


Abstract
Bovine serum albumin (BSA) and fatty acid-free human serum albumin (HSAFAF) reduce the Km values for UGT2B7 substrates by sequestering inhibitory long-chain fatty acids released by incubations of human liver microsomes (HLM) and HEK293 cells expressing this enzyme. However, the scope of the “albumin effect” is unknown. In this investigation we characterized the effects of albumin on the kinetics of 4-methylumbelliferone (4MU) glucuronidation by UDP-glucuronosyltransferase (UGT) 1A1, 1A6, and 1A9, and propofol (PRO) glucuronidation by UGT1A9 and HLM. BSA and HSAFAF, but not human serum albumin, reduced the Km values for 4MU and PRO glucuronidation by UGT1A9. For example, HSAFAF (2%) reduced the Km values for 4MU and PRO glucuronidation from 13.4 to 2.9 and 41 to 7.2 μM, respectively. Similarly, HSAFAF (2%) reduced the Km for PRO glucuronidation by HLM from 127 to 10.6 μM. Arachidonic, linoleic, and oleic acids and a mixture of these decreased the rates of 4MU and PRO glucuronidation by UGT1A9. Km values for these reactions were increased 3- to 6-fold by the fatty acid mixture. Inhibition was reversed by the addition of BSA (2%). Extrapolation of kinetic constants for PRO glucuronidation by HLM in the presence of HSAFAF predicted in vivo hepatic clearance within 15%. Fatty acids had no effect on 4MU glucuronidation by UGT1A1 and UGT1A6 but, paradoxically, all forms of albumin altered the kinetic model for 4MU glucuronidation by UGT1A6 (from Michaelis-Menten to two-site). Only BSA caused a similar effect on 4MU glucuronidation by UGT1A1. It is concluded that BSA and HSAFAF reduce the Km values of only those enzymes inhibited by long-chain unsaturated fatty acids.
Conjugation with glucuronic acid (“glucuronidation”) represents both a clearance and detoxification mechanism for a large number of compounds that include drugs from most therapeutic classes, nondrug xenobiotics, and endogenous compounds (Miners and Mackenzie 1991; Tukey and Strassburg 2000; Miners et al., 2004). Glucuronidation reactions are catalyzed by enzymes of the UDP-glucuronosyltransferase (UGT) superfamily. The individual UGT enzymes exhibit distinct, but overlapping, substrate and inhibitor selectivities and differ in terms of gene regulation, including tissue distribution (Tukey and Strassburg, 2000; Kiang et al., 2005; Kubota et al., 2007). Although 17 functional human UGT proteins have been identified to date (Mackenzie et al., 2005), only 7 of the hepatically expressed enzymes appear to contribute significantly to the elimination of drugs and other xenobiotics: UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15 (Miners et al., 2006).
The ready availability of experimental systems [e.g., human liver microsomes (HLM), isolated hepatocytes, and recombinant enzymes] that enable the kinetic characterization of xenobiotic biotransformation in vitro has resulted in a steady increase in the number of publications that have investigated the kinetics of drug and chemical glucuronidation. Although Km values for some substrates, for example, bilirubin glucuronidation by UGT1A1 (Udomuksorn et al., 2007) and trifluoperazine glucuronidation by UGT1A4 (Uchaipichat et al., 2006a), are in the low micromolar range, reported Km values of many compounds, particularly substrates of UGT2B7, are often in the millimolar range. Notably, Km values for morphine (3- and 6-), codeine, and zidovudine glucuronidation by HLM range from approximately 1.1 to 4.3 mM, even when corrected for nonspecific binding (Boase and Miners 2002; Court et al., 2003). Moreover, the high Km values provide lower than expected estimates of microsomal intrinsic clearance (CLint) that underpredict in vivo hepatic CLint and hepatic clearance (CLH) when used for in vitro-in vivo extrapolation (IV-IVE) (Boase and Miners 2002; Soars et al., 2002; Engtrakul et al., 2005).
The high Km values observed in vitro have led to the general perception that glucuronidation is a “low-affinity” metabolic pathway (e.g., Williams et al., 2004) and that IV-IVE strategies are unlikely to be successful for glucuronidated substrates, especially for kinetic data obtained with HLM and recombinant enzymes (Soars et al., 2002; Engtrakul et al., 2005). However, recent work in this laboratory demonstrated that the addition of bovine serum albumin (BSA) or fatty acid-free human serum albumin (HSAFAF) to incubations of HLM and recombinant UGT2B7 decreased the Km for zidovudine glucuronidation by approximately an order of magnitude (Rowland et al., 2007). Km values obtained in the presence of BSA and HSAFAF (with correction for binding) were 40 to 87 μM, comparable with the Km observed for zidovudine glucuronidation by human hepatocytes. In addition, we demonstrated that extrapolation of the inhibitor constants (Ki) generated in the presence of BSA, with both HLM and UGT2B7 as the enzyme sources, correctly predicted the magnitude of the fluconazole-zidovudine and valproic acid-lamotrigine inhibitory interactions in vivo (Rowland et al., 2006; Uchaipichat et al., 2006b).
The mechanism of the “albumin effect” was shown to involve sequestration of inhibitory long-chain unsaturated fatty acids, particularly oleic, linoleic, and arachidonic acids, released from the microsomal membrane (or from HEK293 cell lysate in the case of recombinant UGT2B7) during the course of an incubation (Rowland et al., 2007). These fatty acids are potent competitive inhibitors of UGT2B7 (Tsoutsikos et al., 2004; Rowland et al., 2007), and hence the Km values of substrates and the Ki values of inhibitors obtained in the absence of BSA or HSAFAF overestimate these parameters. In contrast to the observations with UGT2B7, BSA does not affect the Km for lamotrigine glucuronidation by UGT1A4 (Rowland et al., 2006). This finding suggests that the effects of long-chain unsaturated fatty acids on the kinetics of drug glucuronidation, and hence implications for IV-IVE, vary from enzyme to enzyme.
Here we report the effects of BSA, crude HSA, and HSAFAF on UGT1A1, UGT1A6, and UGT1A9, which, along with UGT1A4, appear to represent distinct clusters of hepatic UGT1A activities. Inhibitory effects of long-chain unsaturated fatty acids on these enzymes were also characterized. Effects on all three enzymes were assessed using the nonselective substrate 4-methylumbelliferone (4MU), whereas subsequent investigation of human liver microsomal UGT1A9 activity used the selective substrate propofol (PRO). Inhibition by fatty acids (and reversal by BSA and HSAFAF) was demonstrated only with UGT1A9, although paradoxical effects of albumin on the kinetic models of 4MU glucuronidation by UGT1A1 and UGT1A6 were observed. Importantly, microsomal CLint values for PRO glucuronidation obtained in the presence of BSA and HSAFAF accurately predicted in vivo CLint.
Materials and Methods
Materials. Alamethicin (from Trichoderma viride), BSA (product number A7906), “crude” HSA (product number A9511), HSAFAF (product number A1887), 4MU, 4-methylumbelliferone β-d-glucuronide (4MUG), fatty acids (as the free acid), PRO, and UDP-glucuronic acid (UDPGA) (trisodium salt) were purchased from Sigma-Aldrich (Sydney, Australia). Solvents and other reagents were of analytical reagent grade.
Human Liver Microsomes and Expression of UGT1A Proteins. Pooled human liver microsomes were prepared by mixing equal amounts of protein from five human livers (H7, 44-year-old female; H10, 67-year-old female; H12, 66-year-old male; H29 45-year-old male; and H40, 54-year-old female), obtained from the human liver bank of the Department of Clinical Pharmacology of Flinders University. Approval for the use of human liver tissue in xenobiotic metabolism studies was obtained from the Flinders Clinical Research Ethics Committee. HLM were prepared by differential centrifugation, as described by Bowalgaha et al. (2005).
cDNAs encoding UGT1A1, UGT1A6, and UGT1A9 were stably expressed in a human embryonic kidney cell line (HEK293) as described previously (Sorich et al., 2002; Uchaipichat et al., 2004). Transfected cells were incubated and harvested according to published methods (Uchaipichat et al., 2004). Lysed cells were centrifuged at 12,000g for 1 min at 4°C, and the supernatant fraction was removed and stored at –80°C until use. Expression of UGT protein was demonstrated by immunoblotting with a nonselective UGT1A antibody according to Uchaipichat et al. (2004) and measurement of 4MU glucuronidation activity (see below).
4MU Glucuronidation Assay. Assay conditions for 4MU glucuronidation by each recombinant UGT were optimized for HEK293 cell lysate protein concentration, incubation time, and 4MU concentration range: UGT1A1, 0.5 mg/ml lysate protein, 90 min, and 5 to 600 μM 4MU; UGT1A6, 0.0025 mg/ml lysate protein, 30 min, and 10 to 600 μM 4MU; and UGT1A9, 0.03 mg/ml lysate protein, 15 min, and 0.5 to 40 μM 4MU, respectively. Incubations, in a total volume of 200 μl, contained phosphate buffer (0.1 M, pH 7.4), MgCl2 (4 mM), albumin (0–2%), HEK293 cell lysate expressing the UGT1A protein, and 4MU (as detailed above). After a 5-min preincubation, reactions were initiated by the addition of UDPGA (5 mM). Incubations were performed at 37°C in a shaking water bath. Reactions were terminated by the addition of perchloric acid (6 μl, 70% v/v). Samples were subsequently centrifuged at 4000g for 10 min, and a 25-μl aliquot of the supernatant fraction was injected directly into the HPLC column. 4MU β-d-glucuronidation by lysate protein from untransfected HEK293 cells was not detectable. Formation of 4MUG was quantified by HPLC as described by Rowland et al. (2007).
PRO Glucuronidation Assay. Incubations, in a total volume of 200 μl, contained phosphate buffer (0.1 M, pH 7.4), MgCl2 (4 mM), HLM (0.1 mg), albumin (0–2%), and PRO (1–500 μM). HLM were fully activated by the addition of the pore-forming polypeptide alamethicin (50 μg/mg of protein) with incubation on ice for 30 min (Boase and Miners, 2002). After a 5-min preincubation, reactions were initiated by the addition of UDPGA (5 mM). Incubations were performed at 37°C in a shaking water bath for 30 min. Reactions were terminated by the addition of 200 μl of ice-cold methanol containing 4% glacial acetic acid. Samples were subsequently centrifuged at 4000g for 10 min, and a 5-μl aliquot of the supernatant fraction was injected directly into the HPLC column.
For reactions performed using recombinant UGT1A9, incubation mixtures contained HEK293 cell lysate (0.05 mg) in place of HLM protein, and the incubation time was decreased to 15 min. Under the reaction conditions used, PRO glucuronidation was linear with respect to incubation time to 90 min and to a protein concentration of 1.5 mg/ml for HLM and 1 mg/ml for HEK293 cell lysate. PRO glucuronidation by lysate from untransfected HEK293 cells was not detectable.
Quantification of PRO-Gluc Formation. HPLC was performed using an 1100 series instrument (Agilent Technologies, Sydney, Australia) fitted with a Synergie Hydro reversed-phase C18 analytical column (150 × 3.0 mm, 4 μm; Phenomenex, Sydney, Australia). PRO-Gluc was separated by gradient elution with a mobile phase containing 20 mM phosphate buffer (pH 4.6) (mobile phase A) and acetonitrile (mobile phase B), at a flow rate of 0.6 ml/min. Initial conditions were 73% mobile phase A and 27% mobile phase B. These conditions were held for 2 min. The proportion of mobile phase B was then increased to 75% over 0.5 min and held for 5.5 min. Column eluant was monitored by UV absorbance at 214 nm. Retention times for PRO-Gluc and PRO were 3.6 and 8.1 min, respectively. The identity of PRO-Gluc was confirmed by hydrolysis with β-glucuronidase (from Escherichia coli) and comparison with blank incubations performed in the absence of UDPGA. Briefly, a 1-ml sample containing 500 μM PRO was incubated overnight in the presence of 2 mg/ml HLM. β-Glucuronidase (5 IU) was added to a 500-μl aliquot of this sample, the pH of the sample was reduced to 5.8, and the sample was incubated for 120 min. Loss of PRO-Gluc and recovery of PRO was determined by comparison with an aliquot of the original sample that was not subjected to β-glucuronidase treatment.
In the absence of an authentic product, PRO-Gluc formation was quantified by comparison of peak areas to those of an PRO standard curve prepared over the concentration range 1 to 40 μM. Vmax and CLint values should therefore be considered “apparent.” Overall within-day assay reproducibility was assessed by measuring PRO-Gluc formation in eight separate incubations of the same batch of pooled HLM. Coefficients of variation were 3.6 and 2.9% for added PRO concentrations of 2 and 500 μM, respectively. The lower limit of quantitation (assessed as three times background noise) for PRO-Gluc was 0.005 μM.
Binding of 4MU and PRO to Albumin, HLM, and HEK293 Cell Lysate. The binding of 4MU to HEK293 cell lysate, albumin (0.1, 1, and 2%), and mixtures of albumin with HEK293 cell lysate was measured by equilibrium dialysis as described previously (Rowland et al., 2007). The binding of PRO to HLM, HEK293 cell lysate, albumin, and mixtures of albumin with the two enzyme sources was determined according to the general procedure of McLure et al. (2000). Binding measurements were performed using a Dianorm equilibrium dialysis apparatus comprising Teflon dialysis cells (capacity of 1.2 ml per side) separated into two compartments with Sigma-Aldrich dialysis membrane (molecular mass cutoff 12 kDa).
One side of the dialysis cell was loaded with 1 ml of a solution of PRO (2–500 μM) in phosphate buffer (0.1 M, pH 7.4). The other compartment was loaded with 1 ml of either a suspension of HLM in phosphate buffer (0.1 M, pH 7.4), HEK293 cell lysate in phosphate buffer (0.1 M, pH 7.4), albumin (0.1, 1, or 2%) in phosphate buffer (0.1 M pH 7.4), or a combination of albumin with each enzyme source in phosphate buffer (0.1 M, pH 7.4). The dialysis cell assembly was immersed in a water bath maintained at 37°C and rotated at 12 rpm for 4 h. Control experiments were also performed with phosphate buffer or albumin on both sides of the dialysis cells at low and high concentrations of each substrate to ensure that equilibrium was attained. A 200-μl aliquot was collected from each compartment, treated with ice-cold methanol containing 4% glacial acetic acid (200 μl), and cooled on ice. Samples were subsequently centrifuged at 4000g for 10 min at 10°C, and an aliquot of the supernatant fraction (5 μl) was analyzed by HPLC.
Quantification of PRO. The HPLC instrument and column used to measure PRO binding was as described for the measurement of PRO-Gluc formation. Separation of PRO was achieved using a 25:75 mixture of mobile phases A and B, as used for the PRO-Gluc assay. The mobile phase flow rate was 0.6 ml/min. Column eluant was monitored at 214 nm. The retention time for PRO under these conditions was 3.6 min. PRO concentrations in dialysis samples were determined by comparison of peak areas to those of a standard curve in the concentration range of 2 to 500 μM. Within-day assay variability was assessed by measuring PRO (10 and 500 μM) (n = 5 for each concentration) in samples containing phosphate buffer (0.1 M, pH 7.4) or BSA in phosphate buffer (0.1 M, pH 7.4). Coefficients of variation were less than 5% in all cases.
Data Analysis. Kinetic data are presented as mean values derived from duplicate experiments with either pooled HLM or recombinant UGT1A proteins. Kinetic constants (Km and Vmax) for PRO glucuronidation by HLM and recombinant UGT1A9 in the presence and absence of albumin were generated by fitting experimental data to the Michaelis-Menten equation. Similarly, kinetic constants (Km and Vmax) for 4MU β-d-glucuronidation by recombinant UGT1A1 and UGT1A9 in the presence and absence of albumin were generated by fitting experimental data to the Michaelis-Menten equation, except for UGT1A1 in the presence of BSA, for which kinetic constants (Km, Vmax, α, and β) were obtained by fitting experimental data to the two-site model described by Houston and Kenworthy (2000) and Uchaipichat et al. (2004):  where Ks represents binding affinity and α and β are modifying factors that reflect changes in Ks and Kp (the catalytic rate constant), respectively.
where Ks represents binding affinity and α and β are modifying factors that reflect changes in Ks and Kp (the catalytic rate constant), respectively.
Similarly, kinetic constants (Km, Vmax, and α) for 4MU β-d-glucuronidation by recombinant UGT1A6 in the presence and absence of albumin were generated by fitting experimental data to the two-site model with β set to 2 (autoactivation) (Uchaipichat et al., 2004). In all cases, fitting was based on unbound substrate concentrations (i.e., corrected for albumin and nonspecific binding) in incubations and performed with EnzFitter (Biosoft, Cambridge, UK). For reactions exhibiting Michaelis-Menten kinetics, intrinsic clearance (CLint) was determined as Vmax/Km.
Results
Binding of PRO to Albumin. The binding of PRO to incubation components was calculated as the concentration of drug in the buffer compartment divided by the concentration of drug in the protein compartment and expressed as the fraction unbound in incubations (fu, inc). In the absence of albumin, the binding of PRO to HEK293 cell lysate (0.25 mg/ml) was negligible (<5%). However, PRO bound significantly to HLM and all forms of albumin. The binding of PRO to HLM was independent of substrate concentration but varied with protein concentration; in the presence of 0.5 mg/ml HLM, the mean fu for PRO was 0.70. Similarly, the binding of PRO to albumin did not vary with substrate concentration or albumin form, but varied with albumin concentration. In the presence of 0.1, 1.0, and 2.0% albumin, mean fu values for PRO were 0.70, 0.25, and 0.20, respectively. Interestingly, the binding of PRO to albumin plus HLM (0.25 mg/ml) and albumin plus HEK293 cell lysate (0.25 mg/ml) was identical to values observed for binding of PRO to the individual albumins alone, indicating that the binding is not additive. Nonadditive binding to BSA and HLM has been observed previously with lamotrigine (Rowland et al., 2006). Recovery of PRO was >95% in all cases. Confirmatory binding studies were also performed with 4MU. The fu, inc values for 4MU binding to 0.1, 1, and 2% BSA, HSA, and HSAFAF were 0.89, 0.49, and 0.27, 0.79, 0.37, and 0.14, and 0.76, 0.34, and 0.09, respectively. These values are in agreement with previous results from this laboratory (Rowland et al., 2007). Where binding was observed, the concentrations of PRO and 4MU present in incubation mixtures were corrected in the calculation of kinetic parameters.

Eadie-Hofstee plots (v versus V/[S]) for 4MU glucuronidation by recombinant UGT1A1 in the presence and absence of BSA (2%). Points are experimentally determined values, and curves are from model fitting.
Effect of Albumin on 4MU Glucuronidation by Recombinant UGT1A Enzymes. The kinetics of 4MUG formation by recombinant UGT1A1, in the presence and absence of albumin, were best described by the Michaelis-Menten equation with the exception of UGT1A1 in the presence of BSA for which kinetic data were consistent with a two-site model that assumes the simultaneous binding of substrate to two equivalent “catalytic” sites (Houston and Kenworthy 2000; Uchaipichat et al., 2004). The Km and Vmax values for 4MU glucuronidation by recombinant UGT1A1 in the absence of albumin were 59 μM and 374 pmol/min · mg, respectively. Addition of BSA (0.1, 1, and 2%) altered the kinetics of 4MU glucuronidation by recombinant UGT1A1 from Michaelis-Menten to two-site (Fig. 1). In contrast, neither HSA nor HSAFAF had an effect on the kinetic model of 4MUG formation by UGT1A1. HSA caused a concentration-dependant increase in the Km for this pathway with no effect on Vmax, whereas HSAFAF did not appreciably affect any of the kinetic parameters (Table 1).
Kinetic parameters for 4MU β-d-glucuronide formation by recombinant UGT1A enzymes
Kinetic constants were generated from fitting experimental data to the Michaelis-Menten equation except UGT1A1 in the presence of BSA and all kinetic data for UGT1A6.
Although the kinetics of 4MUG formation by UGT1A6 in the absence of albumin were adequately described by the Michaelis-Menten equation (Km of 59 μM and Vmax of 86,682 pmol/min · mg), inspection of kinetic plots indicated increasing autoactivation in the presence of all forms of albumin, and hence data were analyzed using the two-site model. By way of example, Fig. 2 shows the kinetics of 4MU glucuronidation by UGT1A6 in the presence of increasing concentrations of BSA. In the absence of albumin the Ks, Vmax, and α values for 4MUG formation by recombinant UGT1A6 were 78 μM, 82,151 pmol/min · mg, and 0.46, respectively. All forms of albumin caused a concentration-dependant decrease in the value of α, consistent with increasing autoactivation. With the exception of higher concentrations of HSAFAF, Ks tended to increase with increasing albumin concentration (Table 1).
4MU glucuronidation kinetics by recombinant UGT1A9, in the presence and absence of albumin, were best described by the Michaelis-Menten equation. In the absence of albumin, the Km and Vmax values for 4MUG formation by recombinant UGT1A9 were 13.4 μM and 8362 pmol/min · mg, respectively. BSA and HSAFAF increased the rate of 4MU glucuronidation by recombinant UGT1A9 in a concentration-dependant manner (Fig. 3) by decreasing the Km for this pathway, without an appreciable effect on Vmax (Table 1). In contrast, crude HSA decreased the rate of 4MU glucuronidation by recombinant UGT1A9, due largely to an increase in Km.
Effect of Albumin on PRO Glucuronidation by HLM and Recombinant UGT1A9. Kinetic data for PRO glucuronidation by HLM and recombinant UGT1A9, in the presence and absence of albumin, were obtained by fitting experimental data to the Michaelis-Menten equation. Kinetic parameters for PRO glucuronidation by HLM and recombinant UGT1A9 (in the presence and absence of albumin) are shown in Table 2. In the absence of albumin, the respective Km and Vmax values for PRO glucuronidation by HLM and recombinant UGT1A9 were 127 μM and 967 pmol/min · mg and 41 μM and 2285 pmol/min · mg. BSA and HSAFAF (0.1, 1, and 2%) increased the rate of PRO glucuronidation by both human liver microsomal and recombinant UGT1A9 in a concentration-dependant manner (Fig. 3) by decreasing the Km for this pathway (Table 2). In contrast, crude HSA decreased the rate of PRO glucuronidation by HLM and recombinant UGT1A9 via an increase in Km (Table 2).
Kinetic parameters for PRO glucuronidation by HLM and recombinant UGT1A9
Inhibition of 4MU and PRO Glucuronidation by Fatty Acids in the Presence and Absence of Albumin. Potential inhibition of UGT1A1-, UGT1A6-, and UGT1A9-catalyzed 4MU glucuronidation by oleic acid (C18:1), linoleic acid (C18:2), and arachidonic acid (C20:4) was measured in the presence and absence of BSA (2%). When added at  of the concentration observed in HLM (Rowland et al., 2007), C18:1 (3 μM), C18:2 (3 μM), and C20:4 (1.5 μM) inhibited the glucuronidation of 4MU by UGT1A9 by 28, 31, and 68%, respectively (Fig. 4A). The addition of BSA (2%) to incubations eliminated the inhibitory effect of each fatty acid (< 2% difference to control values). Inhibition of UGT1A1- and UGT1A6-catalyzed 4MUG formation was negligible (<5%) for all fatty acids, both in the presence and absence of BSA. Inhibition of PRO glucuronidation by C18:1, C18:2, and C20:4 was also measured in the presence and absence of BSA (2%) using recombinant UGT1A9 as the enzyme source. C18:1 (3 μM), C18:2 (3 μM), and C20:4 (1.5 μM) inhibited the UGT1A9-catalyzed glucuronidation of PRO by 25, 26, and 44%, respectively (Fig. 4B). As with HLM, the addition of BSA (2%) in incubations eliminated the inhibitory effects of all fatty acids (<3% difference to control values).
of the concentration observed in HLM (Rowland et al., 2007), C18:1 (3 μM), C18:2 (3 μM), and C20:4 (1.5 μM) inhibited the glucuronidation of 4MU by UGT1A9 by 28, 31, and 68%, respectively (Fig. 4A). The addition of BSA (2%) to incubations eliminated the inhibitory effect of each fatty acid (< 2% difference to control values). Inhibition of UGT1A1- and UGT1A6-catalyzed 4MUG formation was negligible (<5%) for all fatty acids, both in the presence and absence of BSA. Inhibition of PRO glucuronidation by C18:1, C18:2, and C20:4 was also measured in the presence and absence of BSA (2%) using recombinant UGT1A9 as the enzyme source. C18:1 (3 μM), C18:2 (3 μM), and C20:4 (1.5 μM) inhibited the UGT1A9-catalyzed glucuronidation of PRO by 25, 26, and 44%, respectively (Fig. 4B). As with HLM, the addition of BSA (2%) in incubations eliminated the inhibitory effects of all fatty acids (<3% difference to control values).

Eadie-Hofstee plots (v versus V/[S]) for 4MU glucuronidation by recombinant UGT1A6 in the presence of increasing concentrations of BSA (0–2%). Points are experimentally determined values, and curves are from model fitting.
The effect of a mixture of fatty acids (FAM) (comprising 3 μM C18:1, 3 μM C18:2, and 1.5 μM C20:4) on the kinetics of 4MU and PRO glucuronidation by recombinant UGT1A9 was characterized in the presence and absence of BSA (2%). In the absence of BSA (2%), the FAM caused a 6-fold increase in the Km for 4MU glucuronidation by recombinant UGT1A9, from 13 to 89 μM with only a minor effect on Vmax (8920 versus 9304 pmol/min · mg) (Fig. 5). In contrast, the FAM had no effect on the kinetics of 4MU glucuronidation by recombinant UGT1A9 in the presence of BSA (2%). Respective Km and Vmax values obtained for incubations containing BSA were 3.8 and 3.8 μM and 9817 and 10,208 pmol/min · mg in the absence and presence of the FAM (Fig. 5).
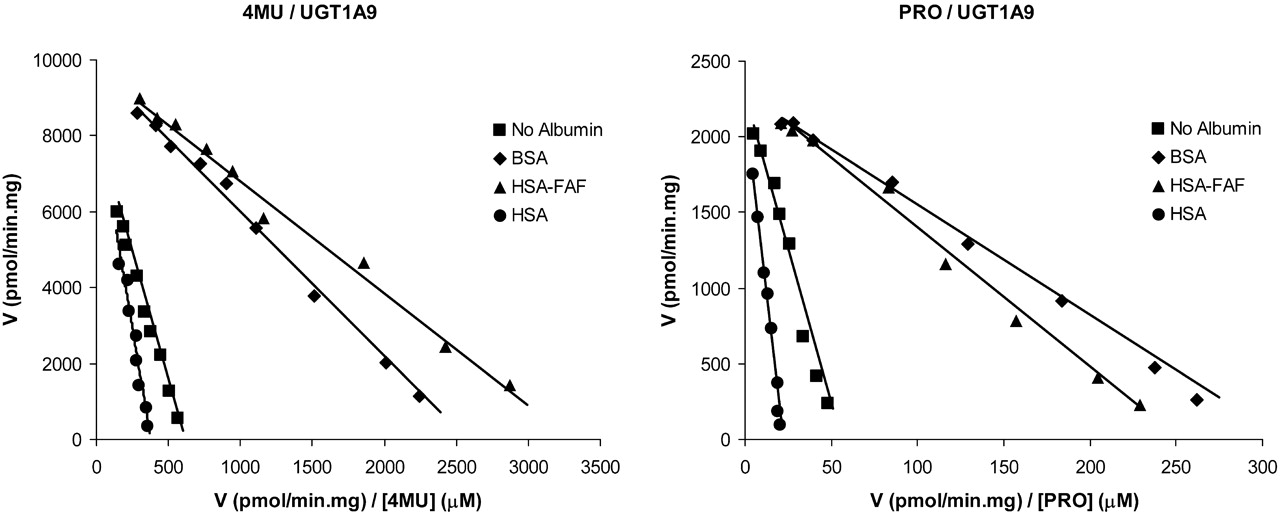
Eadie-Hofstee plots (v versus V/[S]) for 4MU and PRO glucuronidation by recombinant UGT1A9 in the presence and absence of albumin. Points are experimentally determined values, and lines are from model fitting.

The effects of oleic acid (C18:1), linoleic acid (C18:2), and arachidonic acid (C20:4) on the rates of UGT1A9 catalyzed 4MU (A) and PRO (B) glucuronidation. Incubations were performed as described under Materials and Methods, at 4MU and PRO concentrations of 15 and 40 μM, respectively (which correspond to the approximate Km values of these substrates). Bars show the mean of duplicate measurements and “control” refers to the activity in the absence of added fatty acid.
Similarly, the FAM increased the Km for PRO glucuronidation by recombinant UGT1A9, from 41 to 127 μM, with only a minor effect on Vmax (2285 versus 2101 pmol/min · mg) (Fig. 5). In the presence of BSA (2%), the FAM had a negligible effect on the kinetics of PRO glucuronidation by recombinant UGT1A9; respective Km and Vmax values for experiments performed in the absence and presence of the FAM were 7.2 and 7.5 μM, and 2280 and 2316 pmol/min · mg (Fig. 5).
Discussion
Addition of BSA and HSAFAF to incubations of UGT1A9 (expressed in HEK293 cells) caused a concentration-dependent decrease in the Km values for 4MU and PRO glucuronidation. At an albumin concentration of 2%, Km values were decreased by approximately 70 to 80%, with little or no effect on Vmax. Because 4MU is a nonselective UGT substrate, kinetic studies with HLM as the enzyme source were performed only with PRO. Results were generally similar to those observed with recombinant UGT1A9. Km values for PRO glucuronidation by HLM were 88 and 92% lower than control values in the presence of 2% BSA and HSAFAF, respectively, whereas Vmax remained essentially unchanged. In contrast, addition of crude HSA to incubations increased the Km for 4MU and PRO glucuronidation by UGT1A9.
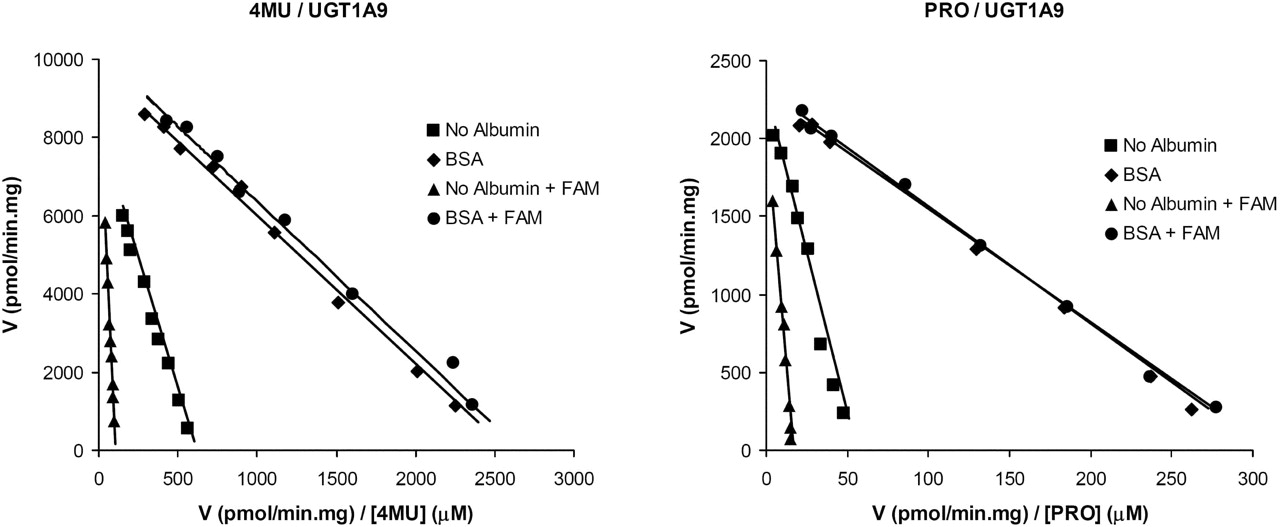
Eadie-Hofstee plots (v versus V/[S]) for 4MU and PRO glucuronidation by recombinant UGT1A9 in the presence and absence of a fatty acid mixture, with and without added BSA. Points are experimentally determined values, and lines are from model fitting.
We have reported previously that BSA and HSAFAF but not HSA decrease the Km values for zidovudine glucuronidation by UGT2B7 and HLM and for phenytoin hydroxylation by CYP2C9 and HLM. The decrease in Km values, approximately 1 order of magnitude, arises from sequestration of long-chain unsaturated fatty acids released from microsomes or cell lysate during the course of an incubation (Rowland et al., 2007, 2008). Long-chain unsaturated fatty acids, particularly arachidonic, linoleic, and oleic, were shown to be potent competitive inhibitors of UGT2B7 and CYP2C9 (Tsoutsikos et al., 2004; Rowland et al., 2007, 2008). The present work indicates that the same mechanism is responsible for the enhanced glucuronidation of UGT1A9 substrates observed in the presence of BSA and HSAFAF. Arachidonic, linoleic, and oleic acids, individually and as a mixture (at concentrations corresponding to approximately  of the content of these fatty acids present in HLM), inhibited UGT1A9-catalyzed 4MU and PRO glucuronidation. Km values for 4MU and PRO glucuronidation were increased 3- to 6-fold by the FAM without a change in Vmax, consistent with competitive inhibition. Inhibition was abolished by the addition of BSA (2%) to incubations. As with UGT2B7 and CYP2C9 (Rowland et al., 2007, 2008), long-chain unsaturated fatty acids are substrates of UGT1A9 (P. Gaganis, K. M. Knights, and J. O. Miners, unpublished data).
of the content of these fatty acids present in HLM), inhibited UGT1A9-catalyzed 4MU and PRO glucuronidation. Km values for 4MU and PRO glucuronidation were increased 3- to 6-fold by the FAM without a change in Vmax, consistent with competitive inhibition. Inhibition was abolished by the addition of BSA (2%) to incubations. As with UGT2B7 and CYP2C9 (Rowland et al., 2007, 2008), long-chain unsaturated fatty acids are substrates of UGT1A9 (P. Gaganis, K. M. Knights, and J. O. Miners, unpublished data).
As noted in the Introduction, there is a perception that glucuronidation is a low-affinity metabolic pathway. However, when taken together with previously published data for zidovudine glucuronidation and the zidovudine-fluconazole and valproic acid-lamotrigine interactions (Uchaipichat et al., 2006; Rowland et al., 2006, 2007), the present study suggests that published Km and Ki values of UGT1A9 and UGT2B7 substrates and inhibitors may be overestimated by as much as 1 order of magnitude because of to the inhibitory effects of fatty acids that are “released” during an incubation. It is noteworthy that low Km values (<5 μM) have been reported for substrates of enzymes not subject to inhibition by long-chain unsaturated fatty acids, for example, bilirubin glucuronidation by UGT1A1 (Udomuksorn et al., 2007) and trifluoperazine glucuronidation by UGT1A4 (Uchaipichat et al., 2006a). The present study further demonstrates that Km (and Ki) values generated using recombinant enzymes are generally lower than those obtained with HLM (Court 2005), presumably because of the lower fatty acid content of expression systems (Rowland et al., 2007, 2008).
Extrapolation of kinetic constants (CLint and Ki) generated in the presence of BSA have been reported to improve the prediction of in vivo CLH for zidovudine (Rowland et al., 2007) and phenytoin (Rowland et al., 2008) and the magnitude of the fluconazole-zidovudine (Uchaipichat et al., 2006b) and valproic acid-lamotrigine interactions (Rowland et al., 2006). PRO disposition in vivo is well characterized. The mean systemic clearance, generally assumed to be almost entirely due to metabolism, taken from four studies is 108 liters/h (Gepts et al., 1987; Servin et al., 1988; Morgan et al., 1990; Bailie et al., 1992). Glucuronidation accounts for 53% of PRO metabolism in humans (Simons et al., 1988); hence clearance via glucuronidation is 57 liters/h.
CLint values obtained for PRO glucuronidation by HLM were extrapolated to a “whole liver” CLint using a microsome yield of 38 mg/g (Rowland et al., 2008) and a liver weight of 1500 g. Extrapolated whole liver CLint values were then substituted in the expression for the well stirred model of hepatic clearance (Houston 1994) assuming that liver blood flow is 90 liters/h and that the fraction of PRO unbound in blood is 0.364. It should be noted that the measured fraction of PRO unbound in blood is 0.014 (Mazoit and Samii, 1999). However, the binding affinity (KD) of the component “free” within erythrocytes (0.35) is very low, approximately 2 mM (Mazoit and Samii, 1999). If it is assumed that this component is diffusible, then the “available” fraction unbound in blood is 0.364. Predicted hepatic clearances based on microsomal CLint values generated in the absence of albumin and in the presence of BSA (2%) and HSAFAF (2%) were 9, 44. and 50 liters/h, respectively.
A previous report from this laboratory demonstrated that lamotrigine glucuronidation by UGT1A4 was unaffected by BSA (Rowland et al., 2006). Here, rates of 4MU glucuronidation by UGT1A1 and UGT1A6 were similarly not enhanced by BSA and HSAFAF. Unexpectedly, BSA, but not HSA or HSAFAF, altered the Eadie-Hofstee plot for 4MU glucuronidation by UGT1A1 from Michaelis-Menten to one resembling substrate inhibition. However, the fit of kinetic data to the substrate inhibition equation was poor (data not shown). Rather, the kinetics of 4MU glucuronidation by UGT1A1 in the presence of BSA were best described by a model that assumes two equivalent interacting substrate binding sites (Houston and Kenworthy, 2000). We have shown previously that the “atypical” (substrate inhibition and sigmoidal) kinetics observed for 4MU and 1-naphthol glucuronidation by several UGT enzymes were well described by the two-site model (Uchaipichat et al., 2004).
Although 4MU glucuronidation by UGT1A6 was adequately modeled by the Michaelis-Menten equation, consistent with Uchaipichat et al. (2004), closer inspection of Eadie-Hofstee plots (Fig. 2) revealed nonlinearity, which became increasingly evident in the presence of increasing concentrations of albumin. Data were consistent with autoactivation, and values of the interaction factor α decreased with increasing albumin concentration. With the exception of HSAFAF (0.1 and 1%), binding affinity tended to decrease (i.e., Ks increased) in the presence of albumin. The mechanism by which the various albumin preparations alter the kinetics of 4MU glucuronidation by UGT1A1 and UGT1A6 is unknown. Interestingly, UGT1A1 has been reported recently to immunocoprecipitate with HSA (Ohta et al., 2005), suggesting the possibility of a direct protein-protein interaction with some UGTs. The current unavailability of selective drug substrates for UGT1A1 and UGT1A6 in vivo precludes assessment of IV-IVE for compounds metabolized by these enzymes.
In summary, BSA and HSAFAF reduced the Km values for 4MU and PRO glucuronidation by UGT1A9 and for PRO glucuronidation by HLM by sequestering inhibitory long-chain unsaturated fatty acids. The approximate 10-fold decrease in the Km for human liver microsomal PRO glucuronidation is similar to the reduction observed for the UGT2B7 substrate zidovudine. Extrapolation of the microsomal CLint obtained in the presence of HSAFAF underpredicted PRO hepatic clearance via glucuronidation by only 13%. In contrast to the effects of BSA and HSAFAF on UGT1A9 and UGT2B7 activities, neither form of albumin enhanced UGT1A1 and UGT1A6 catalyzed 4MU glucuronidation. Paradoxically, the albumin preparations variably altered the kinetic model of 4MU by UGT1A1 and UGT1A6, and the effects of albumin on reactions catalyzed by these enzymes require careful interpretation. However, available evidence indicates that inclusion of BSA or HSAFAF in incubations is likely to improve the accuracy of hepatic clearances predicted from microsomal CLint values for substrates of UGT1A9 and UGT2B7, arguably the most important enzymes involved in drug glucuronidation in humans, and indeed for substrates of cytochromes P450 inhibited by long-chain unsaturated fatty acids (Rowland et al., 2008).
Acknowledgments
Assistance from D. J. Elliot in the establishment of the HPLC method for propofol glucuronidation is gratefully acknowledged.
Footnotes
-
This work was funded by a grant from the National Health and Medical Research Council of Australia. A.R. is the recipient of a Flinders University Postgraduate Research Scholarship.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.021105.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; IV-IVE, in vitro-in vivo extrapolation; HLM, human liver microsomes; BSA, bovine serum albumin; HSAFAF, human serum albumin, fatty acid-free; HSA, human serum albumin; 4MU, 4-methylumbelliferone; PRO, propofol; 4MUG, 4-methylumbelliferone β-D-glucuronide; UDPGA, UDP-glucuronic acid; HPLC, high-performance liquid chromatography; PRO-Gluc, propofol glucuronide; FAM, fatty acid mixture (40% C18:1, 40% C18:2, and 20% C20:4).
- Received February 17, 2008.
- Accepted March 20, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}