


Abstract
Upon characterization of baculovirus-expressed cytochrome P-450 (CYP) 2C19, it was observed that this enzyme metabolized (+/−) bufuralol to 1′hydroxybufuralol, a reaction previously understood to be selectively catalyzed by CYP2D6. The apparentKm for this reaction was 36 μM with recombinant CYP2C19, approximately 7-fold higher than for recombinant CYP2D6. The intrinsic clearance for this reaction was 37-fold higher with CYP2D6 than for CYP2C19. The involvement of human CYP1A2 in bufuralol 1′-hydroxylation was also confirmed using the recombinant enzyme. Using S-mephenytoin as an inhibitor, theKi for inhibition of recombinant CYP2C19-mediated bufuralol hydroxylation was 42 μM, which is the approximate Km for recombinant CYP2C19-mediated S-mephenytoin metabolism. The classic CYP2D6 inhibitors quinidine and quinine showed no inhibition of CYP2C19-catalyzed bufuralol metabolism at concentrations that abolished CYP2D6-mediated bufuralol metabolism. Ticlopidine, a potent inhibitor of CYP2C19 and CYP2D6, inhibited bufuralol 1′-hydroxylation by each of these enzymes equipotently. In human liver microsomes that are known to be deficient in CYP2D6 activity, it was shown that in the presence of quinidine, the Km shifted from 14 to 38 μM. This is consistent with the Kmdetermination for recombinant CYP2C19 of 36 μM. In human liver microsomes that have high CYP2D6 and CYP2C19 activity, theKm shifted to 145 μM in the presence ofS-mephenytoin and quinidine, consistent with theKm determined for CYP1A2. This data suggests that bufuralol, and possibly other CYP2D6 substrates, have the potential to be metabolized by CYP2C19.
There have been many recent investigations into the specificity of several known cytochrome P-450 (CYP)1substrates and inhibitors in human liver microsomes (Newton et al., 1995; Bourrie et al., 1996; Ekins et al., 1997) and in cDNA-expressed human CYPs (Ono et al., 1996; Ekins et al., 1997). One widely used substrate probe, bufuralol, is a β-adrenergic receptor antagonist whose metabolism is thought to be selectively mediated by CYP2D6 (Kronbach et al., 1987). Therefore, bufuralol has been extensively used as a probe substrate for the in vitro study of CYP2D6 and, in particular, polymorphisms associated with this CYP. Bufuralol exists as a racemic mixture and both enantiomers are metabolized by CYP2D6 (Boobis et al., 1985). Although the apparent Km andVmax of the (+) enantiomer was shown to be greater than for the (−) enantiomer with human liver microsomes, the intrinsic clearance (Vmax/Km) for the two enantiomers was shown to be similar (Boobis et al., 1985). It has been known for a considerable time that bufuralol 1′ hydroxylase activity generally exhibits biphasic enzyme kinetic behavior in human liver microsomes that suggests the involvement of more than one CYP (Gut et al., 1986; Kronbach et al., 1987). It was also shown by Gut and coworkers (1986) that there were two different but related CYP2D proteins responsible for bufuralol 1′ hydroxylation in human liver microsomes which were termed ‘buf I’ and ‘buf II’. Buf I was suggested as the high-affinity bufuralol 1′ hydroxylase, with aKm of approximately 30 μM toward (+) bufuralol, whereas buf II corresponded to the low-affinity bufuralol 1′ hydroxylase, with a Km of approximately 200 μM toward either (+) or (−) bufuralol. To date, buf I has been identified as CYP2D6 (Gut et al., 1986), whereas buf II has as yet not been identified. One group has speculated that buf II is CYP1A2 (Yamazaki et al., 1994). Their evidence for this is based on the high Km values for bufuralol obtained byGut et al. (1986) with buf II, which are similar to those obtained with recombinant CYP1A2 in their report (Yamazaki et al., 1994).
While characterizing mixtures of recombinant CYPs for bufuralol 1′ hydroxylase activity, it was shown that the combination of CYP2D6 and CYP2C19 had higher activity than CYP2D6 alone (D.C.M., unpublished observation). At about the same time, Gelboin et al. (1997) showed that recombinant CYP2C19 has significant (+/−) bufuralol 1′hydroxylase activity, which was inhibited by a monoclonal antibody to CYP2C8/9/19. The objective of the present study was to understand the roles of CYP2C19 and CYP2D6 in the metabolism of bufuralol. To this end, complete kinetic characterization using baculovirus-expressed recombinant enzymes was performed. Inhibitor studies were completed with the typical CYP2D6 inhibitors quinidine and quinine (Rendic et al., 1997), the CYP2C19 substrate S-mephenytoin (Wrighton et al., 1992), and in addition, the potent CYP2C19 and CYP2D6 inhibitor ticlopidine (Donahue et al., 1997; Ko et al., 1998). The individual contributions of CYP2D6, CYP2C19, and CYP1A2 to bufuralol metabolism were assessed using quinidine and S-mephenytoin as inhibitors in a human liver microsome lot with known high CYP2C19 and high CYP2D6 activity. This was then compared to a human liver microsome lot with low CYP2D6 and high CYP2C19 activity. The relative contribution of each isoform to bufuralol metabolism will be discussed with respect to the polymorphisms shown for each CYP.
Materials and Methods
Chemicals.
S-mephenytoin, bufuralol, and 1′-hydroxybufuralol were obtained from Gentest (Woburn, MA). All other reagents were obtained from Sigma Chemical Co. (St. Louis, MO) and were of the highest analytical grade available.
Microsomal Preparations.
Recombinant CYP2C19, CYP2D6, and CYP1A2 microsomes were generated at Pfizer using the baculovirus expression system as described by Roussel et al. (1998). These enzymes have human CYP oxidoreductase coexpressed with each CYP at optimized concentrations (Mankowski et al., 1996). Human liver samples were obtained from the International Institute for the Advancement of Medicine (Exton, PA), and microsomes were generated in-house using a published technique (Guengerich et al., 1977). Prepared human liver microsome lots are identified by a numbering system (HL-1008, HL-1062–2). Protein concentrations were measured using a BCA Protein Assay kit (Pierce Chemical Co., Rockford, IL) with BSA as a standard as described by the manufacturer. CYP content was measured by the method of Omura and Sato (1964). It should be noted that measured CYP content in baculovirus microsomal preparations has been shown to be significantly lower than other expression systems (Asseffa et al., 1989). This is therefore an important consideration when using baculovirus-derived CYPs.
Microsomal Incubations and HPLC Analysis.
Bufuralol 1′-hydroxylase activity was determined by a modification of the method described by Kronbach et al. (1987). Briefly, reaction mixtures contained recombinant CYP2C19 (0.3 mg/ml, 5 pmol CYP), CYP2D6 (0.15 mg/ml, 0.9 pmol CYP), CYP1A2 (0.12 mg/ml, 2 pmol CYP), or human liver microsomal protein (0.5 mg/ml, 45 pmol CYP), (+/−) bufuralol at various concentrations, and an NADPH-regenerating system (10 mM MgCl2, 0.44 mM NADP, 5 mMdl-isocitric acid, 0.5 U/ml isocitric dehydrogenase) in 100 mM potassium phosphate buffer, pH 7.4, in a final reaction volume of 0.5 ml. Reactions were initiated by addition of bufuralol (prepared as stock solutions in 100 mM potassium phosphate buffer, pH 7.4) and incubated for 5 to 20 min. The reactions were quenched by the addition of 50 μl of 70% perchloric acid. All reactions were linear with respect to protein and time. Incubates were centrifuged at 11,000 rpm for 5 min, and supernatant (10 μl) was analyzed by HPLC as follows. Bufuralol and its 1′-hydroxy metabolite were separated on a Zorbax SB-C18 column (3.5 micron; 4.6 mm × 7.5 cm) with a mobile phase consisting of 50% 10 mM potassium phosphate buffer (pH 3.0) and 50% methanol, at a flow rate of 1 ml/min. The 1′-hydroxy metabolite was monitored by fluorescence detection using a Shimadzu RF 10 A detector at an excitation wavelength of 252 nm and emission wavelength of 302 nm. Samples were quantitated by injecting known amounts of 1′-hydroxybufuralol in triplicate (100 ng/ml). The Multichrom data acquisition system was used for data collection, analysis, and reporting. For Km determinations, concentrations of bufuralol ranged from 0.5 μM to 2 mM. Kinetic parameters were determined from Eadie-Hofstee plots. Inhibition constants (Ki values) were determined by standard Dixon plots after analysis using multiple substrate and inhibitor concentrations (see text for details). Appropriate solvent controls were included to minimize nonspecific inhibition of CYPs. Any combination of inhibitors (in methanol) added to a specific reaction, that exceeded 1% of the total reaction volume, was evaporated under nitrogen before incubation with human liver microsomes or recombinant enzymes.
Results
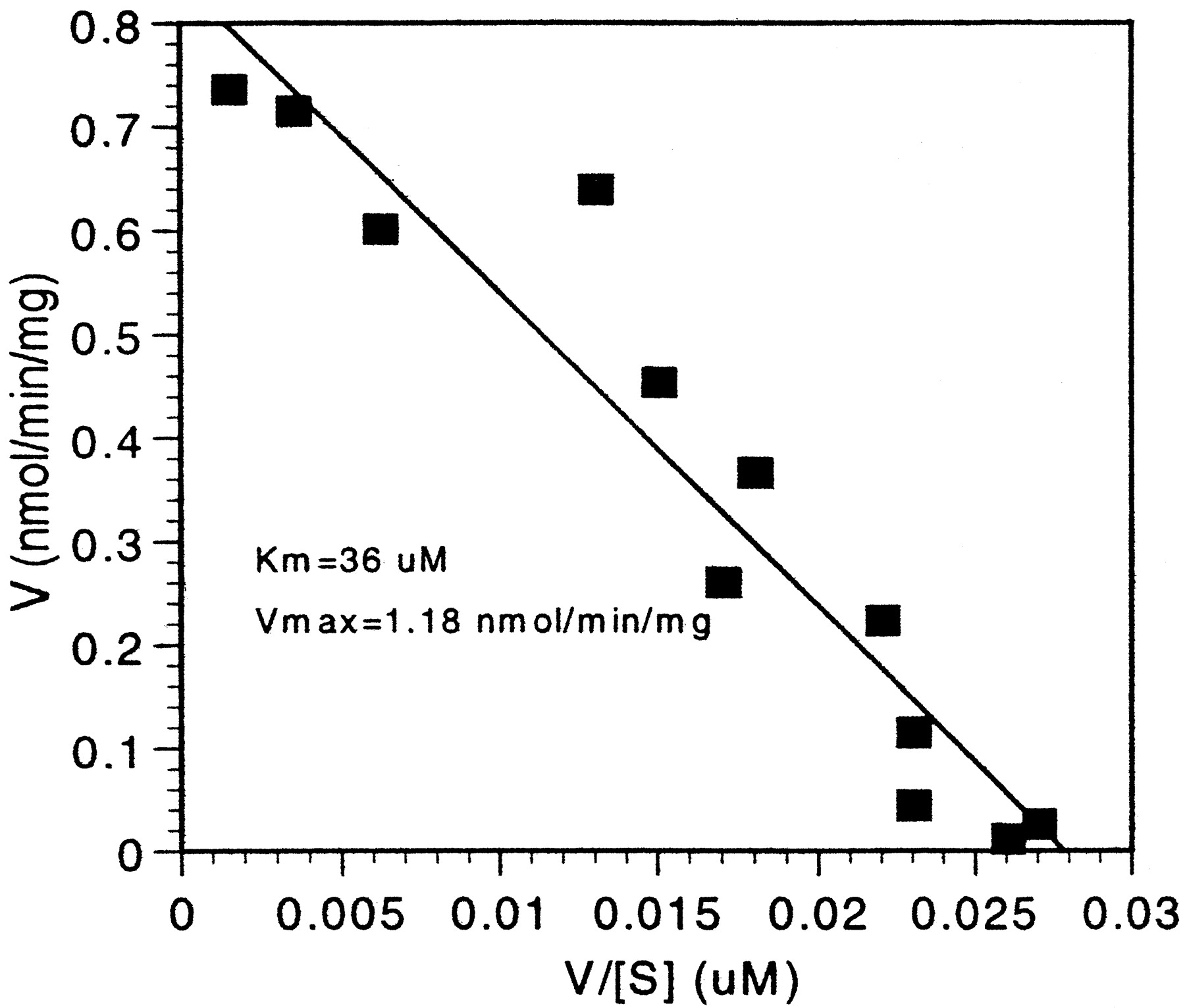
The apparent Km andVmax for bufuralol 1′ hydroxylation by recombinant CYP2C19 were determined to be 36 μM and 1.18 nmol/min/mg (36.9 nmol/min/nmol CYP), respectively (Fig.1). The Kmfor bufuralol 1′ hydroxylation catalyzed by recombinant CYP1A2 was determined to be 176 μM, and the Vmax was 0.178 nmol/min/mg (4.56 nmol/min/nmol CYP), consistent with previously reported values (Yamazaki et al., 1994). The relative contribution of each isoform to bufuralol 1′ hydroxylation intrinsic clearance (Vmax/Km) was determined by applying a CYP abundance normalization factor (Table 1) (Rodrigues, 1999).

Eadie-Hofstee plot of bufuralol 1′-hydroxy formation by baculovirus-expressed CYP2C19.
Method: 0.16 mg recombinant CYP2C19 was incubated with 0.5–500 μM bufuralol for 5 min, in a 0.5 ml final incubation volume containing 100 mM potassium phosphate buffer and an NADPH-regenerating system. 1′OH bufuralol was determined by HPLC with fluorescence detection as described in Materials and Methods. Each point is the average of duplicate determinations.
Determination of the relative contribution of CYP2C19, CYP2D6, and CYP1A2 to bufuralol 1′ hydroxylation in human liver using recombinant data
CYP2C19-mediated bufuralol metabolism was not inhibited by quinidine or quinine, standard potent inhibitors of CYP2D6-mediated bufuralol 1′ hydroxylation, at concentrations up to 100 μM (data not shown). Ticlopidine, a potent CYP2C19 and CYP2D6 inhibitor (Ko et al., 1998), was an equipotent inhibitor of CYP-mediated metabolism of bufuralol (Ki < 1 μM) with both of these enzymes (Fig. 2). S-mephenytoin was also an inhibitor of CYP2C19-mediated bufuralol metabolism (Ki = 42 μM; Fig.3), but not CYP2D6-mediated bufuralol metabolism at concentrations up to 500 μM (data not shown).
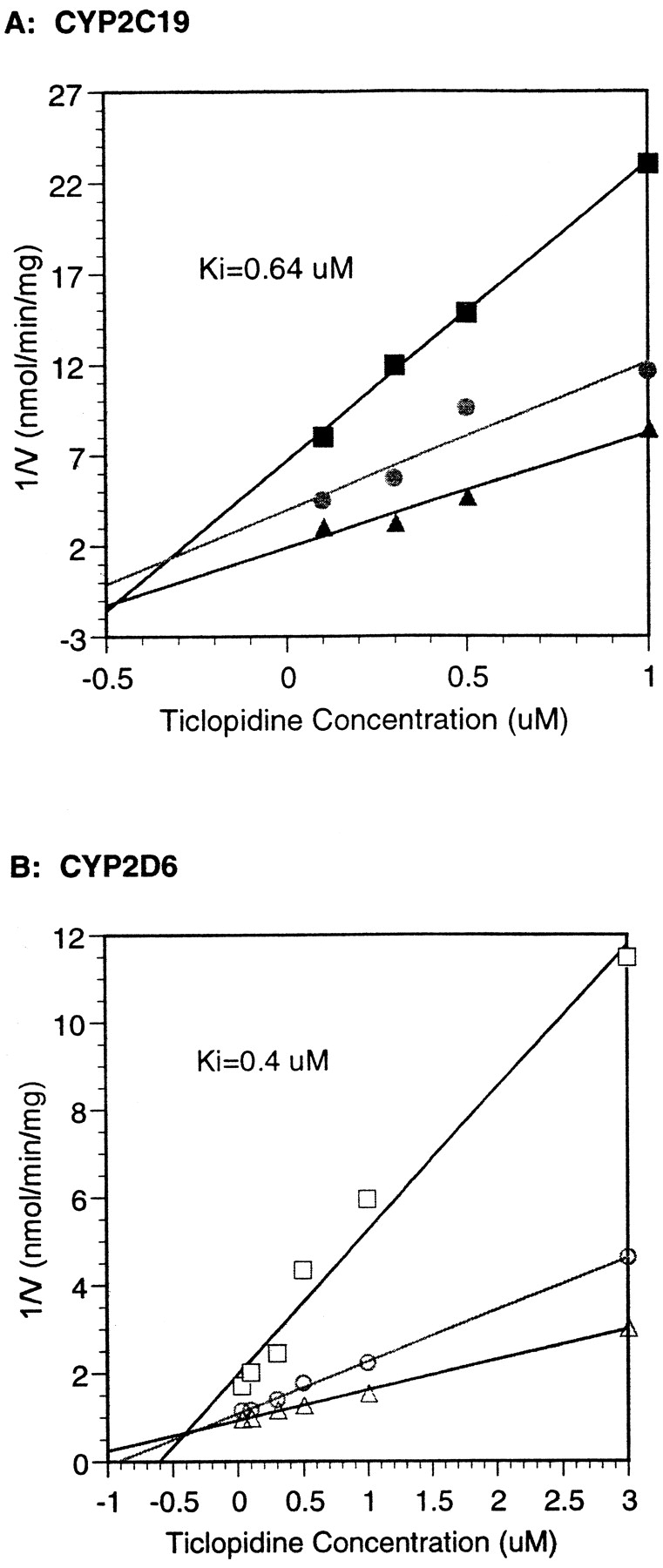
Inhibition of recombinant CYP2C19 and CYP2D6-mediated metabolism of bufuralol by ticlopidine—Kidetermination by Dixon plot.
A: CYP2C19. B: CYP2D6. Method: 0.16 mg recombinant CYP2C19 or 0.08 mg CYP2D6 was incubated with 25 (▪), 50 (●), and 100 μM (▴) bufuralol—CYP2C19; 2 (■), 5 (○), and 10 μM (▵)—CYP2D6 in the presence of 0 to 5 μM ticlopidine for 10 min in a 0.5-ml final volume containing 100 mM potassium phosphate buffer and an NADPH-regenerating system. 1′-OH bufuralol was determined as described in Materials and Methods. Each point is the average of duplicate determinations.

Inhibition of recombinant CYP2C19-mediated metabolism of bufuralol by S-mephenytoin;Ki determination by Dixon plot.
Method: 0.16 mg recombinant CYP2C19 was incubated with bufuralol at 3 concentrations: 25 (▪), 50 (♦), or 100 μM (●) in the presence of 0 to 100 μM S-mephenytoin for 5 min in a 0.5-ml final volume containing 100 mM potassium phosphate buffer and an NADPH-regenerating system. 1′-OH bufuralol was determined as described in Materials and Methods. Each point is the average of duplicate determinations.
Quinidine (1 μM) and S-mephenytoin (50 μM) were then used as selective CYP2D6 and CYP2C19 inhibitors, respectively, to determine the relative contribution of CYP2D6, CYP2C19, and CYP1A2 to bufuralol metabolism in human liver microsomes (Table 2). Using a human liver microsome lot previously phenotyped to be a CYP2D6 poor metabolizer (HL-1008), quinidine (1 μM) was used to block the remaining CYP2D6 contribution and the Kmshifted from 14 to 38 μM, whereas Vmaxdid not change (Table 2). Quinidine (1 μM) andS-mephenytoin (50 μM) were also incubated together in HL-1062–2 (phenotyped with high CYP2D6 and CYP2C19 activities) to determine whether the metabolism would shift to CYP1A2 (Table 2). TheKm increased to 145 μM, approaching that of CYP1A2 reported previously (210 μM; Yamazaki et al., 1994).
Determination of the Km and Vmaxfor bufuralol 1′ hydroxy formation by human liver microsome lots HL-1008 and HL-1062-2 in the presence of quinidine and S-mephenytoin
To attempt to differentiate each CYP’s contribution to total (+/−) bufuralol 1′ hydroxylation in the microsome lot HL-1062–2, 1 μM quinidine, and 50 μM S-mephenytoin were used individually as inhibitors of CYP2D6 and CYP2C19, respectively. Each of the inhibitors was incubated with 1 and 50 μM bufuralol (to approximate the Km value for CYP2D6 and CYP2C19, respectively; Fig. 4). At 1 μM bufuralol, 1 μM quinidine blocked 90% of bufuralol 1′ hydroxylase activity, whereas 50 μM S-mephenytoin blocked only 20% of bufuralol 1′ hydroxylase activity. However, at the higher bufuralol concentration (50 μM), 1 μM quinidine inhibited 70% of bufuralol 1′ hydroxylase activity, whereas 50 μM S-mephenytoin blocked 50% of this activity.

The effect of inhibitors on bufuralol 1′ hydroxylation by human liver microsome lot HL-1062–2 at 1 or 50 μM (+/−) bufuralol.
Method: data taken fromVmax/Kmdeterminations in the presence of inhibitors with human liver microsome lot HL-1062–2. Shaded columns, 50 μM bufuralol; Open columns, 1 μM bufuralol.
Discussion
CYP2D6 and CYP2C19 are two CYPs that are polymorphically expressed in different populations (Kivisto and Kroemer, 1997). Poor metabolizers of CYP2D6 substrates such as debrisoquine and sparteine occur with higher frequency (8–10%) in Caucasian populations, whereas poor metabolizers of S- mephenytoin (CYP2C19) are relatively rare (2–5%) in this population (Nakamura et al., 1985). In contrast, the Asian population has a very low incidence of phenotyped CYP2D6 poor metabolizers (1–2%), whereas CYP2C19 phenotyped poor metabolizers can reach up to 25% of the population (Nakamura et al., 1985; Kimura. et al., 1998). Therefore, it is important in a drug discovery setting to characterize the metabolism of new chemical entities by these enzymes with a number of different approaches.
(+/−) Bufuralol 1′ hydroxylation is a widely used in vitro probe substrate for CYP2D6 activity. It was recognized early on, however, that more than one enzyme was responsible for bufuralol metabolism (Gut et al., 1986). At high substrate concentrations (>100 μM), CYP1A2 was shown to play a role in its metabolism to 1′-, 4′-, and 6′-hydroxylated metabolites (Yamazaki et al., 1994). The possibility that CYP2C19 catalyzes bufuralol 1′-hydroxylation was first shown by Gelboin et al. (1997), using a single concentration (50 μM) of bufuralol. This study was primarily aimed at analysis of a monoclonal antibody to CYP2D6. When complete inhibition was not achieved using this antibody in human liver microsomes, bufuralol metabolism was examined using recombinant CYP2C19, along with other recombinant CYPs. In the study by Yamazaki et al. (1994), a reconstituted system containing CYP2C19 purified from Escherichia coli was also used to determine isoform identity with respect to bufuralol metabolism, and the authors state that no metabolite production was observed. However, even at concentrations of bufuralol at or below 10 μM, CYP2C19 coexpressed with CYP-oxidoreductase in the baculovirus expression system appears to play a significant role in the formation of 1′-hydroxy bufuralol, the major metabolite formed by CYP2D6 (Gut et al., 1986). This is in comparison to data previously generated with recombinant CYP2D6 (Km andVmax values of 3.4 μM and 1.54 nmol/min/mg, respectively; Roussel et al., 1998). Based on individual isoform abundance values (Rodrigues, 1999), normalized CLint from individual recombinant enzyme incubations with bufuralol show that CYP2D6 is responsible for 95% of bufuralol clearance, whereas CYP2C19 is responsible for 5% of the intrinsic clearance (Table 1). CYP1A2 has a small contribution to bufuralol clearance (<1%). The percentage of drug metabolized by CYP2C19 could increase significantly in a population with a high incidence of CYP2D6 polymorphisms, such as the Caucasian population (Nakamura et al., 1985). Indeed, when human liver microsomes characterized with low CYP2D6 activity (HL-1008) were incubated with bufuralol and quinidine (to block the CYP2D6 contribution), CYP2C19 was predominantly responsible for bufuralol metabolism, as evidenced by the shift in theKm value (Table2).
Previous in vivo studies with (+/−) bufuralol (Dayer et al., 1985;Meyer et al., 1986) compared ratios of (−) to (+) bufuralol plasma ratios in CYP2D6-poor versus -extensive metabolizers. It was shown that the (−/+) ratio increased in the poor metabolizer group. This study was confounded by the fact that (+) bufuralol is directly glucuronidated and would decrease the ratio by this mechanism. It would be tempting to speculate that the ratio increase shown in the CYP2D6 poor metabolizer group was due to CYP2C19 involvement in bufuralol metabolism. In the present study, only the major metabolite of bufuralol, 1′-hydroxybufuralol, was monitored with each CYP. Experiments are currently underway to determine whether recombinant CYP2C19 forms other bufuralol metabolites in addition to the known 1′-hydroxy metabolite.
CYP2C19-mediated 1′-hydroxybufuralol formation is not inhibited by quinidine or quinine at concentrations up to 100 μM (data not shown). This is in contrast to the potent inhibition of CYP2D6-mediated bufuralol 1′OH formation by quinidine and quinine (approximately 1 μM; Ching et al., 1995; Roussel et al., 1998). Quinidine is therefore a useful probe to determine the contribution of CYP2C19 to bufuralol metabolism in human liver microsomes by selectively blocking the CYP2D6 contribution to metabolism (Table 2). By using both quinidine andS-mephenytoin as inhibitors of CYP2D6 and CYP2C19 in human liver microsomes, it was possible to show that at different concentrations of bufuralol, both CYP2D6 and CYP2C19 contributed to bufuralol 1′hydroxylation (Fig. 4). Ticlopidine, a recently identified potent CYP2C19 inhibitor (Donahue et al., 1997), blocked both CYP2D6- and CYP2C19-catalyzed metabolism of bufuralol to 1′-hydroxybufuralol equipotently (Ki < 1 μM; Fig. 2).
It is of interest to determine whether other classical CYP2D6 substrates are also substrates for CYP2C19. Substrates such as dextromethorphan (Kronbach et al., 1987), debrisoquine (Boobis et al., 1985), and sparteine (Gut et al., 1986) have been shown in this lab not to be metabolized by CYP2C19 at concentrations up to 100 μM (data not shown). However, it has been shown that the in vivo metabolic clearance of propranolol, another CYP2D6 substrate and β-adrenergic receptor antagonist, is also dependent on CYP2C19 (Ward et al., 1989). In this study, patients who were deficient in CYP2C19 activity showed an impaired clearance of propranolol, as evidenced by a decreased urinary excretion of the deisopropylated metabolite, α-napthoxylactic acid (Ward et al., 1989). In contrast, a study by Sowinski et al. (1997)looked at propranolol metabolism in CYP2D6-poor and -extensive metabolizers that were both phenotyped as extensive CYP2C19 metabolizers, and found no change in oral clearance of propranolol in either group. They hypothesized that CYP2C19 did not play a role, because there was no change in the excreted ratio of the stereoisomers of propranolol. Studies with propranolol metabolism in human liver microsomes (Otton et al., 1990) showed that S-mephenytoin did not inhibit the deisopropylation reaction. Additional studies byMasubuchi et al. (1994) in human liver microsomes, and Yoshimoto et al. (1995) using recombinant enzymes, identified CYP1A2 as also playing a role in the deisopropylation reaction. The metabolism of propranolol appears to differ from that of bufuralol, in that each CYP metabolizes propranolol to different metabolites. Preliminary data from our lab shows that incubation of R- or S-propranolol with recombinant CYP2C19 in the presence of an NADPH-regenerating system results in the formation of 4′- and 5′-hydroxy metabolites for both isomers with an apparent Km of 40 μM (D.C.M., unpublished observations). The observation that two drugs in the same pharmacological class of known CYP2D6 substrates are also CYP2C19 substrates is of importance in the drug discovery setting. Many potential CYP2D6 substrates are discarded, and this knowledge argues strongly for the concurrent screening of these compounds with CYP2C19.
In conclusion, the contribution of CYP2C19 to bufuralol 1′-hydroxylation in vitro indicates that metabolism of bufuralol in vivo could potentially be catalyzed by CYP2C19. This will be important when CYP2D6 is decreased, inhibited, or absent. This latter observation would suggest an important role of CYP2C19 in CYP2D6 poor metabolizers, a role that should be further explored.
Acknowledgments
I thank Donna Hiller for bufuralol assay HPLC method development, Dr. Michael P. Lawton for critical review and support of these studies, and Drs. Sean Ekins and R. Scott Obach for valuable discussions and review of this manuscript.
Footnotes
-
Send reprint requests to: Dayna C. Mankowski, Pfizer Central Research, Eastern Point Rd., Box 825, Groton, CT 06340. E-mail: Dayna_C_Mankowski{at}groton.pfizer.com
- Abbreviation used is::
- CYP
- cytochrome P-450
- Received February 23, 1999.
- Accepted June 15, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}