


Abstract
The metabolism of diclofenac to its 5-hydroxylated derivative in humans is catalyzed by cytochrome P450 (CYP)3A4. We report herein that in vitro this biotransformation pathway is stimulated by quinidine. When diclofenac was incubated with human liver microsomes in the presence of quinidine, the formation of 5-hydroxydiclofenac increased ∼6-fold relative to controls. Similar phenomena were observed with diastereoisomers of quinidine, including quinine and thethreo epimers, which produced an enhancement in the formation of 5-hydroxydiclofenac in the order of 6- to 9-fold. This stimulation of diclofenac metabolism was diminished when human liver microsomes were pretreated with a monoclonal inhibitory antibody against CYP3A4. In contrast, neither cytochromeb5 nor CYP oxidoreductase appeared to mediate the stimulation of diclofenac metabolism by quinidine, suggesting that the effect of quinidine is mediated through CYP3A4 protein. Further kinetic analyses indicated thatVmax values for the conversion of diclofenac to its 5-hydroxy derivative increased 4.5-fold from 13.2 to 57.6 nmol/min/nmol of CYP with little change inKm (71–56 μM) over a quinidine concentration range of 0 to 30 μM. Conversely, the metabolism of quinidine was not affected by the presence of diclofenac; theKm value estimated for the formation of 3-hydroxyquinidine was ∼1.5 μM, similar to the quinidine concentration required to produce 50% of the maximum stimulatory effect on diclofenac metabolism. It appears that the enhancement of diclofenac metabolism does not interfere with quinidine's access to the ferriheme-oxygen complex, implicating the presence of both compounds in the active site of CYP3A4 at the same time. Finally, a ∼4-fold increase in 5-hydroxydiclofenac formation was observed in human hepatocyte suspensions containing diclofenac and quinidine, demonstrating that this type of drug-drug interaction occurs in intact cells.
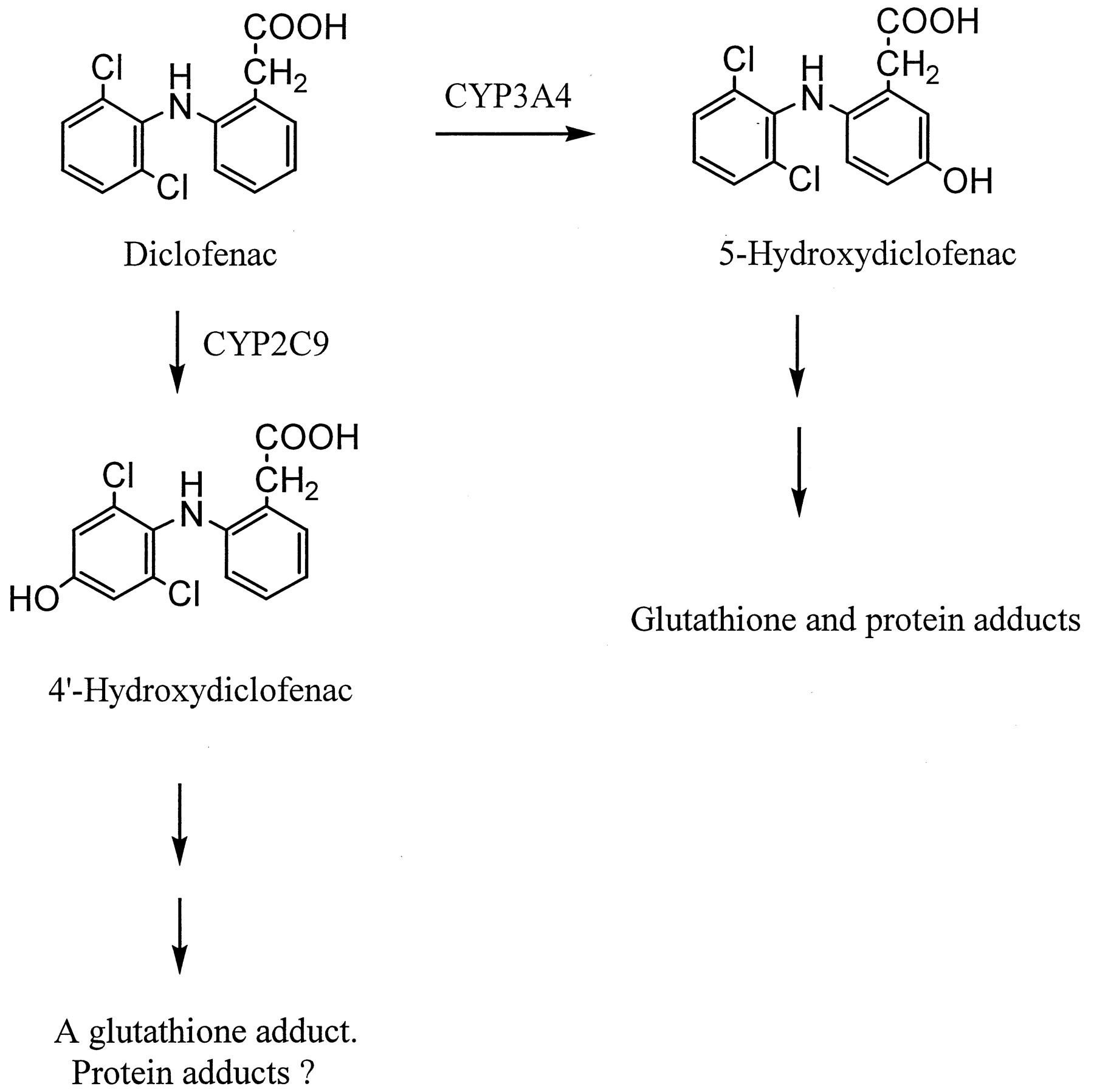
The use of diclofenac, a nonsteroidal anti-inflammatory drug, has been associated with rare but sometimes severe liver injury (Breen et al., 1986;Helfgott et al., 1990; Purcell et al., 1991). Analyses of more than 100 clinical cases suggest that the hepatotoxicity is idiosyncratic in nature and possibly related to the formation of reactive metabolite(s) (Banks et al., 1995; Boelsterli et al., 1995). Earlier metabolic studies in preclinical species and humans indicated that diclofenac biotransformation consisted mainly of two pathways, namely glucuronidation of the carboxylic acid group and hydroxylation of the two phenyl rings (Stierlin et al., 1979; Stierlin and Faigle 1979). Diclofenac acyl-glucuronide appears to be reactive, in that it covalently modifies cellular proteins, presumably via acyl migration followed by imine formation with free amino groups of proteins (Hargus et al., 1994; Kretz-Rommel and Boelsterli, 1994). On the other hand, the oxidative metabolism of diclofenac is catalyzed by cytochrome P450 (CYP),1 resulting primarily in 4′-hydroxy and 5-hydroxy derivatives (Fig. 1; Leemann et al., 1993; Shen et al., 1999; Tang et al., 1999c; Transon et al., 1996). In the urine of human subjects treated with diclofenac by oral dosing, the ratio of 4′-hydroxydiclofenac versus its 5-hydroxy analog was found to be approximately 3 to 4 (Stierlin and Faigle, 1979). Both 4′- and 5-hydroxydiclofenac have been implicated in the formation of benzoquinone imine intermediates that react further with glutathione or microsomal proteins (Fig. 1; Shen et al., 1999; Tang et al., 1999a,c).

CYP-catalyzed metabolism of diclofenac in humans.
The formation of 5-hydroxydiclofenac is stimulated in vitro by quinidine.
The 5-hydroxylation of diclofenac is unique in that, in certain cases, it can be stimulated by the presence of quinidine (Tang et al., 1999b,c). For example, this pathway in rhesus monkeys was shown to be CYP3A-dependent and exhibited a 2- to 3-fold increase in incubations with monkey liver microsomes or hepatocytes containing quinidine (Tang et al., 1999b). Consistent with these in vitro data, hepatic clearance of diclofenac in monkeys was enhanced after coadministration of quinidine (Tang et al., 1999b). In humans, the 5-hydroxylation of diclofenac is mainly catalyzed by CYP3A4 and to a much less extent by CYP2C9 and other isoforms (Shen et al., 1999; Tang et al., 1999c). The CYP3A4-catalyzed formation of 5-hydroxydiclofenac was observed to be stimulated more than 4-fold in human liver microsomal incubations containing quinidine (Tang et al., 1999c). In this report, we demonstrate that the CYP3A4-mediated interaction of diclofenac and quinidine stimulates the 5-hydroxylation of diclofenac but has no apparent effect on quinidine metabolism. This interaction was investigated in incubations with human liver microsomes and with human hepatocytes, and a kinetic model was proposed based on data generated with recombinant CYP3A4.
Experimental Procedures
Materials.
7,8-Benzoflavone, cinchonidine, cinchonine, chloroquine, cytochromec (from horse heart), diclofenac, glutathione (reduced form), mefenamic acid, NADPH, quinidine, and quinine were purchased from Sigma Chemical Co. (St. Louis, MO); 4′-hydroxydiclofenac was from Gentest Co. (Woburn, MA); 9-epiquinidine and 9-epiquinine were from Buchler GmbH (Braunschweig, Germany); hydroquinidine and hydroquinine were from Aldrich Chemical Co. (Milwaukee, WI); and cytochromeb5 was from PanVera Co. (Madison, WI). Recombinant CYP3A4, coexpressed with NADPH-CYP oxidoreductase (OR) and cytochrome b5(CYP3A4+OR+b5) or coexpressed with OR but no b5 (CYP3A4+OR), and 3-hydroxyquinidine were purchased from Gentest. According to the manufacture, the ratio of CYP3A4:b5 in the CYP3A4+OR+b5 system was 1:6 and the cytochrome c reductase activity was 1920 nmol/min/mg of protein. The cytochrome c reductase activity in the CYP3A4+OR system was 770 nmol/min/mg of protein. BondElut C18 extraction cartridge columns were obtained from Varian Chromatography Systems (Walnut Creek, CA), and Oasis MCX extraction plates were from Waters Co. (Milford, MA).
5-Hydroxydiclofenac was synthesized via conversion of 5-iododiclofenac to the corresponding arylboronic ester followed by oxidation with oxone in aqueous acetone (Tang et al., 1999a). Quinidine N-oxide was synthesized through oxidation of quinidine by 6% hydrogen peroxide (Guentert et al., 1982).
A monoclonal inhibitory antibody against human hepatic CYP3A4 was prepared in mice by immunization with baculovirus-expressed CYP3A4 (Mei et al., 1999).
Instrumentation.
Liquid chromatography-tandem mass spectrometry (LC/MS/MS) was carried out on a Perkin-Elmer Sciex API 3000 tandem mass spectrometer (Toronto, Canada) interfaced to an HPLC system consisting of a Perkin-Elmer Series 200 quaternary pump and a Series 200 autosampler (Norwalk, CT). LC/MS/MS experiments were performed using either a Heated Nebulizer interface or a Turbo IonSpray interface with positive ion detection.
With the Heated Nebulizer interface, the source temperature was set at 450°C, corona discharge at 3.0 μA, orifice potential at 39 V, and collision energy at 40 eV. The collision gas was nitrogen.
With the Turbo IonSpray interface, the source temperature was set at 150°C, ionization voltage at 5 kV, orifice potential at 50 V, and collision energy at 35 eV. The collision gas was nitrogen.
Incubations with Human Liver Microsomes and Recombinant CYP3A4.
Human liver samples were obtained from the Pennsylvania Regional Tissue Bank (Exton, PA). The medical history of the donors, including disease state, alcohol consumption, and substance abuse, were provided in an earlier report (Tang et al., 1999c). Liver microsomes were isolated from individual livers by differential centrifugation (Raucy and Lasker, 1991). Aliquots from each preparation were pooled on the basis of equivalent protein concentrations. Specific CYP activities in these microsomal preparations were reported previously (Newton et al., 1995).
Diclofenac and glutathione in phosphate buffer (pH 7.4) and quinidine or its analogs in methanol were added to human liver microsomes (0.5 nmol of CYP/ml) or recombinant CYP3A4 (CYP3A4+OR+b5, 10 pmol of CYP/ml) suspended in phosphate buffer (0.1 M, pH 7.4) containing EDTA (1 mM). Diclofenac concentrations ranged from 0 to 500 μM and quinidine from 0 to 200 μM. The concentration of glutathione was 5 mM, and that of methanol was 0.2% (v/v). Controls lacked quinidine but contained the same amount of methanol. The mixture was incubated at 37°C for 5 min, and then NADPH in phosphate buffer was added to a final concentration of 1 mg/ml. After an additional 5-min incubation, the reaction was quenched with 10% aqueous trifluoroacetic acid.
Similar incubations were performed with recombinant CYP3A4 expressed without cytochrome b5 (CYP3A4+OR, 0.1 nmol of CYP/ml). Comparisons were made between incubations containing cytochrome b5(b5/CYP ratio of 1) with those lacking this protein.
Immunoinhibition experiments were carried out with a monoclonal antibody against CYP3A4. Liver microsomes or recombinant CYP3A4 were preincubated with the antibody (5–40 mg of IgG/nmol of CYP) for 15 min at room temperature. Control incubations contained IgG from untreated animals. Diclofenac, quinidine, glutathione, and NADPH were added thereafter, and the incubations were performed in a manner similar to that described above.
All experiments were performed in duplicate.
Incubations with Human Hepatocytes.
The human liver samples were obtained from the Pennsylvania Regional Tissue Bank. The death of one donor, a 25-year-old male, was caused by an accident; the second donor, a 63-year-old female, died from subarachnoid hemorrhage; the third donor was a 41-year-old male who died from head trauma. Hepatocytes were isolated based on a two-step perfusion procedure (Pang et al., 1997) and exhibited a viability of greater than 80% as determined by the trypan blue exclusion test. The cells then were suspended in Krebs-bicarbonate buffer. Diclofenac in Krebs-bicarbonate buffer was added to the suspension to provide a final drug concentration of 100 μM, following which quinidine in aqueous solution was added to give a concentration range of 25 to 200 μM. Control incubations lacked quinidine. After incubation for 2 h at 37°C, the suspension medium was acidified with 10% aqueous trifluoroacetic acid. The experiment was performed in duplicate.
Detection and Quantification of Metabolites of Diclofenac and Quinidine.
Aliquots (50 μl) from incubations with recombinant CYP3A4 were mixed with mefenamic acid, cinchonine (50 ng, internal standards), and 4 M urea (1 ml) and applied to an Oasis MCX extraction plate, which was prewashed with methanol and water. The cartridge was washed consecutively with water and 90% aqueous acetonitrile (300 μl) containing 0.1% trifluoroacetic acid, and the acetonitrile eluate was analyzed by LC/MS/MS.
For analyses of 5-hydroxydiclofenac and 3-hydroxyquinidine, LC/MS/MS was operated using the Heated Nebulizer interface with multiple reaction monitoring. Transitions monitored were m/z 312.2 → 230.3 (5-hydroxydiclofenac), 340.6 → 159.6 (3-hydroxyquinidine), 242.3 → 208.8 (mefenamic acid), and 294.6 → 165.6 (cinchonine). Chromatography was performed on a Keystone Scientific (Wilmington, DE) Betasil C8 column (4.6 × 50 mm, 5 μm), and samples were delivered at a flow rate of 1 ml/min. The mobile phase consisted of 90% aqueous acetonitrile containing 1 mM ammonium acetate and 0.1% trifluoroacetic acid. Standard curves were generated over a range of 10 to 10,000 ng/ml.
For analyses of quinidine N-oxide, LC/MS/MS was operated in the IonSpray mode with multiple reaction monitoring. Transitions monitored were m/z 341.0 → 136.0 (quinidineN-oxide), 341.0 → 226.0 (3-hydroxyquinidine), and 295.0 → 166.0 (cinchonine). Chromatography was performed on a Jones Chromatography (Lakewood, CO) Genesis C8 column (4.6 × 150 mm, 3 μm), and samples were delivered at a flow rate of 1 ml/min with a 1:25 split to the mass spectrometer. The mobile phase consisted of 40% aqueous methanol containing 6 mM ammonium acetate and 0.06% trifluoroacetic acid. Standard curves were generated over a range of 100 to 10,000 ng/ml.
Aliquots (50 μl) of samples from incubations with human liver microsomes or hepatocytes were mixed with mefenamic acid (50 ng, internal standard) and applied to a BondElut C18 extraction cartridge column, that was prewashed with methanol and water. The column was washed consecutively with water and methanol. The methanol eluate was evaporated to dryness under a stream of nitrogen, the residue was reconstituted in 60% aqueous acetonitrile (300 μl) containing 0.05% trifluoroacetic acid, and the resulting sample was analyzed by LC/MS/MS (Heated Nebulizer interface) with multiple reaction monitoring. Transitions monitored were m/z 312.2 → 230.3 (5-hydroxydiclofenac) and 242.3 → 208.8 (mefenamic acid). Chromatography was performed on a DuPont Zorbax (Wilmington, DE) Rx-C8 column (4.6 × 250 mm, 5 μm), and samples were delivered at a flow rate of 1 ml/min. The mobile phase consisted of aqueous acetonitrile containing 10% methanol and 0.05% trifluoroacetic acid; a linear gradient was employed in which the acetonitrile content increased from 10 to 70% during a 30-min period. Data are expressed relative to controls.
Determination of OR Activities in Recombinant Systems.
Cytochrome c (50 μM) and the recombinant microsomes (CYP3A4+OR; 0.1 mg of protein/ml) were suspended in phosphate buffer (1 M, pH 7.7). Quinidine in methanol was added such that the final concentration of quinidine was 100 μM and that of methanol was 0.2% (v/v). Controls contained no quinidine. NADPH at a final concentration of 100 μM was added to initiate the reaction. The reduction of cytochrome c was monitored at 550 nm, and enzyme activities were calculated based on the extinction coefficient of 2.1 × 104 M−1cm−1 (Van Gelder and Slater, 1962). The experiment was performed in duplicate.
Kinetic Calculations.
Apparent Km andVmax values were calculated according to the Michaelis-Menten equation.
Kinetic parameters also were calculated based on a model containing two distinct binding domains in the active site of the CYP enzyme (Fig.2). The active site was defined as the area within which substrates interact with activated oxygen (Shou et al., 1994). The velocity equations derived from this model, assuming rapid equilibrium between the enzyme and substrates, were as follows:

Proposed kinetic model with two substrate binding sites in the active site of CYP3A4 for interactions of diclofenac and quinidine with the enzyme.
Results
Diclofenac Metabolism in Incubations with Human Liver Microsomes.
Monohydroxylated metabolites of diclofenac were identified by LC/MS/MS based on coincidence of HPLC retention times and product ion spectra with those of the authentic standards. The formation of 5-hydroxydiclofenac in incubations with human liver microsomes was linear over a period of 15 min, and all studies were performed therefore following 5-min incubations.
The formation of 5-hydroxydiclofenac increased 6-fold relative to controls when incubations were performed in the presence of quinidine (Table 1). Similar stimulation was observed with quinine, epiquinidine, epiquinine, cinchonidine, and cinchonine (Table 1). Quinidine and quinine represent a pair oferythro diastereoisomers and epiquinidine and epiquinine are the corresponding threo epimers. Cinchonidine and cinchonine are another pair of diastereoisomers. Diclofenac metabolism also was enhanced by 3-hydroxyquinidine, a metabolite of quinidine, but was not enhanced by chloroquine, which may be viewed as a distant structural analog of quinidine. Interestingly, the formation of 5-hydroxydiclofenac was inhibited by 7,8-benzoflavone (Table 1), although this flavone derivative is known to be an activator for a number of CYP3A4-mediated reactions (Buening et al., 1981; Kerr et al., 1994). The structure of the CYP3A4 modulators are shown in Fig.3.
Effects of quinidine and quinidine analogs on the formation of 5-hydroxydiclofenac in incubations with human liver microsomes or recombinant CYP3A41-a

Structures of the CYP3A4 modulators examined for their effect on diclofenac metabolism.
The conversion of diclofenac to its 5-hydroxy derivative was inhibited approximately 80% by pretreatment of human liver microsomes with a monoclonal antibody against CYP3A4, suggesting that CYP3A4 was a major contributor to that metabolic pathway (Table2). The 5-hydroxylation of diclofenac also was inhibited by the monoclonal antibody against CYP3A4 when incubations were performed in the presence of quinidine (Table 2). In fact, the amount of 5-hydroxydiclofenac formed in incubations containing both the antibody and quinidine was essentially the same as that formed in incubations containing only the antibody (data not shown). It appeared, therefore, that diclofenac metabolism was not influenced by quinidine once CYP3A4 was inhibited.
Inhibition of diclofenac metabolism in incubations with human liver microsomes or recombinant CYP3A42-a
Diclofenac Metabolism in Incubations with Recombinant CYP3A4.
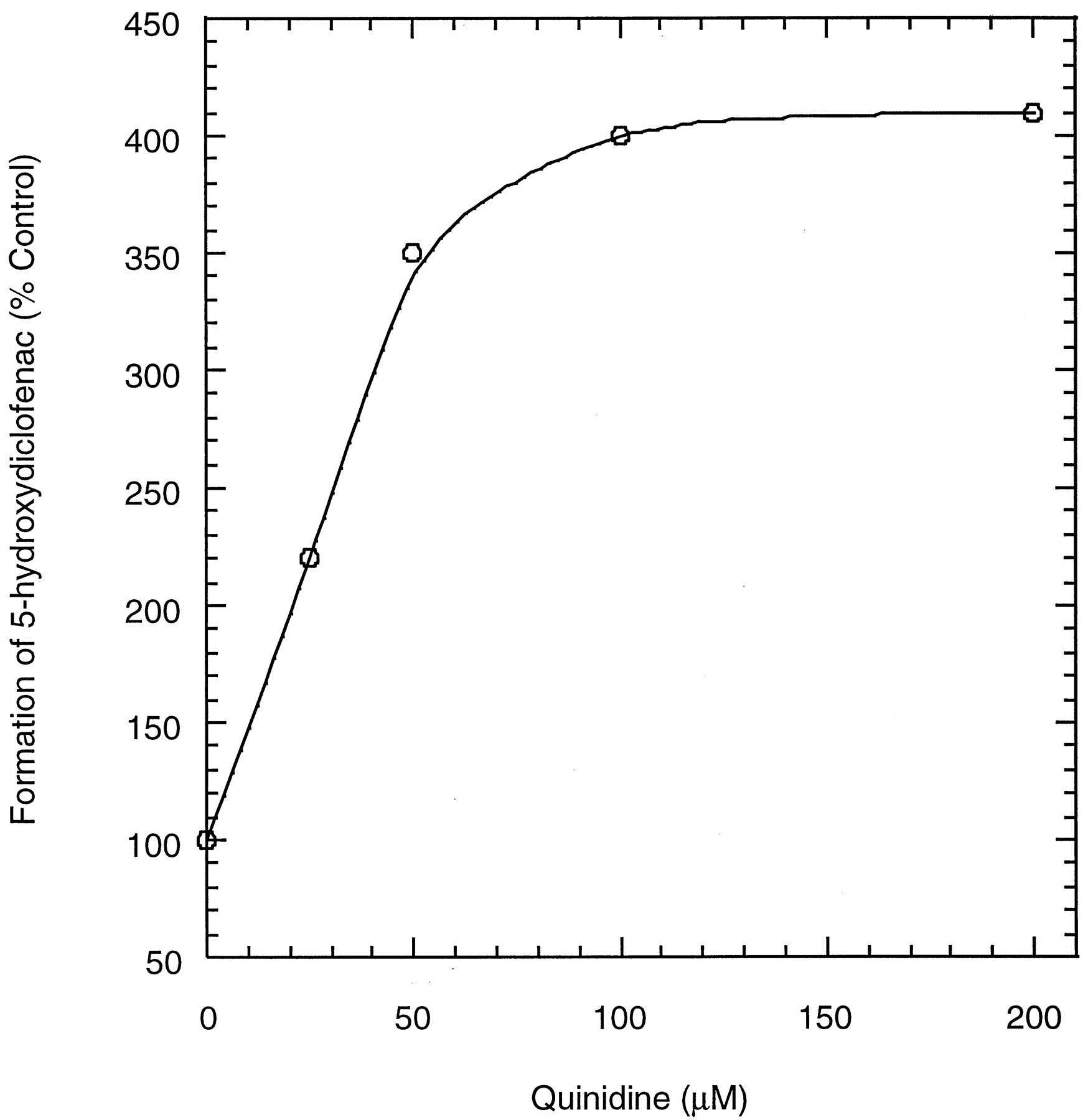
Diclofenac metabolism in incubations with recombinant CYP3A4 coexpressed with cytochrome b5(CYP3A4+OR+b5) produced only 5-hydroxydiclofenac. This biotransformation was stimulated when incubations were performed in the presence of quinidine, such that the formation of 5-hydroxydiclofenac increased with increasing quinidine concentrations (Fig. 4). Based on these profiles, the concentration of quinidine required to produce 50% of the maximum stimulatory effect (EC50) was estimated to be ∼1 μM.

Formation of 5-hydroxyldiclofenac in incubations of diclofenac with recombinant CYP3A4(+OR+b5) at various quinidine concentrations.
Diclofenac concentrations were 30 and 100 μM.
Subsequent kinetic studies of diclofenac metabolism were carried out over a quinidine concentration range of 0 to 30 μM. In the absence of quinidine, the Km andVmax values for the formation of 5-hydroxydiclofenac were estimated at 71 μM and 13.2 nmol/min/nmol of CYP. When quinidine was included in incubations, the Vmax of the reaction increased and maximized at ∼56 nmol/min/nmol of CYP, which represented a 4.5-fold elevation over controls (Table3). The Kmvalue, on the other hand, changed little, ranging from 44 to 69 μM (Table 3).
Influence of quinidine on diclofenac metabolism versus influence of diclofenac on quinidine metabolism in incubations with recombinant CYP3A43-a
Incubations also were performed with recombinant CYP3A4 expressed without cytochrome b5 (CYP3A4+OR), and a comparison was made for the effect of quinidine between incubations containing cytochrome b5 versus those that lacked b5. The rate of 5-hydroxydiclofenac formation in these incubations, when normalized based on CYP concentration, was ∼50-fold lower than in incubations with CYP3A4 coexpressed with cytochrome b5 (data not shown). Although addition of cytochrome b5to the incubations produced more 5-hydroxydiclofenac, the increase was only 2-fold that of controls (data not shown). On the other hand, the same biotransformation was stimulated by the presence of quinidine, resulting in a 9-fold increase in metabolite formation. However, the enhancement of diclofenac metabolism in those incubations containing both cytochrome b5 and quinidine was nearly equivalent to the combined effect of b5 and quinidine (Table 4). In fact, the stimulatory effect of quinidine appeared to be weaker in incubations containing CYP3A4 coexpressed with cytochromeb5 than in those containing CYP3A4 expressed without b5 (Table 4). These data suggest that cytochrome b5 is not required for stimulation of diclofenac metabolism by quinidine.
Influence of quinidine on diclofenac metabolism in the presence or absence of cytochrome b54-a
Quinidine Metabolism in Incubations with Recombinant CYP3A4.
Quinidine metabolites were identified by LC/MS/MS (IonSpray). 3-Hydroxyquinidine and quinidine N-oxide were the major products of quinidine metabolism in incubations with recombinant CYP3A4, with the Km andVmax values estimated at 1.3 μM and 5.1 nmol/min/nmol of CYP and 54.1 μM and 3.8 nmol/min/nmol of CYP, respectively. These data indicate that 3-hydroxylation represents the primary route for quinidine metabolism, particularly in a low substrate concentration range (less than 30 μM) wherein the maximum stimulation of diclofenac metabolism was observed. Therefore, a subsequent investigation of the effect of diclofenac on quinidine metabolism was carried out by examining the 3-hydroxylation pathway.
Kinetic experiments of quinidine 3-hydroxylation were performed in the presence of diclofenac over a concentration range of 20 to 600 μM. In contrast to the stimulatory effect of quinidine on diclofenac metabolism, no effect of diclofenac was observed on the 3-hydroxylation of quinidine. Neither Km (∼1.5 μM) norVmax (∼5.5 nmol/min/nmol of CYP) changed significantly relative to controls (Table 3).
CYP Oxidoreductase Activities in Recombinant Systems.
The OR activities in recombinant CYP3A4 coexpressed with OR (CYP3A4+OR) were measured by cytochrome c reduction. The enzyme activity was estimated at 576 nmol/min/mg of protein. In the presence of 100 μM quinidine, the OR activity was little influenced, being 496 nmol/min/mg of protein.
Kinetic Model for the Interaction of Diclofenac and Quinidine with CYP3A4.
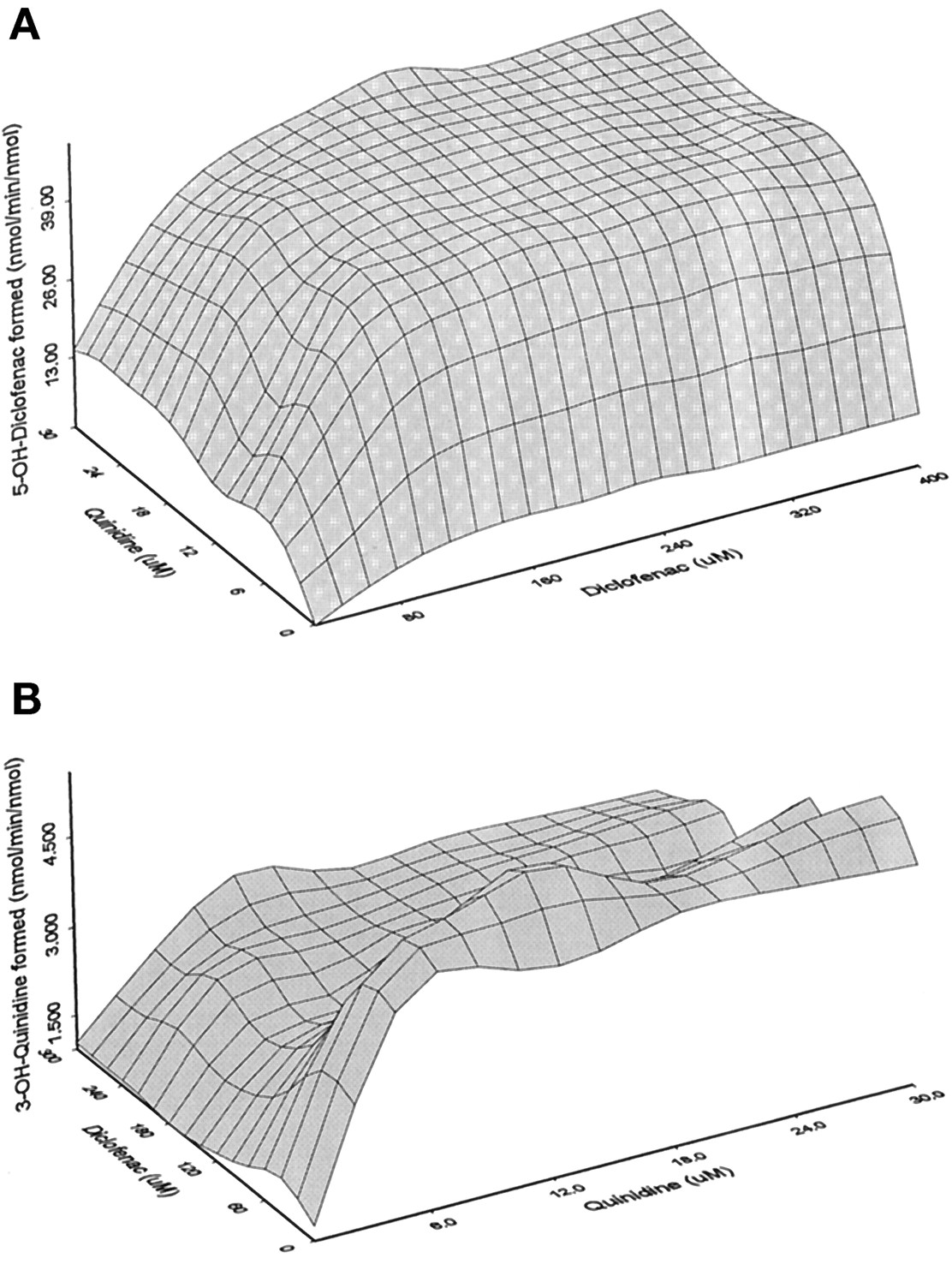
The proposed kinetic scheme for the interaction of diclofenac and quinidine with CYP3A4 (Fig. 2) was a simplified version of the Two Substrates-Two Binding Sites model reported earlier (Korzekwa et al., 1998), assuming 1) rapid equilibrium; 2) fast release of product(s); and 3) two independent substrate binding sites, one for diclofenac and another for quinidine. Experimental data were fit to the equations derived from the model and resulting velocities (z axis) were plotted against both substrate (x axis) and effector (y axis) concentrations (Fig.5). On this basis, the dissociation constants were estimated at 60 μM for the equilibrium between diclofenac and CYP3A4 and 1.3 μM for the equilibrium between quinidine and the enzyme (Table 5). These constants remained unchanged in the presence of a second substrate or an effector (ES1 ⇌S2ES1 ⇌S2E; Figs. 2 and 5). The velocity for the formation of 3-hydroxyquinidine from the quinidine-CYP3A4-diclofenac complex was similar to that from the quinidine-CYP3A4 complex; in other words, the metabolism of quinidine was not influenced by the presence of diclofenac (Fig. 5B and Table 5). The formation of 5-hydroxydiclofenac, on the other hand, not only was dependent on substrate (diclofenac) concentrations but also increased with increasing effector (quinidine) concentrations (Fig. 5A). The maximal velocity for the formation of 5-hydroxydiclofenac from the diclofenac-CYP3A4-quinidine complex increased 6-fold compared with that from the diclofenac-CYP3A4 complex (Table 5).

Parameters generated from a kinetic model for the interaction of diclofenac and quinidine with CYP3A45-a
Stimulation of Diclofenac Metabolism in Human Hepatocyte Suspensions.
Two monohydroxylated metabolites, namely, 4′-hydroxydiclofenac and 5-hydroxydiclofenac, were identified by LC/MS/MS in human hepatocyte suspensions with diclofenac. When incubations were performed in the presence of quinidine, the formation of the 5-hydroxylated derivative increased with increasing quinidine concentrations, and an approximately 4-fold increase, which appeared to represent the maximal augmentation, was reached at 100 μM quinidine (Fig.6). The formation of 4′-hydroxydiclofenac in human hepatocyte suspensions was not affected by the presence of quinidine (data not shown).
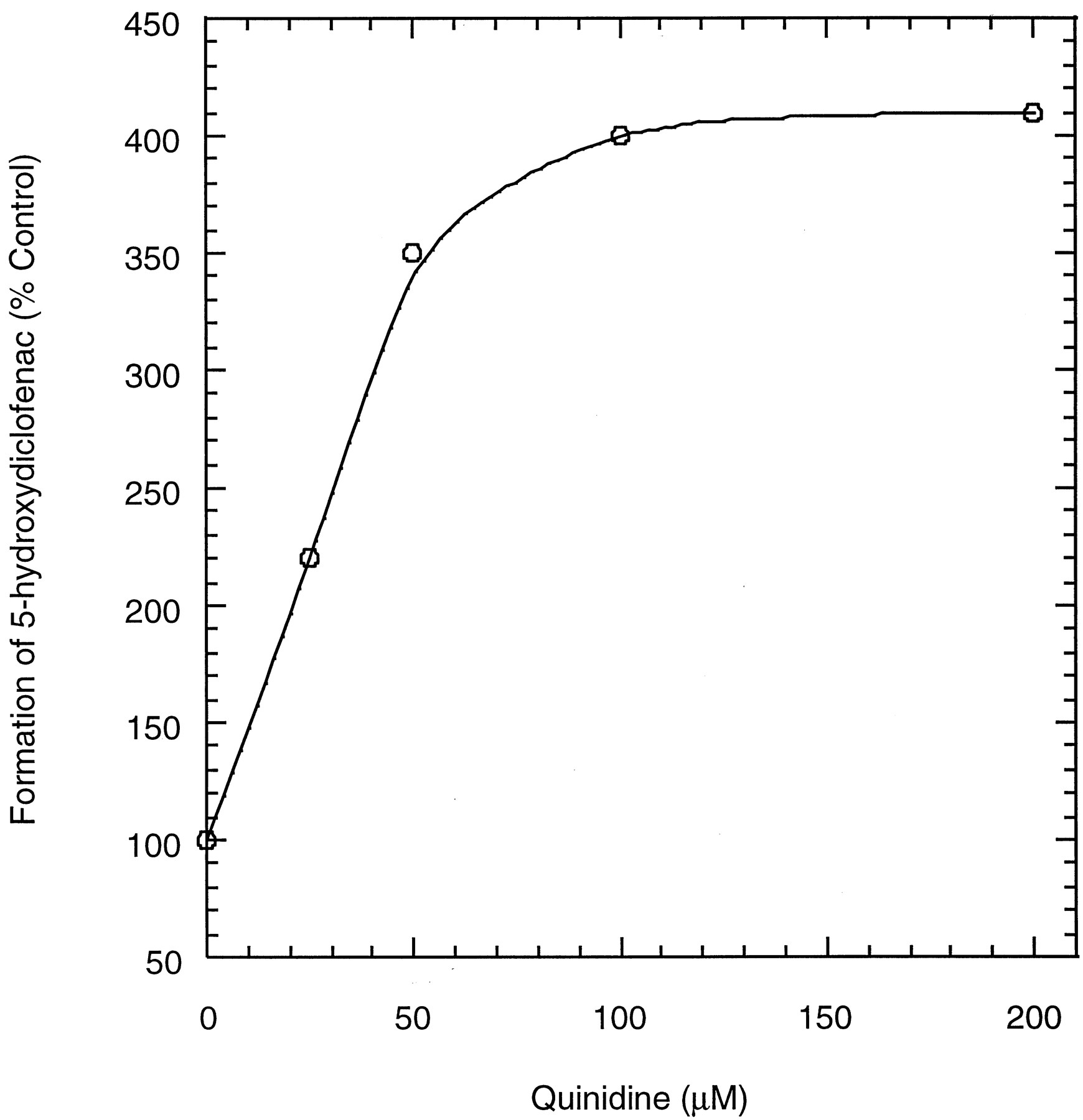
Formation of 5-hydroxydiclofenac from diclofenac in human hepatocyte suspensions containing quinidine.
The diclofenac concentration was 100 μM. Data are expressed relative to controls.
Discussion
It is known that CYP-mediated drug metabolism can be stimulated by the presence of certain xenobiotic compounds (Guengerich, 1997; Szklarz and Halpert, 1998). For example, the oxidation of aflatoxin B1, benzo(a)pyrene, and carbamazepine was shown to be enhanced by flavonoids (Buening et al., 1981; Kerr et al., 1994). A similar effect with caffeine was observed on acetaminophen bioactivation in incubations with rat liver microsomes (Lee et al., 1991). The data presented here provide another example of this phenomenon, wherein diclofenac metabolism was stimulated by quinidine.
In this case, the formation of 5-hydroxydiclofenac increased 6-fold when incubations with human liver microsomes were performed in the presence of quinidine. Such an enhancement appeared to be mediated by the CYP3A4 isoform, because 1) the stimulation was observed in incubations with recombinant CYP3A4 and 2) the stimulatory effect was diminished when human liver microsomes or recombinant CYP3A4 was pretreated with an inhibitory antibody against CYP3A4. In addition, it was possible to exclude a role for cytochromeb5 in the stimulation, because the effect of quinidine was maximal in the absence of cytochromeb5. With respect to OR, its activity, as measured by cytochrome c reduction, was not influenced by quinidine. Thus, it appears that the stimulatory effect of quinidine on diclofenac metabolism is produced through the interaction of quinidine with CYP(3A4) protein. A similar stimulation also was observed with diastereoisomers of quinidine, including quinine and thethreo epimers, indicating that the augmentation of CYP3A4 activities was insensitive to activator stereochemistry in these cases.
Subsequent kinetic analyses of diclofenac metabolism in recombinant CYP3A4 systems indicated that Vmax values for the formation of the 5-hydroxylated derivative increased 4.5-fold (from 13.2 to 57.6 nmol/min/nmol of CYP) with little change inKm (71–56 μM) over a quinidine concentration range within which the stimulation was maximized. Conversely, the CYP3A4-catalyzed metabolism of quinidine was not affected by the presence of diclofenac. The estimatedKm value for the formation of 3-hydroxyquinidine (1.5 μM) was similar to the quinidine concentration required to produce 50% of the maximum stimulatory effect on diclofenac metabolism. Taken together, these results suggest that the catalytic capacity of CYP3A4 for diclofenac metabolism is enhanced by quinidine, whereas diclofenac does not interfere with quinidine's access to the ferriheme-oxygen complex. The binding site in CYP3A4 for quinidine 3-hydroxylation probably is identical with that for its stimulatory effect, and both diclofenac and quinidine may be present in the active site of CYP3A4 at the same time.
Accordingly, a kinetic model with two distinct substrate binding domains in the active site was developed to illustrate the interaction of diclofenac and quinidine with CYP3A4. In this model, the binding of diclofenac to CYP3A4 was characterized as independent from quinidine and vice versa; the dissociation constant for either substrate remained unchanged when the second substrate (which also was an effector) was bound to the enzyme. The production of 5-hydroxydiclofenac was enhanced once quinidine occupied its binding site on CYP3A4, whereas the formation of 3-hydroxyquinidine was unaffected when diclofenac was present in the active site.
This model is similar to that proposed in earlier reports for the CYP3A4-mediated interaction of 7,8-benzoflavone and phenanthrene, wherein both substrates were postulated to be present simultaneously at the active site of the enzyme (Shou et al., 1994; Korzekwa et al., 1998). An analogous model containing a substrate binding domain and an effector binding domain in the active site of CYP3A4 also was proposed (Ueng et al., 1997; Harlow and Halpert, 1998), and the supporting data were provided by site-directed mutagenesis studies (Harlow and Halpert, 1998). On the other hand, an allosteric model with two distinct binding sites was employed to rationalize the interaction between 7,8-benzoflavone and aflatoxin B1 (Ueng et al., 1997). These models are not mutually exclusive, their major difference being the proximity of two binding sites (Ueng et al., 1997). Although our data are not conclusively in favor of one model over the other, the effector quinidine in this case appeared to have access to the reactive oxygen from its binding domain, and this access is not hindered by the presence of diclofenac. There is also a possibility that two binding sites exist with similar affinities for quinidine; one in the active site for quinidine metabolism and the other for the allosteric effect. Such a scenario would be similar to a triple occupancy model involving two substrate and one effector binding domains (Domanski et al., 2000). The remaining issue is the localization of the effector. It ought to be noted that 7,8-benzoflavone is an activator in those models dealing with CYP3A4 cooperativity but is an inhibitor for the 5-hydroxylation of diclofenac. This may be indicative of the distinction between the flavone and quinidine in terms of their mechanisms for stimulation.
The stimulatory effect of quinidine on diclofenac metabolism may be clinically relevant, because a ∼4-fold increase in 5-hydroxydiclofenac formation was observed in human hepatocyte suspensions containing diclofenac and quinidine. It has been reported that the stimulation of diclofenac metabolism in monkey hepatocyte suspensions correlated with an in vivo increase in hepatic clearance of the drug following coadministration with quinidine (Tang et al., 1999b). Thus, this type of drug-drug interaction, namely enhancement of the metabolism of drug A due to the presence of drug B, may occur in patients who are on multiple medications.
In summary, the CYP3A4-mediated metabolism of diclofenac in vitro is stimulated by quinidine. This stimulation is characterized by increases in Vmax with little change inKm for the formation of 5-hydroxydiclofenac. The metabolism of quinidine, however, is not influenced by diclofenac. Because quinidine is an inhibitor of CYP3A4 in many cases, current studies are underway to investigate the underlying mechanism of the interactions between quinidine and CYP3A4.
Acknowledgments
We thank J. Pang for isolation of human hepatocytes, and R. Wang, Drs. S.-H. Chiu. and S. Huskey for critical reading of the manuscript. We also thank Dr. A. Lu (Rutgers University) for valuable discussions.
Footnotes
-
Send reprint requests to: Wei Tang, Ph.D., Department of Drug Metabolism, Merck & Co., PO Box 2000, RY800-B211, Rahway, NJ 07065. E-mail: wei_tang{at}merck.com
- Abbreviations used are::
- CYP
- cytochrome P450
- LC/MS/MS
- liquid chromatography-tandem mass spectrometry
- OR
- NADPH-cytochrome P450 oxidoreductase
- Received April 3, 2000.
- Accepted June 1, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}