specifically stated. The pemetrexed model assumptions were the following: 1) pemetrexed was not metabolized, and the only site of elimination was the kidney; 2) the passive permeability of pemetrexed in the vector control cell was representative of the passive permeability of pemetrexed in the kidney cells; 3) OAT3 is homogeneously expressed in the basolateral membrane of the three segments of the proximal tubular cells, and it only transports pemetrexed in one direction, from the blood into the cells; 4) the efflux of pemetrexed from the proximal tubular cells is governed by only one transporter, OAT4, which is expressed uniformly on the apical membrane of the three segments of proximal tubular cells and transports pemetrexed only in one direction, from inside the cells out to into the tubular fluid compartment.
Ibuprofen PBPK Model.
A full PBPK model was built for ibuprofen in the Simcyp Simulator (version 12.1) using the Rodgers and Rowland (2006) method with rapid equilibrium between plasma and tissues. The parameters used in the PBPK model for ibuprofen can be found in Table 2. The dosing regimen of ibuprofen consisted of nine doses of 400 mg, taken every 6 hours for 2 days before the administration of pemetrexed, and a tenth dose of ibuprofen taken 1 hour before pemetrexed infusion (Sweeney et al., 2006). The model was based on the physicochemical properties and pharmacokinetic parameters of ibuprofen collected from published data (Yee, 1997; Davies, 1998; Chen et al., 2012). The absorption of ibuprofen was modeled using the advanced absorption and dissolution model in the Simcyp Simulator, where the human effective jejunal permeability (Peff) was predicted from Caco-2 permeability values (Yee, 1997). The intrinsic solubility of ibuprofen was 0.06 mg/ml, and the model assumed an immediate release formulation (Shaw et al., 2005).
Ibuprofen physicochemical and biochemical parameters used for the PBPK model
Virtual Population.
The virtual population used in the simulations was built to replicate the cancer population in pemetrexed published clinical studies (Sweeney et al., 2006) (Table 3). The population was based on the north European Caucasian with an average body weight of 76.8 kg, age ranging from 34 to 80 years old, and 30% of the subjects were female. The relative abundances and corresponding coefficient of variation (60%) of the transporters in the three segments of the proximal tubules were the same. No genetic polymorphisms were considered.
Characteristics of the population used in the simulation
Parameter Sensitivity Analysis and Optimization.
A parameter sensitivity analysis was performed to evaluate the effect of model parameter values on the AUC0–inf and Cmax of pemetrexed. The first analysis was run for those parameters with high uncertainty or variability, such as the difference in expression or activity of the transporters in vitro versus in vivo. The model was optimized according to the results of the sensitivity analysis using the plasma concentration-time profiles of 25 patients who were administered 500 mg/m2 of pemetrexed supplemented with folic acid and vitamin B12. The parameter estimation was performed using the hybrid minimization method in the Simcyp Simulator and weighted least squares as the objective function.
Model Verification.
The PBPK model predictions for AUC0–inf and Cmax over a range of pemetrexed doses from 50 to 600 mg/m2 were compared with the observed ones in published clinical studies (Latz et al., 2006; Mita et al., 2006; Nakagawa et al., 2006; Sweeney et al., 2006). Predicted AUC0–inf was calculated by dividing the dose by the predicted clearance. The observed pharmacokinetic parameters were calculated using noncompartmental analysis, according to the published clinical studies.
Drug-Drug Interaction Predictions.
The interaction between ibuprofen and pemetrexed occurred at both OAT3 and OAT4. The Ki and fu,inc values were obtained from in vitro experiments (discussed earlier) and are listed in Table 2. To evaluate the effect of the interaction between pemetrexed and ibuprofen, we calculated and compared the observed and predicted AUC0–inf and Cmax ratios.
The AUC0–inf ratio is defined as the pemetrexed AUC0–inf when pemetrexed is coadministered with ibuprofen, over the AUC0–inf of pemetrexed when pemetrexed is administered alone. Likewise, the Cmax ratio is defined as the maximum concentration of pemetrexed when ibuprofen is coadministered over the pemetrexed Cmax the when pemetrexed is administered alone.
For the interaction model, the assumptions were the following. 1) The interaction occurs via OAT3 and OAT4 in the proximal tubular cells of the kidney. 2) Ibuprofen exerts its inhibition in a competitive manner for both OAT3 and OAT4. 3) Both the S and R enantiomers of ibuprofen have the same affinity for OAT transporters. 5) Ibuprofen metabolites do not interact with OAT3 and OAT4.
Results
Preliminary In Vitro Screen.
[14C]Pemetrexed uptake was significantly higher in OAT3 and OAT4 transfected HeLa cells compared with the control (Fig. 1). The uptake of [14C]pemetrexed by all other transporters tested was not significantly different than the control. Based on this, all subsequent in vitro uptake and inhibition studies were performed in OAT3, OAT4, and vector control transfected cells.

Results of the in vitro screening assay. Expression of drug uptake transporters was performed using a recombinant vaccinia system in HeLa cells. Uptake of [14C]pemetrexed, 60 µM (n = 3–6), at the 10-minute time point. The relative uptake was compared with HeLa cells transfected with a vector lacking transporter cDNA (vector control). Data are presented as mean ± S.D. (n = 3). ***P < 0.001.
In Vitro Transporter Uptake and Inhibition Studies.
The results of the qualification experiments, shown in Fig. 2A, demonstrated that the transfected HEK-PEAK hOAT3 and hOAT4 were functional, and the uptake of E3S, a known substrate for these transporters, is at least 100-fold higher than the vector control cells. When the OAT3 and OAT4 transfected cells were cotreated with probenecid, the uptake of E3S via OAT3 and OAT4 was substantially reduced by 97 and 74%, respectively.

In vitro uptake studies. (A) The uptake of [3H]estrone-3-sulfate (E3S) with or without probenecid, into vector control (VC), OAT3, and OAT4-transfected HEK-PEAK cells. The cell cultures were treated with 1 μM E3S, alone (□), or in combination with 200 μM probenecid (▪) for 2 minutes at room temperature. Data are presented as mean ± S.D. (n = 3). (B) Time-dependent uptake of pemetrexed (39 μM) in VC (▴), OAT3 (▪), and OAT4 (●) transfected HEK-PEAK cells. Experiments were performed at room temperature. Data are presented as mean ± S.D. (n = 3). (C) Uptake of pemetrexed into OAT3-transfected HEK-PEAK cells treated with increasing concentrations of pemetrexed for 5 minutes. (▪) Mean ± S.D. of the uptake in HEK OAT3; (▴) mean ± S.D. of the uptake in vector control cells. (D) Uptake of pemetrexed into OAT4-transfected HEK-PEAK cells treated with increasing concentrations of pemetrexed for 5 minutes; (▪) mean ± S.D. of the uptake in HEK OAT4; (▴) mean ± S.D. of the uptake in vector control cells.
To probe the active and passive uptake of pemetrexed, kinetic studies were performed in HEK-PEAK vector control, OAT3, and OAT4 cells over a concentration range of 1.6 and 117 μM of pemetrexed. As seen in Fig. 2B, the uptake of pemetrexed was linear up to 10 minutes in the conditions tested. Figure 2C shows saturable OAT3-mediated uptake of pemetrexed with a Vmax and Km of 185 pmol/mg per million cells and 18.2 μM, respectively. The passive intrinsic clearance of pemetrexed was estimated to be 3.98 × 10−3 μl/min per million cells. The uptake of pemetrexed in OAT4-transfected cells did not reach saturation in the conditions tested, so the Vmax and Km values could not be calculated (Fig. 2D). As a result, the intrinsic uptake clearance of OAT4 (CLint,T) was calculated by dividing the initial uptake of pemetrexed by its corresponding concentration, and was estimated to be 0.99 μl/min per million cells.
The inhibition of pemetrexed uptake by several NSAIDs was studied over a concentration range of 0.1 to 200 μM for the NSAIDs and 19.5 μM for pemetrexed. Table 4 shows the IC50 values for OAT3 and OAT4 for all the tested inhibitors. Table 5 shows the reported maximum plasma concentration of the tested inhibitors, fraction unbound in plasma (fu), and calculated [Iu]/IC50 values for the NSAIDs (Goodman and Gilman, 1941). The U.S. Food and Drug Administration’s 2012 draft guideline on drug-drug interactions (DDIs) (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf) concludes that an [Iu]/IC50 value higher than 0.1 indicates a high risk of clinical drug interaction and low potential for false negative results. Ibuprofen was the only tested NSAID with an [Iu]/IC50 value above 0.1, so PBPK models were not built for the other NSAIDs.
IC50 values of the inhibition of pemetrexed OAT-mediated uptake by different NSAIDs in OAT3 and OAT4 transfected HEK-PEAK cells
The results are expressed as mean (% coefficient of variation), n = 3.
DDI indexes for OAT3 and OAT4 used to rank and predict the risk of the interaction between pemetrexed and different NSAIDs
Cmax values and unbound fractions were taken were taken from Goodman and Gilman (1941).
Pemetrexed PBPK Model.
The bottom-up pemetrexed PBPK model was built using the measured in vitro transport kinetic data without any scaling factors. The simulations predicted an AUC0–inf of 362 µg/ml∙h (2.2-fold higher than the observed value), and a clearance of 2.5 l/h (55% smaller than the observed value). Because the difference between the in vitro and the in vivo amount and activity (relative expression factor/relative activity factor, REF/RAF) of the transporters is unknown, the model was optimized by estimating these parameters using clinical data. The REF/RAF value for OAT3 was estimated to 5.3 using human pemetrexed plasma concentration-time profiles published by Sweeney et al. (2006). The REF/RAF for OAT4 was set to 5.3 as well, which resulted in a lack of cellular accumulation and immediate excretion of pemetrexed in the urine. The optimized PBPK model was able to recover the observed plasma concentration-time profiles of pemetrexed.
Figure 3 shows the mean plasma concentrations of the predicted and the observed individual plasma concentration-time profiles of pemetrexed after a single intravenous 10-minute infusion. The lines for the 5 and 95% confidence intervals are also shown in the graph. The observed/prediction ratios for the Cmax, AUC0–inf , and clearance were all within 0.91 and 1.04. For AUC0–inf, the observed and predicted geometric means were 164.7 and 158 µg/ml per hour, respectively. The observed and predicted geometric means for Cmax were 102.1 and 111.6 µg/ml, respectively. For plasma clearance, the observed and predicted geometric mean were 5.6 and 5.7 l/h, respectively.
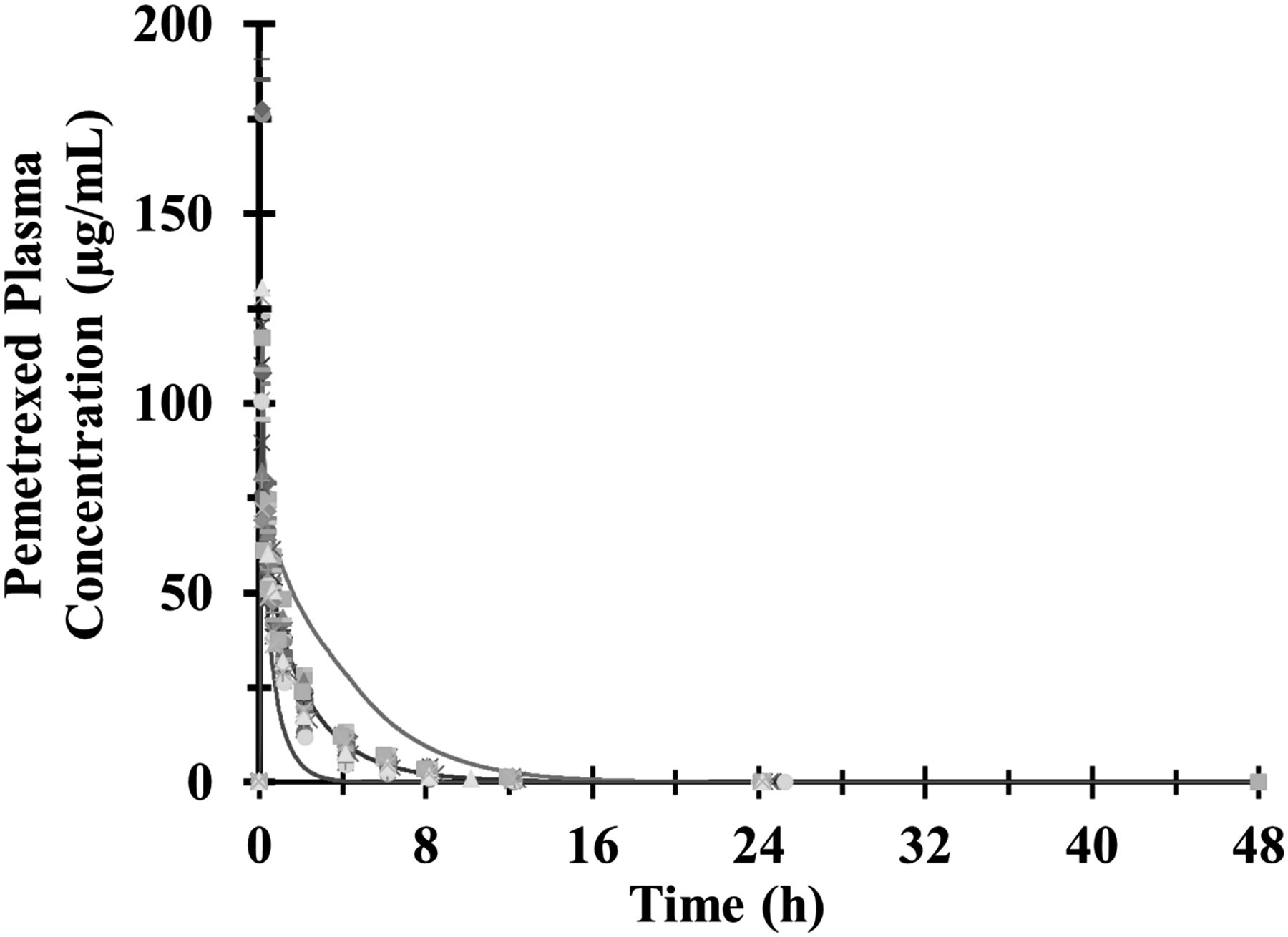
Results of the pemetrexed PBPK model simulation. Observed and predicted plasma concentration-time profiles after the administration of a single intravenous infusion of 500 mg/m2 of pemetrexed. The points represent the individual measured plasma concentrations of pemetrexed. The solid black line represents the predicted mean concentrations, and the thin gray lines represent the 5 and 95% confidence intervals of the prediction.
Impact of OAT3 and OAT4 on the PBPK Model Predictions.
The impact of adding the kidney transporters to the PBPK model was evaluated by comparing the predicted against the observed pemetrexed pharmacokinetic parameters (Rinaldi et al., 1999). As shown on Table 6, when OAT3 is not added to the basolateral membrane of the proximal tubular cells in the model, the clearance of pemetrexed was underpredicted by 74% and the predicted AUC0–inf was 3.8-fold higher. When OAT3 was present but OAT4 was not added to the apical membrane of the proximal tubular cells in the kidney model, the plasma concentrations were not affected, but the fraction excreted in urine unchanged (fe,0–24) was underpredicted by 70%. The predicted maximum intracellular drug concentration in the proximal tubular cells was 1000-fold in the absence of OAT4 compared with the model that had this transporter on the apical membrane.
Effect of the addition of OAT3 and OAT4 to the kidney model
Observed values are from Sweeney et al. (2006).
Model Verification.
The model was tested by comparing the predicted versus the observed Cmax and AUC0–inf for a range of pemetrexed doses between 50 and 700 mg/m2. The observed pharmacokinetic values were extracted from a phase 1 single-dose escalation study (Rinaldi et al., 1999). Figure 4A shows the predicted and observed AUC0–inf versus pemetrexed dose in mg/m2. In both the predicted and observed settings, the AUC0–inf increased linearly with dose. The slopes of the lines are very similar, 0.4283 and 0.4896 for the predicted and observed values, respectively (observed/predicted ratio = 1.14). Figure 4B shows the predicted and observed maximum plasma concentration versus pemetrexed dose in mg/m2. Again, the Cmax changed linearly with the dose, and the model predicts a similar slope of 0.243 and 0.219 for the observed and predicted values, respectively (observed/predicted ratio = 1.11).
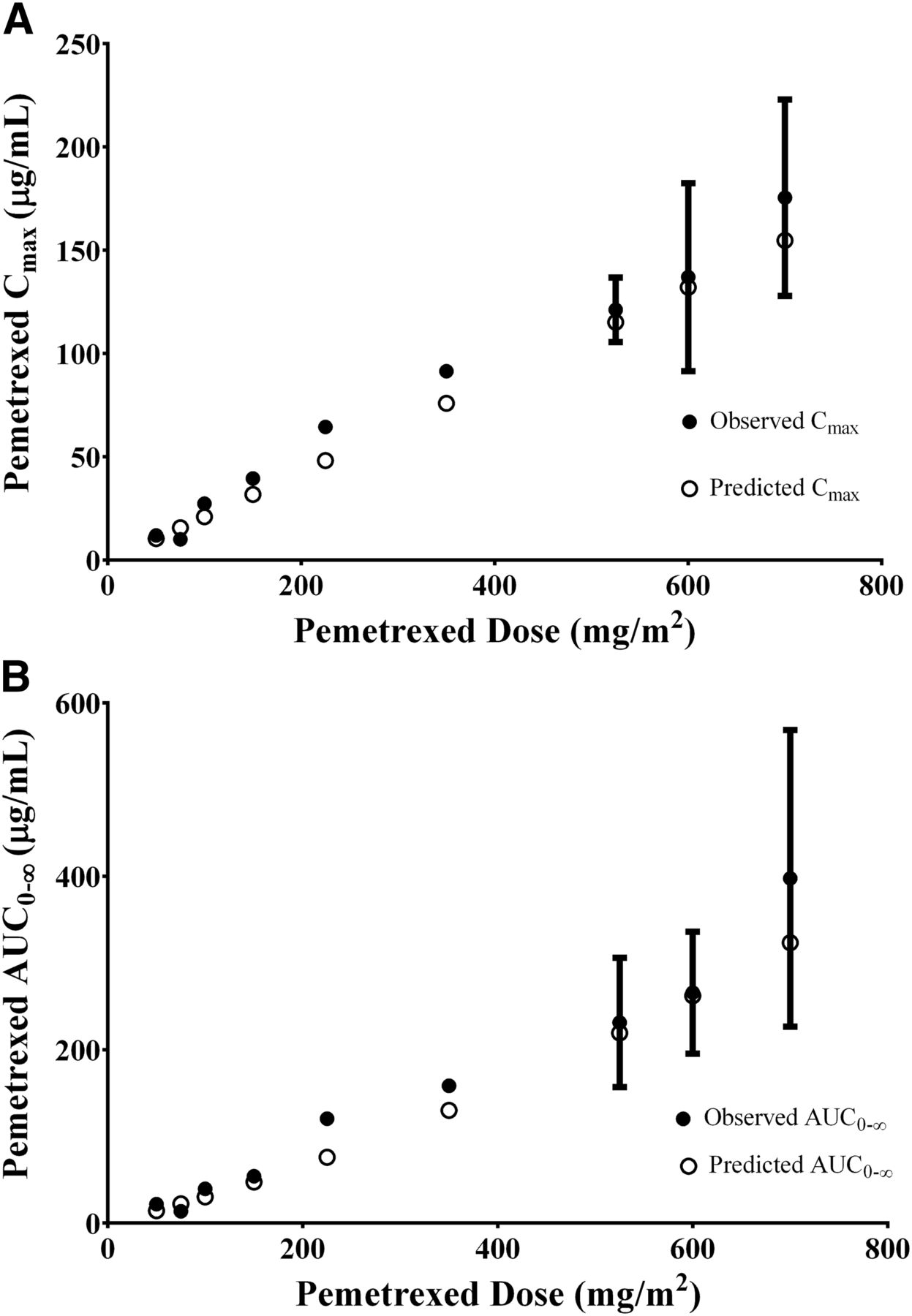
(A) Effect of increasing dose on pemetrexed plasma AUC0–inf. The open circles represent the predicted mean AUC0–inf, and the solid circles represent the observed values. Data are represented as mean ± S.D. (B) Effect of increasing dose on pemetrexed plasma Cmax. (○) Predicted mean plasma Cmax values and (●) observed values. Data are represented as mean ± S.D.
Drug-Drug Interaction Predictions.
Ibuprofen plasma concentrations were not measured during the clinical study, so the predicted plasma concentration-time profiles of ibuprofen at the time of pemetrexed administration were compared against data from the literature (Aarons et al., 1983a,b; Grennan et al., 1983; Lockwood et al., 1983a,b; Albert et al., 1984; Wagner et al., 1984; Davies, 1998). The pharmacokinetics of ibuprofen depend on several factors (i.e., formulation, fasted/fed state, physiology, and disease state), so a meta-analysis was performed to extract pharmacokinetic parameters from studies where ibuprofen was dosed at 400 mg in both healthy subjects and patients. Table 7 lists the reported and predicted steady-state Cmax, time of maximum concentration (Tmax), and AUC values for ibuprofen dosed at 400 mg every 6 hours. The predicted Tmax and Cmax of ibuprofen at steady state were 3 and 7%, respectively, lower than the average of the values found in the literature (Aarons et al., 1983a; Grennan et al., 1983; Ragni et al., 1992; Davies, 1998). The predicted plasma exposure of ibuprofen between 42 and 48 hours (AUC42–48h) was 1.01-fold higher than the reported value.
Observed and predicted ibuprofen pharmacokinetic parameters at steady state
Steady-state (ss) values obtained via Aarons et al. (1983a).
Figure 5A shows the predicted mean plasma concentrations and the 5 and 95% confidence intervals of ibuprofen. The results of the interaction model between ibuprofen and pemetrexed are shown in Fig. 5B. The pemetrexed AUC ratio was predicted to be 1.23 compared with 1.2 (1.11–1.31) observed, and the predicted Cmax ratio was 1.01 compared with 1.15 (1.03–1.28) observed in patients.
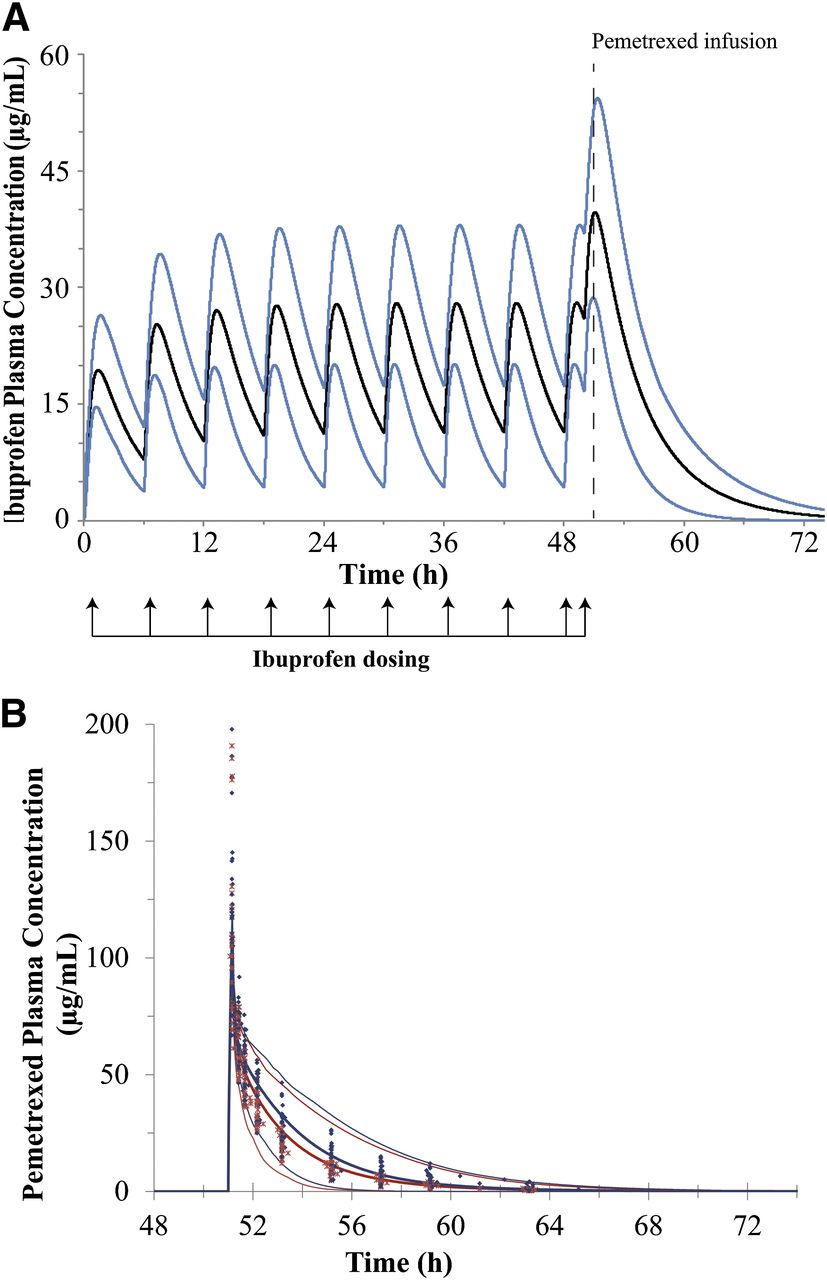
Results of the PBPK simulations for ibuprofen and for interaction between pemetrexed and ibuprofen. (A) Predicted plasma concentration-time profiles of ibuprofen after oral administration of multiple doses of 400 mg the NSAID. The thick black line represents the predicted mean concentrations in plasma, and the thin blue lines represent the 5 and 95% confidence intervals of the predictions. (B) Predicted pemetrexed plasma concentration-time profiles after a single intravenous 10-minute infusion of 500 mg/m2 of pemetrexed without (red lines) and with (blue lines) coadministration of ibuprofen in clinical subjects. The thick lines represent the mean values and the thin dotted lines represent the 5 and 95% confidence intervals, respectively. Blue closed diamonds represent the observed pemetrexed concentration-time points when pemetrexed was coadministered with ibuprofen. The red crosses represent the observed concentration-time points when pemetrexed was given alone.
Discussion
The goal of this study was to develop a strategy to predict clinically relevant transporter-mediated DDIs for pemetrexed using in vitro data, and to build confidence for the future prediction of drug interactions for renally cleared drugs. The first step in the strategy involved the in vitro identification of the transporters responsible for the active tubular secretion of pemetrexed. This was performed using a single time and single concentration assay in transiently transfected HeLa cells. OAT3 and OAT4 were identified as the transporters likely to be involved in the active tubular secretion of pemetrexed.
Under initial rate conditions in HEK-PEAK cells stably transfected with OAT3 and OAT4, we determined the Vmax, Km, and CLint, and employed these parameters in a PBPK model to reproduce the disposition of pemetrexed in humans. The initial bottom-up PBPK model performed well and predicted the shape of the concentration-time curve, but the predicted systemic clearance was 55% lower than the observed. A parameter sensitivity analysis was conducted to identify possible causes of the under prediction of pemetrexed clearance.
Based on the sensitivity analysis and the expected uncertainties, we hypothesized that the REF/RAF of OAT3 was the most likely contributor to this underprediction. The REF/RAF scaling factor was estimated to be 5.3 using a hybrid minimization method and weighted least squares as the objective function. The middle-out approach that employed the scaling factor recovered the shape of the concentration-time profile and systemic clearance accurately (Table 6).
The need of scaling factors for transport parameters obtained in vitro is well recognized in the prediction of drug absorption, distribution, and elimination via transporters (Bouzom et al., 2012; Tsamandouras et al., 2013; Hsu et al., 2014). In some cases, the scaling factor may represent the differences in transporter protein abundance between in vitro and in vivo, but it may also reflect the dissimilarities in activity per milligram of protein owing to several factors, including membrane environment or differences in posttranslational modifications (Straumann et al., 2006; Croset et al., 2012). Is important to note that in this study the Vmax and Km values were estimated from in vitro experiments performed at room temperature and not at 37°C. Rates of transport at room temperature are generally lower than those observed at 37°C, so the scaling factor should be lower when parameters are obtained at 37°C. This was demonstrated by Jackson and Halestrap (1996) for the active uptake of l-lactate into hepatocytes by the hepatic monocarboxylate transporter.
The model encompassed several important assumptions. First, it was assumed that the OATs identified in the transporter screen were the only transporters involved in the tubular secretion of pemetrexed. We assumed that the uptake of pemetrexed into the tubular cells was unidirectional and driven by blood concentrations and OAT3 intrinsic clearance. It was also assumed that the concentrations of the cotransported dicarboxylates, such as α-ketoglutarate and succinate, were not rate limiting. We also assumed unidirectional flow of pemetrexed from the cells into the tubular fluid mediated by OAT4. Despite the fact that pemetrexed has been shown to be a substrate of MRP2 and MRP5, OAT4 was chosen as the only mechanism of active transport across the apical membrane given that its pemetrexed intrinsic clearance (2.5 μl/min per mg protein) is at least 10-fold higher than the reported ones for MRP2 (0.27 μl/min per mg protein) and MRP5 (0.04 μl/min per mg protein) (Pratt et al., 2002). It was also assumed that the passive permeability of pemetrexed was negligible based on the in vitro measurements. Under these conditions the OAT3-mediated step in the tubular secretion was an identifiable parameter and could be estimated using plasma curves. In comparison, the OAT4-mediated step in the model is nonidentifiable because we do not have tubular concentrations nor do we have urinary excretion curves with samples collected over short intervals. Therefore, OAT4 was not scaled, resulting in complete urinary excretion and no cellular accumulation of pemetrexed. This is consistent with the absence of renal cytotoxicity and the 70–90% observed urinary recovery of pemetrexed (Rinaldi, 1999; Rinaldi et al., 1999).
One of the objectives of the current work was to test whether the in vitro inhibition potency of OATs could be confidently employed to predict the DDI observed in vivo. Using the transfected OAT3 and OAT4 HEK-PEAK cells, we determined the IC50 values for commonly used NSAIDs. Of the NSAIDs tested, ibuprofen was identified as the most likely to cause a DDI with pemetrexed using the ratio of the unbound plasma Cmax to the IC50 (Table 5). To foresee the effect of ibuprofen on the tubular secretion of pemetrexed, a PBPK model was constructed for ibuprofen based on pharmacokinetic data from the literature (Table 7). The combination of the predicted unbound ibuprofen plasma concentration-time profile and the in vitro inhibition constants (Ki) for OAT3 and OAT4 resulted in an accurate prediction of the increase in pemetrexed exposure.
It is important to note that although the model included the inhibition of both OAT3 and OAT4, the predicted plasma clearance of pemetrexed was only affected by inhibition of OAT3. The inhibition of OAT4 by ibuprofen was shown to have no effect on the plasma clearance of pemetrexed (Fig. 6). This is not surprising because the plasma clearance will be dependent on the uptake into the proximal tubular cells and not on the efflux of the drug from the cells into the tubules (Fig. 6). Another important assumption of the current in vitro to in vivo extrapolation approach was that only the parent drug was responsible for inhibiting the active transport, and that ibuprofen metabolites played no significant role in the inhibition. We believe this assumption is valid because the levels of ibuprofen in plasma are considerably higher (approximately 10-fold) than the levels of its two primary metabolites (Mills et al., 1973).
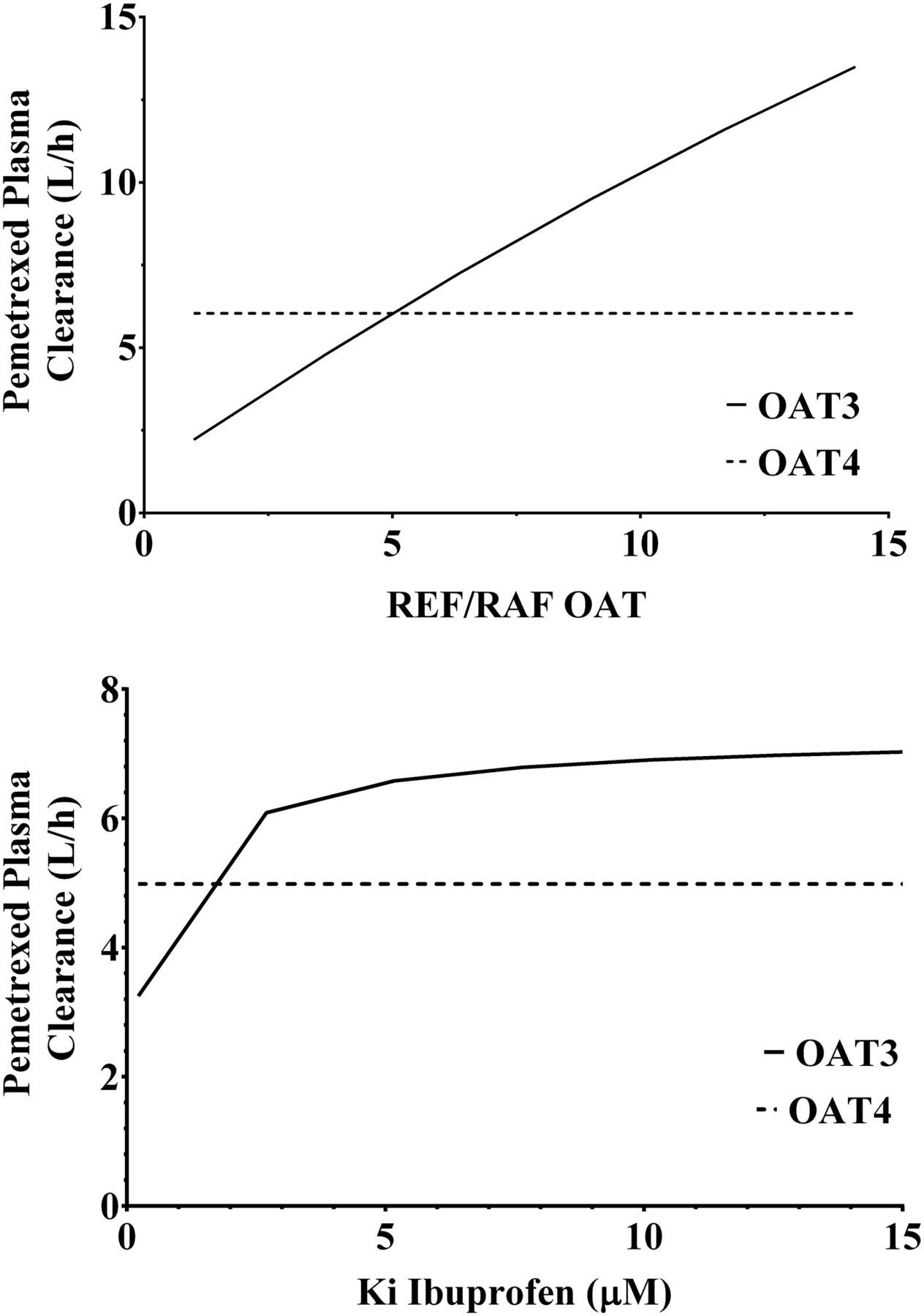
Parameter sensitivity analyses for pemetrexed plasma clearance. (A) Effect of the relative expression factors/activities of OAT3 and OAT4 on the plasma clearance of pemetrexed. (B) Effect of the inhibition constant of ibuprofen (Ki) OAT3 and OAT4 on the plasma clearance of pemetrexed.
An important strength of mechanistic PBPK modeling is the ability to simulate the outcome of different physiologic or pathologic changes that have not been specifically studied. This is illustrated in the case of pemetrexed by examining the effect of renal impairment on the plasma clearance. Under the assumptions that the glomerular filtration rate and active tubular secretion intrinsic clearance decrease proportionally in renal insufficiency and that the plasma binding of pemetrexed is not altered, we predict a plasma clearance of 53.8 ml/min in patients with moderate renal insufficiency (glomerular filtration rate = 40–59 ml/min). However, in this case, a clinical study had been performed to determine the effect of renal insufficiency on pemetrexed pharmacokinetics in cancer patients (Mita et al., 2006). In that study, the observed plasma clearance of pemetrexed in the moderate renal insufficiency group was 54.7 ml/min (34% coefficient of variation), which is in good agreement with the prediction. The recruitment of the severe renal insufficiency (glomerular filtration rate <20 ml/min) group was problematic in that study, resulting in a single patient being studied. This is an example where mechanistic PBPK modeling could be used to estimate the effect of altered physiology/pathology in situations that are difficult to study. Similarly, scenarios involving the combinations of DDIs and disease states could be studied.
One the main challenges of mechanistic PBPK modeling of renal elimination is the lack of information on the intrinsic clearance of efflux transporters such as OAT4 because plasma concentration-time curves are insensitive to this parameter (Fig. 6). To resolve this shortcoming, the measurement of pemetrexed concentrations in tubular cells or tubular fluid is necessary. A potential surrogate would be urine concentrations if the study design were customized for this purpose and urine is collected quantitatively, at sufficient frequency, and at early time intervals, but this is rarely achieved in a clinical study. To date, the successful implementation of this approach is lacking; consequently, the quantitation of renal tubular efflux transporter intrinsic clearance will usually be poorly defined.
In conclusion, we demonstrated for the first time that pemetrexed is transported by OAT3 and OAT4, and not by other solute carrier transporters that are involved in active secretion in the kidney. Importantly, we detail how the in vitro transport parameters for OAT3 and OAT4 can define the structure of a PBPK model that will predict the renal clearance of pemetrexed within 2-fold of the observed value. Once the structure of the model was defined, we demonstrated for the first time that the decrease in the pemetrexed OAT3-mediated uptake and therefore the renal clearance could be predicted using in vitro inhibition parameters for OAT3. In this translational framework, bottom-up PBPK models are a powerful tool for the prediction of pharmacokinetics in the presence of transporter-mediated drug interactions and will be enhanced when the accurate quantitation of protein concentration in in vitro and in vivo systems is available. However, it is likely the in vitro systems will not completely reproduce the transport rates and affinities that exist in vivo and the middle-out parameter optimization approach will be a necessary step in model verification. Ongoing work is designed to determine whether the current model paradigm accurately predicts the pharmacokinetics of other OAT substrates and the interactions with inhibitors.
Authorship Contributions
Participated in research design: Posada, Bacon, Kim, Hall, Hillgren.
Conducted experiments: Posada, Bacon, Tirona, Pak.
Performed data analysis: Posada, Bacon, Pak, Higgins, Schneck.
Wrote or contributed to the writing of the manuscript: Posada, Bacon, Tirona, Hall, Hillgren,
Footnotes
- Received July 21, 2014.
- Accepted December 12, 2014.
Abbreviations
- AUC
- area under the plasma-concentration time curve
- DDI
- drug-drug interaction
- E3S
- [3H]estrone-3-sulfate
- LC-MS/MS
- liquid chromatography with tandem mass spectrometry
- NSAID
- nonsteroidal anti-inflammatory drug
- OAT
- organic anion transporter
- PBPK
- physiologically based pharmacokinetic
- REF/RAF
- relative expression factor/ relative activity factor
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}