


Abstract
Fa2N-4 cells have been proposed as a tool to identify CYP3A4 inducers. To evaluate whether Fa2N-4 cells are a reliable surrogate for cryopreserved human hepatocytes, we assessed the basal mRNA expression of 64 drug disposition genes in Fa2N-4 cells. Significant differences were found in the expression of major drug-metabolizing enzymes, nuclear receptors, and transporters between both cell types. Importantly, the expression of constitutive androstane receptor (CAR) and several hepatic uptake transporters was significantly lower (>50-fold) in Fa2N-4 cells, whereas the expression of pregnane X-receptor (PXR) and aryl hydrocarbon receptor (AhR) was similar between Fa2N-4 cells and human hepatocytes. By using an optimized induction assay for Fa2N-4 cells, CYP3A4 induction by rifampicin, the prototypical PXR activator, increased from 1.5- to 7-fold at the level of functional activity. With nine selected compounds, which are known inducers of CYP3A4 either via activation of PXR, CAR, or both, we evaluated CYP3A4 and CYP2B6 mRNA induction using Fa2N-4 cells and human hepatocytes. No response was observed in Fa2N-4 cells treated with the selective CAR activators 6-(4-chlorophenyl)imidazo[2,1-b][1,3]-thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime and artemisinin. CYP3A4 and CYP2B6 induction in Fa2N-4 cells were also low for phenytoin, phenobarbital, and efavirenz, which are dual activators of PXR/CAR. This finding was in agreement with the lack of expression of CAR. The EC50 value for rifampicin-mediated CYP3A4 induction was 10-fold higher than that in human hepatocytes. This result could be attributed to the low expression of hepatic organic anion-transporting polypeptides OATP1B1 and OATP1B3 in Fa2N-4 cells. In summary, our findings identify limitations of Fa2N-4 cells as a predictive induction model.
Drug metabolism occurs primarily in the liver via cytochrome P450 (P450) enzymes. CYP3A4 is one of the predominant P450 isoforms expressed in adult liver. It catalyzes the metabolism of more than 50% of clinically used drugs (Schuetz, 2004). Induction of CYP3A4 can result in clinically significant drug-drug interactions or autoinduction (Lin, 2006) and, therefore, is a major concern for the pharmaceutical industry.
Significant progress has been made in understanding the regulation of drug-metabolizing enzymes. Two members of the gene superfamily of nuclear hormone receptors (Schuetz et al., 1993; Mangelsdorf et al., 1995), namely the pregnane X receptor (PXR) and the constitutive androstane receptor (CAR), were identified as key transcription factors in hepatic P450 induction by xenobiotics (Blumberg et al., 1998; Kliewer et al., 1999). These nuclear receptors function as ligand-dependent transcription factors by binding to specific DNA sequences called response elements within the promoter region of genes such as CYP3A4. PXR and CAR heterodimerize with the retinoid X receptor and bind to the above response elements and activate target gene expression (Mangelsdorf et al., 1990). Recent studies demonstrated a considerable overlap of these two receptors both in terms of the spectrum of genes regulated and their affinity to DNA-response elements (Chen et al., 2005).
A number of in vitro models have been developed to assess the induction potential of drug candidates. Nuclear receptor-based assays, such as PXR assays, have the potential of screening large numbers of compounds that are activators of PXR (Ogg et al., 1997). However, they run the risk of false-negative results because multiple mechanisms, e.g., non-PXR-mediated activation, may contribute to the induction of drugs or induction caused by metabolites not formed in cell lines. Primary or cryopreserved human hepatocytes have been considered as the standard in vitro models to evaluate the propensity of drug candidates to cause induction (Hewitt et al., 2007). The utility of such hepatocytes is, however, restricted by the limited and erratic supply, and the significant interindividual variability in the expression of drug-metabolizing enzymes (Hewitt et al., 2007). These limitations have led to the search for alternative systems.
Recently, Fa2N-4, a Simian virus 40 immortalized human hepatocyte cell line, has been developed by MultiCell Technologies (Lincoln, RI) (Mills et al., 2004). Initial studies by Mills et al. (2004) have shown that multiple P450 enzymes, including CYP3A4, CYP1A2, and CYP2C9, are inducible in Fa2N-4 cells after exposure to several prototypical inducers. In addition, expression of PXR and AhR, which are important transcription factors involved in the regulation of drug-metabolizing enzymes and transporters, have been detected in Fa2N-4 cells (Mills et al., 2004). Ripp et al. (2006) further characterized Fa2N-4 cells by evaluating the effect of selected compounds on CYP3A4 induction and comparing in vitro induction data with in vivo induction response. It was suggested that Fa2N-4 cells could be used to identify CYP3A4 inducers as well as to predict the magnitude of clinical drug-drug interactions. Given that Fa2N-4 cells have a morphology similar to that of human hepatocytes, maintain the inducibility of various drug-metabolizing enzymes, and are easily maintained and propagated, Fa2N-4 cells have been proposed as a tool to identify CYP3A4 inducers in drug discovery and development. In the draft Guidance for Industry issued by the U.S. Food and Drug Administration (http://www.fda.gov/cder/guidance/6695dft.pdf), the use of immortalized cells, as a P450 induction model, is considered acceptable provided that it can be demonstrated with positive controls that CYP3A4 is inducible in these cell lines.
However, it remains unclear whether Fa2N-4 cells are a reliable surrogate for primary or cryopreserved human hepatocytes. For instance, CYP3A4 induction by selective CAR activators was not evaluated in Fa2N-4 cells (Ripp et al., 2006). Furthermore, the expression of major drug-metabolizing enzymes, nuclear receptors, and hepatic transporters in these cells are also not known. This information is critical to understand the molecular mechanism of CYP3A4 induction in Fa2N-4 cells.
The objectives of our studies were 1) to compare the basal expression of drug disposition genes in Fa2N-4 cells and cryopreserved human hepatocytes, 2) to optimize the culture and induction conditions to attain a good CYP3A4 functional activity window, and 3) to compare CYP3A4 and CYP2B6 induction potency in Fa2N-4 cells and human hepatocytes for selected compounds that are known inducers of CYP3A4 either via potential activation of PXR or CAR or both.
Materials and Methods
Materials. Rifampicin, artemisinin, phenytoin, phenobarbital, and CITCO were purchased from Sigma-Aldrich (St. Louis, MO). All other compounds were synthesized by the Merck Chemistry Department (West Point, PA) and were of the highest purity. RNeasy 96 Kits were purchased from QIAGEN (Valencia, CA). The Quant-iT Ribogreen RNA Reagent Kit was purchased from Molecular Probes, Invitrogen Detection Technologies (Carlsbad, CA). Universal PCR Master Mix and Low Density Microarrays were purchased from Applied Biosystems (Foster City, CA). Collagen-coated 48-well plates were purchased from BD Biosciences (San Jose, CA). All other reagents and chemicals were of the highest grade and purchased from either Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich.
Cells. Cryopreserved Fa2N-4 cells, MFE support medium, and MFE plating medium were purchased from XenoTech LLC (Lenexa, KS). Cryopreserved human hepatocytes (LHO, female Caucasian 68 years old; 455, female Caucasian 37 years old; 527, female Caucasian 36 years old; and DMQ, African-American female 59 years old), antibiotic/antimycotic Torpedo Mix, In Vitro-GRO-CP media and In Vitro-GRO-HI incubation media were obtained from In Vitro Technologies (Baltimore, MD) or XenoTech LLC, respectively.
Optimization of CYP3A4 and CYP1A2 Induction in Fa2N-4 Cells. CYP3A4 and CYP1A2 induction studies in Fa2N-4 cells were optimized from the manufacturer's protocol (http://www.multicelltech.com/pdfs/5.pdf). CYP3A4 and CYP1A2 induction was assessed on the basis of both mRNA expression and enzymatic activity. Rifampicin and omeprazole, prototypical PXR and AhR activators (LeCluyse, 2001; Yueh et al., 2005), were used to evaluate CYP3A4 or CYP1A2 induction. Testosterone (250 μM) or phenacetin (100 μM) was selected as the probe substrate to measure CYP3A4-mediated testosterone 6β-hydroxylation and CYP1A2-mediated phenacetin de-ethylation, respectively. In our studies, the following conditions were evaluated at both mRNA and activity levels to increase the induction window: A) cells were plated at different plating densities ranging from 75,000 to 130,000 cells/well; B) the probe substrate incubation period was assessed over a period ranging from 1 to 4 h; C) P450 induction was assessed in the presence and absence of the 48-h adaptation period; and D) functional activity was compared between 48 and 72 h of drug treatment. The optimized protocol is described in detail below.
Cell Culture and Induction Studies of Fa2N-4 Cells and Cryopreserved Human Hepatocytes. Fa2N-4 cells (95,000 cells/well) or human hepatocytes (120,000 cells/well) were plated in collagen-coated 48-well plates. Cells were maintained at 37°C with 95% humidity and 5% CO2. After cell attachment (approximately 3 h), serum-containing plating medium was replaced with serum-free culture medium. Fa2N-4 cells were incubated for 48 h (adaptation period) before treating with test compounds. This adaptation period was necessary for the cells to exhibit a robust induction response. Cryopreserved human hepatocytes were allowed an adaptation period of 24 h. Stock solutions of test compounds were prepared in dimethyl sulfoxide (DMSO) and diluted 1000-fold with serum-free medium for drug treatment. After 24 h of treatment with test compounds or vehicle control (0.1% DMSO, v/v), medium was discarded and replaced with fresh medium containing test compounds. This was repeated every 24 h during the 48-h drug incubation period for human hepatocytes and the 72-h drug incubation period for Fa2N-4 cells. At the end of the incubation, drug-containing medium was discarded, and the cells were washed with drug-free medium. Cells were then exposed to the probe substrates 250 μM testosterone (CYP3A4) or 100 μM phenacetin (CYP1A2) for 1 h. The media were collected, and the metabolites produced by testosterone 6β-hydroxylation or phenacetin de-ethylation were quantitated using liquid chromatography/tandem mass spectrometry as described previously (Prueksaritanont et al., 2005). The remaining cells were used to isolate mRNA, which was subjected to quantitative PCR to assess changes in CY3A4, CYP2B6, and CYP1A2 mRNA expression. Rifampicin (10 μM) was used as the positive control for both CYP3A4 and CYP2B6, whereas omeprazole (50 μM) functioned as the positive control for CYP1A2.
Cytotoxicity Assessment. Cytotoxicity studies were performed in parallel with the WST-1 Cell Proliferation kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's protocol. Briefly, after the 72-h drug treatment period, WST1 reagent was added to cell culture medium (1 part reagent to 10 parts culture medium), and mixed gently on an orbital shaker for 1 min. Cells were then returned to the incubator for 2 h (37°C at 5% CO2). Thereafter, plates were shaken gently on an orbital shaker for 1 min, and absorbance was measured at 450 nm. Wells containing culture medium but no cells were used to determine background values. Treatments were deemed toxic if there was a significant decrease in absorbance compared with vehicle treatment.
Reverse Transcription Quantitative PCR. Total RNA was isolated using the RNeasy 96 Kit and quantitated using the Quant-iT Ribogreen RNA Reagent Kit. A two-step reverse transcriptase-PCR reaction was conducted by reverse transcribing 50 ng of total RNA to cDNA using TaqMan reverse transcription reagents, according to the TaqMan Universal PCR Master Mix protocol. PCR reactions were then prepared by adding an aliquot of cDNA (3 μl) to a reaction mixture containing the TaqMan Fast Universal PCR Master Mix solution, primers, and probes for CYP3A4/CY2B6/CYP1A2. Low-density microarray studies to assess the basal expression of drug disposition genes were conducted by adding cDNA (50 μl) to TaqMan Universal PCR Master Mix solution. Low-density microarrays (microfluidic cards) were custom-made and validated by Applied Biosystems and contained probes in triplicate for the detection of 64 genes (Tables 1, 2, 3). PCR-amplified cDNAs were detected by real-time fluorescence on an ABI PRISM 7900 Fast Sequence Detection System (PerkinElmer Life and Analytical Sciences, Wellesley, MA).
Comparison of phase I and phase II enzyme gene expression between Fa2N-4 cells and cryopreserved human hepatocytes
The gene expression levels were evaluated in human hepatocytes and Fa2N-4 cells after the 24- and 48-h adaptation periods, respectively. Data are expressed as means ± S.E.-fold change in human hepatocytes compared with Fa2N-4 cells determined from three independent experiments, as described under Materials and Methods.
Comparison of transcription factor gene expression between Fa2N-4 cells and cryopreserved human hepatocytes
The gene expression levels were evaluated in human hepatocytes and Fa2N-4 cells after the 24- and 48-h adaptation periods, respectively. Data are expressed as means ± S.E.-fold change in human hepatocytes compared with Fa2N-4 cells determined from three independent experiments, as described under Materials and Methods.
Comparison of transporter gene expression between Fa2N-4 cells and cryopreserved human hepatocytes
The gene expression levels were evaluated in human hepatocytes and Fa2N-4 cells after the 24- and 48-h adaptation periods, respectively. Data are expressed as means ± S.E.-fold change in human hepatocytes compared with Fa2N-4 cells determined from three independent experiments, as described under Materials and Methods.
Data Analysis. Quantitation of the target cDNAs to assess basal gene expression were normalized to 18S ribosomal RNA (Cttarget – Ct18S =ΔCt), and the difference in expression for each target cDNA in the human hepatocytes was expressed relative to the amount in Fa2N-4 cells (ΔCthuman hepatocytes – ΔCtFa2N =ΔΔCt). Fold changes in target gene expression were determined by taking 2 to the power of this number (2–ΔΔCt). Statistical analysis of the differences in basal gene expression between Fa2N-4 cells and human hepatocytes were based upon differences in the ΔCt values (Pfaffl, 2001).
Student's t tests were used to determine the significance of differences between measurements. Differences with p values < 0.05 were considered significant.
To determine the EC50 and Emax values, the data from concentration-response curves were fitted to a three-parameter sigmoid (Hill) model, according to the following equation:  All curve-fitting was carried out using SigmaPlot 10.0 (Systat Software, Inc., Chicago, IL). The studies conducted with Fa2N-4 cells represent the mean of three independent experiments done in triplicate. Studies conducted in human hepatocytes (LHO, 455, and 527) represent the mean of triplicate wells, whereas studies conducted in DMQ represent the mean of duplicate wells.
All curve-fitting was carried out using SigmaPlot 10.0 (Systat Software, Inc., Chicago, IL). The studies conducted with Fa2N-4 cells represent the mean of three independent experiments done in triplicate. Studies conducted in human hepatocytes (LHO, 455, and 527) represent the mean of triplicate wells, whereas studies conducted in DMQ represent the mean of duplicate wells.
Results
Changes in the Expression of Drug Disposition Genes in Cultured Fa2N-4 Cells and Cryopreserved Human Hepatocytes after an Adaptation Period. Given the fact that some drug disposition genes, including uptake and efflux transporters, as well as transcription factors could influence the induction of CYP3A4 (Lam et al., 2006), we evaluated the basal expressions of 64 drug disposition genes (listed in Tables 1, 2, 3) in Fa2N-4 cells and compared those to four batches of cryopreserved human hepatocytes. Genes analyzed included phase I and II drug-metabolizing enzymes, hepatic transporters, transcription factors, and the coactivators and corepressors involved in the regulation of the aforementioned genes.
We first assessed whether gene expression levels changed during the 48- and 24-h adaptation periods for Fa2N-4 cells and cryopreserved human hepatocytes, respectively. In Fa2N-4 cells, the expression levels of all 64 genes increased after the 48-h adaptation period. We observed a >10-fold increase for 19 genes (Tables 1, 2, 3, denoted by footnote a). The largest increases in expression were observed for CYP2B6 (∼67-fold) and GSTA2 (∼82-fold). In contrast, in most cases, gene expression levels in human hepatocytes decreased (2- to 4-fold) or remained the same after the 24-h adaptation period (data not shown). In human hepatocytes, the genes that showed larger than 10-fold increases were limited to ABCC2, CYP3A5, and UGT1A6 for LHO; SLCO1B1, CYP2C19, and NR1H4 for DMQ; and ABCG2 for 455 (Tables 1, 2, 3, denoted by footnote b).
Basal Expression of Drug Disposition Genes in Fa2N-4 CellsCompared with Human Hepatocytes. After determining the impact of the adaptation period on gene expression in Fa2N-4 cells and human hepatocytes, we assessed the differences in gene expression before the initiation of drug treatment, which was defined as basal expression between the human hepatocytes and Fa2N-4 cells. Significant differences were observed for all categories of genes including phase I and II enzymes, transporters, and transcription factors and coactivators/corepressors (Tables 1, 2, 3).
As shown in Table 1, in four batches of cryopreserved hepatocytes tested, the CYP3A4 expression levels, compared with those in Fa2N-4 cells, ranged from 0.3- to 18-fold. CYP1A2 expression levels were significantly higher in all four batches of hepatocytes (19.3- to >100-fold) compared with those in Fa2N-4 cells. Similarly, CYP2D6, CYP2E1, CYP1A1, UGT1A1, UGT1A6, UGT2B15, and UGT2B4 expression levels were significantly higher in human hepatocytes compared with those in Fa2N-4 cells.
Except for NROB2 [small heterodimer partner (SHP)], NR1I3 (CAR), and VDR (vitamin D receptor), Fa2N-4 cells appear to have higher expression of most transcription factors and coactivators/corepressors that have been associated with PXR- or CAR-mediated enzyme induction (Moore et al., 2006) (Table 2). The expression of NR1I2 (PXR) was slightly higher in Fa2N-4 cells compared with three batches of hepatocytes (455, 527, and LHO), but lower than that in DMQ (∼2.5-fold) (Table 2). Interestingly, the expression of NR1I3 (CAR) in Fa2N-4 cells (Ct value ∼35) was nearly 2 orders of magnitude lower than expression in all four batches of human hepatocytes (Table 2). The expression of AhR and NR3C1 (glucocorticoid receptor), which are involved in the regulation of CYP1A2 and CYP3A4 (Tirona and Kim, 2005), respectively, were significantly lower in all four batches of human hepatocytes compared with Fa2N-4 cells. NROB2 (SHP) showed much higher expression in all four batches of hepatocytes than in Fa2N-4 cells. Conversely, the expression of TCF1 (hepatocyte nuclear factor 1), NCOR1 (nuclear receptor corepressor 1), NCOR2 (nuclear receptor corepressor 2), and NR1H2 (liver X receptor) were lower in human hepatocytes than those in Fa2N-4 cells (Table 2).
Expression levels of hepatic transporters were also compared between Fa2N-4 cells and cryopreserved hepatocytes (Table 3). Fa2N-4 cells demonstrated a significantly lower basal expression of several major hepatic uptake transporters, including SLC10A1 (sodium/bile acid cotransporter 10A1, NTCP), SLC22A1 (solute carrier organic cation transporter 22A1, OCT1), SLCO1B1 [solute carrier organic anion transporter (OATP) 1B1], and SLCO1B3 (solute carrier OATP1B3). The most significant differences were observed for SLC10A1, SLCO1B1, and SLCO1B3 (Ct value in Fa2N-4 cells was >35), in which the expression of these genes were 50-fold higher in all four batches of hepatocytes compared with Fa2N-4 cells (Table 3). Except for SLCO1A2, the basal expression levels of all uptake transporters were highest in DMQ compared with the other three batches of human hepatocytes tested. The basal expression of several hepatic efflux transporters was also evaluated. The expression of ABCB1 (the multidrug resistance gene, P-glycoprotein) was significantly lower in all four batches of human hepatocytes than that in Fa2N-4 cells, whereas the expression of ABCC2 (the multidrug resistance protein 2) was ∼2- to 5-fold (p < 0.05) higher. The expression of ABCB11 [bile salt exporter protein (BSEP)] was approximately 2 orders of magnitude greater in all four batches of hepatocytes than that in Fa2N-4 cells. The expression of ABCG2 (breast cancer resistance protein) appears to be similar in Fa2N-4 cells and human hepatocytes. Variable gene expression was observed for all of the other uptake and efflux transporters among the four batches of human hepatocytes compared with Fa2N-4 cells (Table 3).

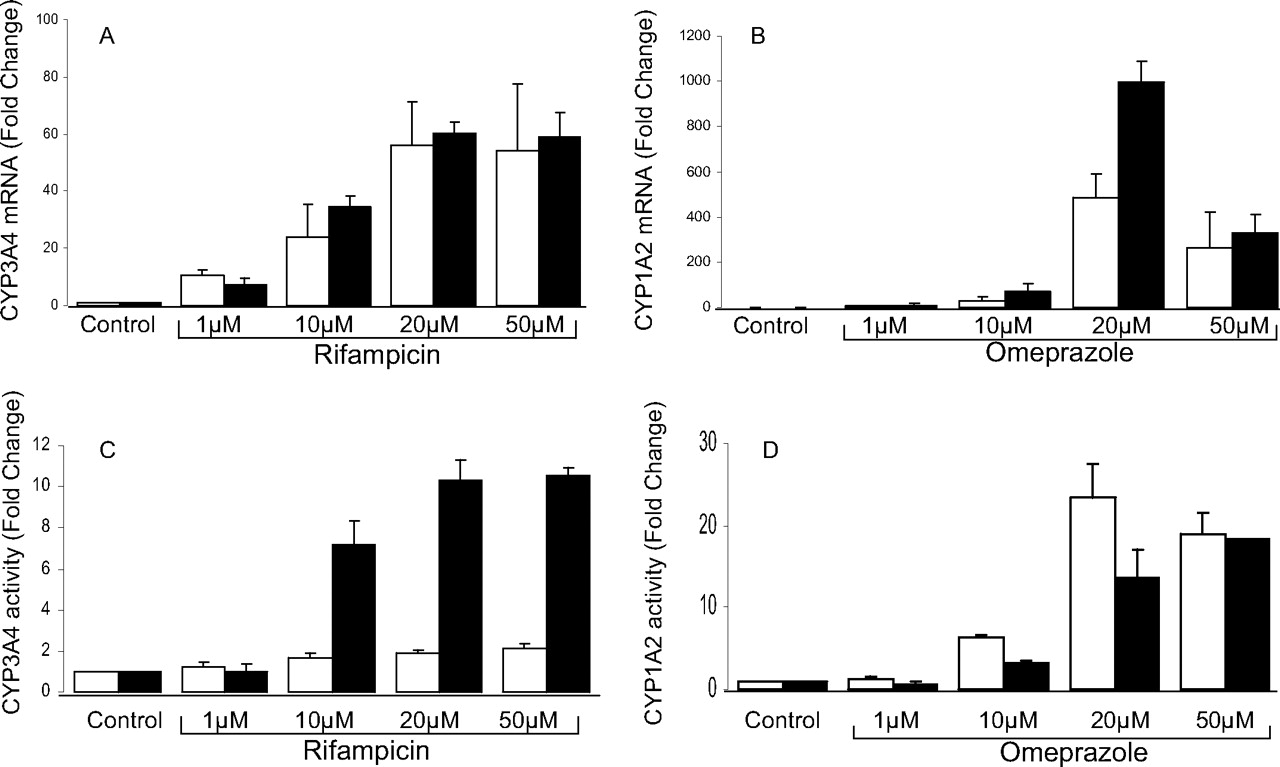
Induction of CYP3A4 and CYP1A2 mRNA and activity in Fa2N-4 cells. Rifampicin-mediated CYP3A4 and omeprazole-mediated CYP1A2 inductions were compared using the protocol recommended by the manufacturer (XenoTech LLC) (□) and our optimized assay conditions (▪) as described under Materials and Methods. CYP3A4 mRNA (A), CYP1A2 mRNA (B), CYP3A4-mediated testosterone 6β-hydroxylation (C), and CYP1A2-mediated phenacetin O-deethylation (D) after treatment with various concentrations of rifampicin and omeprazole, respectively. The data were expressed as mean ± S.E. of the fold changes compared with 0.1% DMSO vehicle control and obtained from three independent experiments.
Induction of CYP3A4 and CYP1A2 in Fa2N-4 Cells under Optimized Culture Conditions. By using the protocol recommended by the distributor (XenoTech LLC), CYP3A4 functional activity was less than 2-fold in Fa2N-4 cells treated with 1–50 μM rifampicin (Fig. 1C), although we observed a concentration-dependent increase in CYP3A4 mRNA expression (1–20 μM rifampicin), which ranged from 10- to 60-fold (Fig. 1A). We therefore optimized the culture and induction conditions to increase the CYP3A4 induction window at the level of functional activity as described under Materials and Methods.
With the optimized protocol, a dose-dependent increase in CYP3A4 functional activity was observed, which ranged from 1- to 10-fold after the cells were treated with 1–20 μM rifampicin (Fig. 1C), whereas CYP3A4 induction at the mRNA level was comparable with the data obtained using the manufacturer-recommended protocol (Fig. 1A). A dose-dependent increase in CYP1A2 mRNA and activity were observed in both methods when Fa2N-4 cells were treated with omeprazole (1–20 μM) (Fig. 1, B and D). At 50 μM omeprazole (the U.S. Food and Drug Administration-recommended CYP1A2 positive control concentration), CYP1A2 induction at both mRNA and activity levels were comparable between these two methods (Fig. 1, B and D).
Comparison of CYP3A4 Induction in Fa2N-4 Cells and Human Hepatocytes. To assess whether Fa2N-4 cells could be used as a predictive model to evaluate CY3A4 induction, nine compounds were selected to measure concentration-dependent CYP3A4 induction in Fa2N-4 cells and cryopreserved human hepatocytes (Figs. 2 and 3). Among the compounds tested, rifampicin, bosentan, moricizine, and ritonavir are reported as selective PXR-activators (LeCluyse, 2001; Luo et al., 2002; van Giersbergen et al., 2002), phenobarbital, phenytoin, and efavirenz are PXR/CAR dual activators (Hariparsad et al., 2004; Wang et al., 2004; Trubetskoy et al., 2005; Bell and Michalopoulos, 2006; Faucette et al., 2007), and CITCO and artemisinin are selective CAR activators (Maglich et al., 2003; Simonsson et al., 2006).

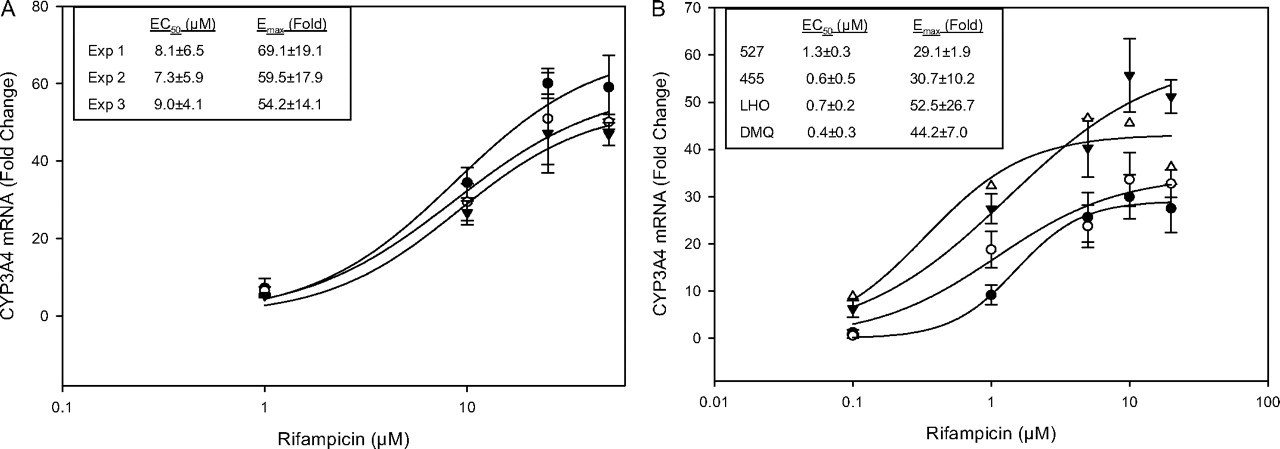
Concentration-response curves for CYP3A4 mRNA induction in Fa2N-4 cells and human hepatocytes treated with rifampicin. Concentration-response curves obtained from (A) Fa2N-4 cells (•, experiment 1; ○. experiment 2; ▴, experiment 3) and (B) human hepatocytes (•, 527; ○, 455, ▴, LHO; ▵, DMQ) were fitted to a sigmoidal model as described under Materials and Methods, 527, 455, and LHO represent the mean ± S.E. of one experiment performed in triplicate. The data from DMQ represent the mean of one experiment performed in duplicate.
Rifampicin-mediated CYP3A4 induction was conducted in Fa2N-4 cells and four batches of human hepatocytes (DMQ, 527, 455, and LHO). Dose-response curves in Fa2N-4 cells (Fig. 2A) obtained from three independent experiments were fitted to the sigmoidal model with EC50 values ranging between 7 and 9 μM and Emax ranging between 54- and 69-fold. In comparison, EC50 values in human hepatocytes (DMQ, 527, 455, and LHO) ranged from 0.4 to 1.3 μM and Emax values from 29- to 53-fold (Fig. 2B). Rifampicin-mediated CYP3A4 induction was also evaluated at the level of CYP3A4 functional activity in Fa2N-4 cells and human hepatocytes (DMQ) (data not shown). The EC50 values obtained were 6.2 ± 3.1 μM for Fa2N-4 cells and 0.3 ± 0.2 μM for human hepatocytes (DMQ), respectively, and the corresponding Emax values were 12.7 ± 2.2- and 12.3 ± 2.0-fold.
Dose-response studies for CYP3A4 induction were also conducted for additional compounds in Fa2N-4 cells, and results were compared with the data obtained in one batch of human hepatocytes (DMQ) (Fig. 3). Because of the poor correlation that we observed for most compounds between changes in CYP3A4 mRNA expression and functional activity, we compared the responses between both cell types on the basis of CYP3A4 mRNA expression. As shown in Fig. 3 and Table 4, the EC50 and Emax values for phenytoin and moricizine were roughly within a 2-fold range between the two cell systems. Interestingly, although the EC50 values for bosentan were reasonably similar between the two cell systems, the Emax in hepatocytes was 4-fold higher in hepatocytes than in Fa2N4 cells.
EC50 and Emax values for induction of CYP3A4 and CYP2B6 mRNA for compounds tested in Fa2N-4 cells and human hepatocytes
EC50 and Emax are expressed as mean ± calculated S.E. using SigmaPlot version 10 as described under Materials and Methods. Studies conducted with Fa2N-4 cells represent the mean of three independent experiments performed in triplicate. Studies conducted in human hepatocytes (DMQ) represent the mean of one experiment done in duplicate.
The EC50 values of two selective CAR activators, artemisinin and CITCO, were 21.4 and 0.8 μM in human hepatocytes (Table 4). However, the induction response in Fa2N-4 cells was too low to be fitted to the sigmoidal model (Fig. 2, D and E).
Dose-response curves for ritonavir could not be fitted to a sigmoidal model because of the bell-shaped response (Fig. 2C). A dose-dependent CYP3A4 induction was observed in both Fa2N-4 cells and human hepatocytes treated with ritonavir with a maximal response observed between 5 and 10 μM, after which the fold change decreased sharply. The reason for the ritonavir bell-shaped response is currently not clearly understood (Ripp et al., 2006). Data for phenobarbital also could not be fitted to the sigmoidal model because no saturable response was observed in either Fa2N-4 cells or human hepatocytes (DMQ) within the concentration range tested (Fig. 2F). Human hepatocytes treated with phenobarbital at 200, 500, or 1000 μM produced 20-, 40- and 90-fold CYP3A4 mRNA induction, whereas 4.7-, 10.4- and 28.3-fold induction was observed in Fa2N-4 cells treated with the same concentrations of phenobarbital. A bell-shaped response was observed in Fa2N-4 cells and human hepatocytes (DMQ) treated with efavirenz (Fig. 2H). The lower induction observed at higher concentrations of efavirenz (20–50 μM) was caused by cytotoxicity. At a 5 μM concentration, 1.8- and 22.4-fold induction was observed in Fa2N-4 cells and human hepatocytes (DMQ) treated with efavirenz.
Comparison of CYP2B6 mRNA Induction in Fa2N-4 Cells and Human Hepatocytes (DMQ). To further confirm the absence of CAR in Fa2N-4 cells, CYP2B6 mRNA induction was compared between human hepatocytes and Fa2N-4 cells treated with selected compounds. Rifampicin-mediated CYP2B6 induction was fitted to the sigmoidal model with an EC50 of 8.0 μM and an Emax of 4.1-fold for Fa2N-4 cells and an EC50 of 2.3 μM and an Emax of 12.9-fold for human hepatocytes, respectively (Fig. 4A; Table 4). In Fa2N-4 cells, the EC50 and Emax values for phenobarbital were 204.4 μM and 5.0-fold, respectively. Correspondingly, the EC50 and Emax values in human hepatocytes were 60.4 μM and 31.3-fold, respectively (Fig. 4B; Table 4). Bosentan-mediated CYP2B6 induction was low in Fa2N-4 cells, but produced an EC50 value of 0.4 μM and an Emax of 3.8-fold in human hepatocytes (Fig. 4F; Table 4). Fa2N-4 cells treated with the selective CAR agonists artemisinin and CITCO or the dual PXR/CAR activator phenytoin produced a low response in Fa2N-4 cells and could not be fitted to the sigmoidal model. Data from human hepatocytes (DMQ) showed a concentration-dependent response for phenytoin, artemisinin, and CITCO (Fig. 4, C–E; Table 4). These data supported the conclusion that the CAR pathway is deficient in Fa2N-4 cells.
Discussion
In this report, we performed a comprehensive assessment of the utility of Fa2N-4 cells as a CYP3A4 induction model. Our studies indicate that Fa2N-4 cells possess comparable levels of PXR and AhR, which are major pathways involved in the regulation of CYP3A4 and CYP1A2, respectively, in human hepatocytes (Table 2; Fig. 1). However, significant differences exist in the basal mRNA expression of several drug disposition genes in Fa2N-4 cells compared with that in human hepatocytes. In particular, very low expression of CAR and several hepatic uptake transporters was observed in Fa2N-4 cells. This could potentially limit the application of Fa2N-4 cells as an in vitro tool for the (quantitative) prediction of CYP3A4 induction in the clinic.
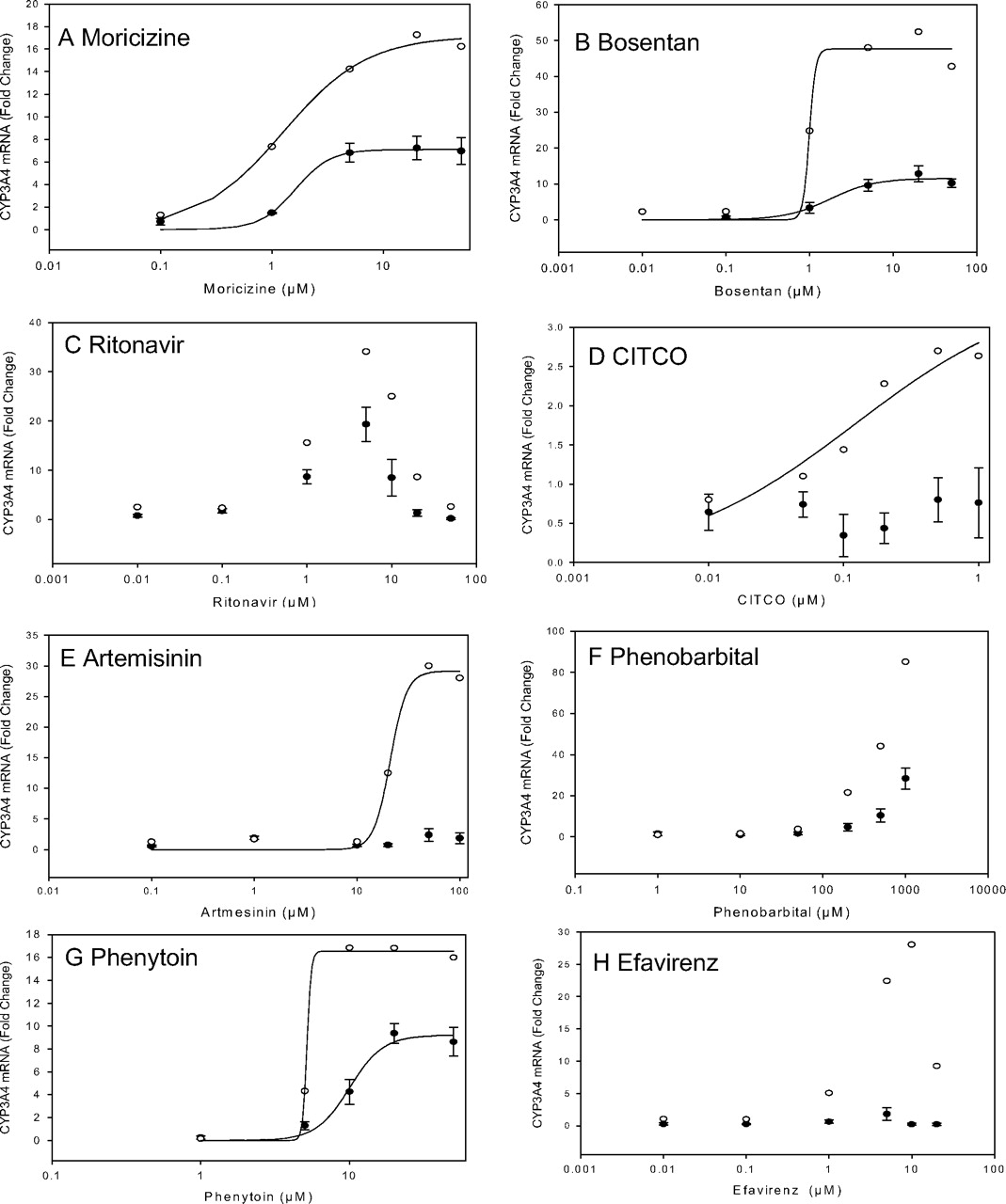
Concentration-response curves for CYP3A4 mRNA induction in Fa2N-4 cells and human hepatocytes treated with several compounds. Concentration-response curves obtained from both Fa2N-4 cells (•) and human hepatocytes (DMQ) (○) were fitted to a sigmoidal model as described under Materials and Methods, CITCO in Fa2N-4 cells, efavirenz, phenobarbital, or ritonavir in both Fa2N-4 cells and human hepatocytes were not fitted to the model because of the very low CYP3A4 induction, lack of a saturable response, or a bell-shaped concentration-response relationship. The data obtained from Fa2N-4 cells represent the mean ± S.E. of three independent experiments performed in triplicate. The data from human hepatocytes (DMQ) represent the mean of one experiment performed in duplicate.
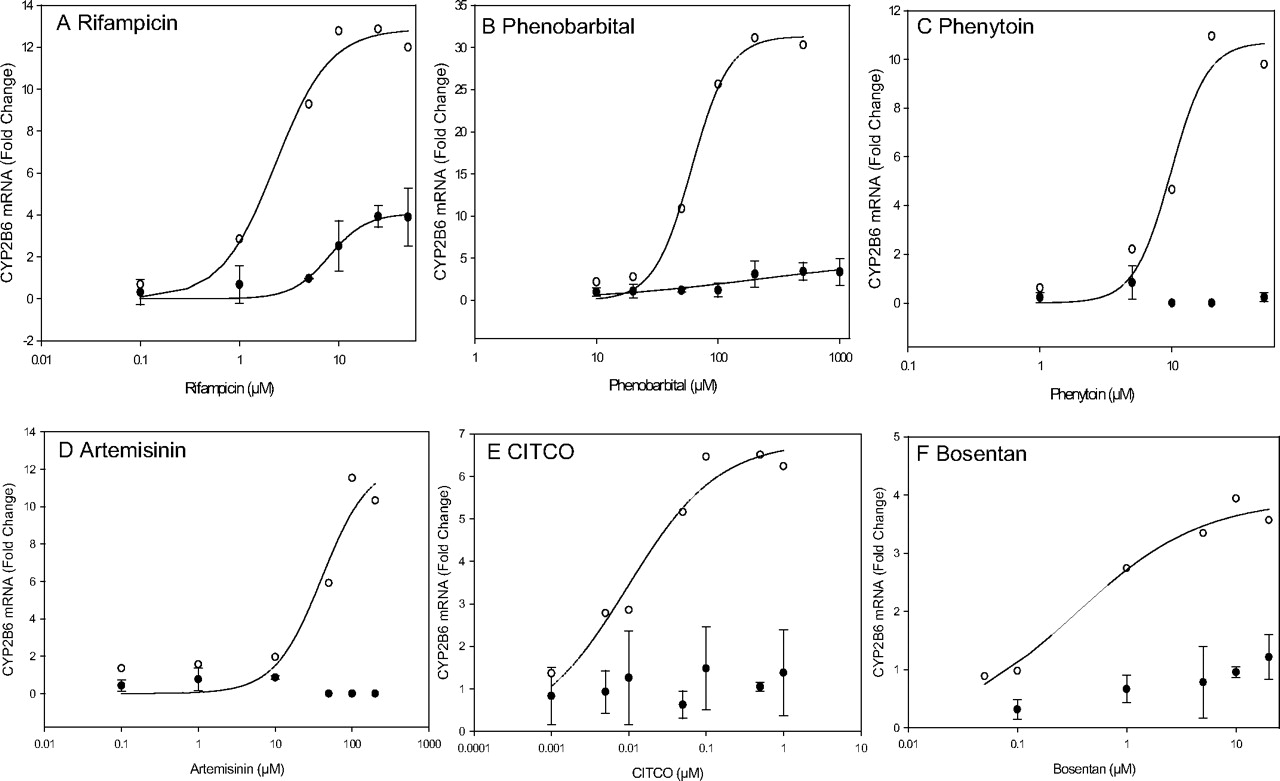
Concentration-response curves for CYP2B6 mRNA induction in Fa2N-4 cells and human hepatocytes treated with several tested compounds. Concentration-response curves obtained from both Fa2N-4 cells (•) and human hepatocytes (○) were fitted to a sigmoidal model as described under Materials and Methods. In Fa2N-4 cells, dose-response data for phenytoin, artemisinin, CITCO, and bosentan could not be fitted to the sigmoidal model because of the very low observed response. The data obtained from Fa2N-4 cells represent the mean of three independent experiments performed in triplicate. The data from human hepatocytes (DMQ) represent the mean of one experiment performed in duplicate.
The expression levels of PXR (NR1I2), the primary transcription factor involved in the regulation of CYP3A4 (Tirona and Kim, 2005), were similar between Fa2N-4 cells and human hepatocytes (Table 2). In addition, several coactivators and corepressors of PXR (Moore et al., 2006) such as the small heterodimer partner (SHP/NCOB2), nuclear receptor corepressor 2 (NCOR2/SMRT), steroid receptor coactivators 1 (SRC1/NCOA1) and 2 (SRC2/GRIP1), nuclear receptor interacting protein 1 (NRIP1/RIP140), peroxisome proliferator-activated receptor-γ coactivator, and Forkhead transcription factor FKHR (FOXO1) were also expressed in Fa2N-4 cells. Interestingly, the expression levels of CAR were dramatically lower (p < 0.05) in Fa2N-4 cells than those in all four batches of cryopreserved hepatocytes (Table 2). This will have a significant impact on the induction of genes such as CYP3A4 and CYP2B6, given that there is considerable overlap of PXR and CAR both in terms of the spectrum of genes regulated and their affinity to DNA-response elements (Chen et al., 2005).
Basal expression levels of several hepatic uptake transporters, including OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), NTCP (SLC10A1), and OCT1 (SLC22A1), were significantly lower in Fa2N-4 cells than those in all four batches of human hepatocytes. Uptake of drugs into hepatocytes is achieved by both passive diffusion and carrier-mediated transport (van Montfoort et al., 2003; Shitara et al., 2006). OATP1B1 and OATP1B3, which are specifically expressed in the hepatocyte sinusoidal membrane, transport several clinically used drugs, including some potent CYP3A4 inducers, such as rifampicin (Tirona and Kim, 2002). Hepatic uptake transporters could affect the intracellular exposure of their substrates in the liver and, therefore, modulate the induction by P450 enzymes, when transporter-mediated hepatic uptake is the rate-limiting step for uptake of drugs into the liver. Given the low hepatic transporter expression in Fa2N-4 cells, the induction potential of substrates for these transporters probably will be underestimated. Basal expression of hepatic efflux transporter ABCB11 (BSEP) was also significantly lower in Fa2N-4 cells (Table 3). The relevance of the low BSEP expression to CYP3A4 induction is unclear as most BSEP substrates are not inducers of CYP3A4.
To validate whether Fa2N-4 cells could be used as a reliable model for CYP3A4 induction, we first compared the CYP3A4 mRNA induction by rifampicin, a prototypical activator of PXR, in Fa2N-4 cells and human hepatocytes (Fig. 2). The Emax obtained from Fa2N-4 cells (∼60-fold) was comparable with that in four batches of human hepatocytes (∼53-fold); however, the EC50 in Fa2N-4 cells (∼8 μM) was ∼10-fold higher than that in human hepatocytes (∼0.8 μM) (Fig. 2). Similar results were also observed at the level of CYP3A4 activity (data not shown). The increased EC50 could be attributed to the low OATP1B1/1B3 expression in Fa2N-4 cells as rifampicin is a substrate of these transporters (Tirona et al., 2002). Tirona et al. (2002) have demonstrated that increased expression of OATP1B1 in HeLa cells significantly enhanced rifampicin-mediated PXR activation. Such studies would also be needed in Fa2N-4 cells to confirm our hypothesis. Interestingly, bosentan, a PXR agonist and a substrate for OATP1B1 (Treiber et al., 2007), showed an EC50 value of 1.8 μMin Fa2N-4 cells, which was less than 2-fold higher than that in human hepatocytes (1 μM in DMQ), whereas the Emax in Fa2N-4 cells (12-fold induction) was nearly 5-fold lower than that in human hepatocytes (48-fold induction). The mechanism explaining this difference in Emax is not clear, but it might be explained by the involvement of other nuclear receptors such as, for instance, CAR. More experiments will be needed to understand this further.
The lack of CYP3A4 and CYP2B6 induction for the selective CAR agonists CITCO and artemisinin in Fa2N-4 cells confirms our observation that CAR expression is very low in Fa2N-4 cells (Figs. 2 and 3; Table 2). In human hepatocytes (DMQ), the EC50 value for CITCO-mediated CYP2B6 induction (0.01 μM) was much lower than that for CYP3A4 (0.8 μM). In addition, CITCO showed a much lower Emax for CYP3A4 induction than rifampicin. This finding concurs with the current literature that CYP2B6 is regulated primarily via CAR (Faucette et al., 2006).
We also evaluated CYP3A4 and CYP2B6 induction with several dual PXR and CAR activators, including phenytoin, efavirenz, and phenobarbital (Figs. 3 and 4; Table 4). Phenytoin showed a relatively low CYP3A4 induction response in Fa2N-4 cells compared with human hepatocytes (Fig. 3G). Furthermore, a dose-dependent CYP2B6 induction was observed in human hepatocytes (EC50 = 9.8 μM) but not in Fa2N-4 cells. Studies conducted by Luo et al. (2002) and Wang et al. (2004) indicated that phenytoin is a potent CAR but a weak PXR activator. Therefore, the low inductive response of phenytoin in Fa2N-4 cells is probably due to the low CAR expression. Similarly, efavirenz a relatively potent CAR but weak PXR activator (Faucette et al., 2007) also showed low CYP3A4 induction in Fa2N-4 cells (Fig. 3H). Although the dose-response curves for CYP3A4 mRNA induction by phenobarbital were not saturable at the maximum concentration tested (1000 μM), the CYP3A4 induction in Fa2N-4 cells at phenobarbital concentrations of 200, 500, and 1000 μM was much lower than those in human hepatocytes (Fig. 3F). Phenobarbital caused a low CYP2B6 induction in Fa2N-4 cells (Table 4). It was not unexpected that phenobarbital induced CYP2B6 in Fa2N-4 cells, even in the absence of CAR expression. This observation could be explained by the cross-talk between PXR and CAR in the regulation of drug disposition genes (Pascussi et al., 2008). Faucette et al. (2006) observed that PXR activators induced both CYP3A4 and CYP2B6, whereas CAR activators induced CYP2B6 but not CYP3A4. Thus, the expression of PXR in Fa2N-4 cells and human hepatocytes could cause the induction of CYP2B6 mRNA in Fa2N-4 cells treated with phenobarbital as this compound is also a PXR activator (Bell and Michalopoulos, 2006). This may also explain why rifampicin, which is a selective PXR activator, induced CYP2B6 (EC50 = 8.0 μM and Emax = 4.1-fold) in Fa2N-4 cells (LeCluyse, 2001). Interestingly, the EC50 (2.3 μM) value generated in human hepatocytes was significantly lower than that observed in Fa2N-4 cells, whereas the Emax value (12.9-fold) was significantly higher (Table 4). This finding further emphasizes the importance of CAR in the regulation of CYP2B6 (Faucette et al., 2006, 2007).
In conclusion, we observed very low expression of CAR, a key nuclear hormone receptor for CYP3A4 and CYP2B6 induction, in Fa2N-4 cells. In addition, we also found that there is a significantly lower expression of several hepatic uptake transporters in Fa2N-4 cells. This could result in false-negative results or a low level of induction for CAR activators or substrates of hepatic uptake transporters. Our studies indicate that Fa2N-4 cells cannot replace primary or cryopreserved human hepatocytes as an in vitro model system to prospectively predict the induction potential of drug candidates in the clinic.
Acknowledgments
We thank Drs. D. Nicoll-Griffith and B. R. Smith for critically reading the manuscript and for helpful suggestions. We also thank Alisha Norcross for technical assistance with the human hepatocyte induction studies.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.020677.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; CAR, constitutive androstane receptor; CITCO, 6-(4-chlorophenyl)imidazo-[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; PCR, polymerase chain reaction; DMSO, dimethylsulfoxide; OATP, organic anion-transporting polypeptide; BSEP, bile salt exporter protein.
- Received January 29, 2008.
- Accepted March 5, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}