


Abstract
2-Acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (prasugrel) is a novel thienopyridine prodrug with demonstrated inhibition of platelet aggregation and activation. The biotransformation of prasugrel to its active metabolite, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid (R-138727), requires ester bond hydrolysis, forming the thiolactone 2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone(R-95913), followed by cytochrome P450–mediated metabolism to the active metabolite. The presumed role of the human liver- and intestinal-dominant carboxylesterases, hCE1 and hCE2, respectively, in the conversion of prasugrel to R-95913 was determined using expressed and purified enzymes. The hydrolysis of prasugrel is at least 25 times greater with hCE2 than hCE1. Hydrolysis of prasugrel by hCE1 showed Michaelis-Menten kinetics yielding an apparent Km of 9.25 μM and an apparent Vmax of 0.725 nmol product/min/μg protein. Hydrolysis of prasugrel by hCE2 showed a mixture of Hill kinetics at low substrate concentrations and substrate inhibition at high concentrations. At low concentrations, prasugrel hydrolysis by hCE2 yielded an apparent Ks of 11.1 μM, an apparent Vmax of 19.0 nmol/min/μg, and an apparent Hill coefficient of 1.42, whereas at high concentrations, an apparent IC50 of 76.5 μM was obtained. In humans, no in vivo evidence of inhibition exists. In vitro transport studies using the intestinal Caco-2 epithelial cell model showed a high in vivo absorption potential for prasugrel and rapid conversion to R-95913. In conclusion, the human carboxylesterases efficiently mediate the conversion of prasugrel to R-95913. These data help explain the rapid appearance of R-138727 in human plasma, where maximum concentrations are observed 0.5 h after a prasugrel p.o. dose, and the rapid onset of action of prasugrel.
Platelet aggregation and activation are serious concerns for patients who undergo percutaneous coronary intervention and stent placement. The underlying mechanism of platelet aggregation is mediated through two G protein-coupled P2 receptors, P2Y1 and P2Y12 (Gachet, 2001). P2Y1 activation leads to a transient aggregation, whereas P2Y12 activation maintains a sustained aggregation. To reduce platelet aggregation, the development of P2Y12-selective inhibitors has yielded the thienopyridine prodrugs, which include ticlopidine, clopidogrel (structures available in Farid et al., 2008), and prasugrel (Fig. 1), a novel thienopyridine currently in clinical development.
Although the active metabolites for prasugrel and clopidogrel have equipotency at the P2Y12 receptor in vitro (Sugidachi et al., 2007), p.o. administered prasugrel is 10 and 100 times more effective on an equal-dose basis in inhibiting platelet aggregation than clopidogrel and ticlopidine, respectively (Niitsu et al., 2005). Clopidogrel and prasugrel differ markedly in the biotransformation pathways leading to their activation. Prasugrel (Fig. 1) has a single dominant metabolic pathway leading to the active metabolite (Farid et al., 2007a). However, clopidogrel has two competing metabolic pathways for the parent compound, with the major pathway leading to the formation of an inactive metabolite, clopidogrel carboxylic acid derivative (Caplain et al., 1999). The clopidogrel carboxylic acid derivative is formed through ester hydrolysis by the human carboxylesterase (hCE) 1 (Tang et al., 2006). The minor pathway in clopidogrel metabolism yielding the active metabolite requires two sequential steps of cytochrome P450 (P450) biotransformation (Kurihara et al., 2005), whereas prasugrel bioactivation requires the hydrolysis of the ester and then oxidation of the formed thiolactone, R-95913 (Farid et al., 2007a) (Fig. 1), to form the active metabolite of prasugrel. The oxidation of R-95913 has been shown to be mediated by several P450 enzymes but primarily by CYP3A and CYP2B6 (Rehmel et al., 2006).
The carboxylesterases are a multigene family that hydrolyze compounds containing an ester, amide, or thioester linkage. Carboxylesterases are broadly expressed throughout the body with two major forms in humans, hCE1 and hCE2. Although both forms have high mRNA expression in the liver, hCE1 levels exceed those of hCE2 (Satoh et al., 2002). Importantly, the extrahepatic expression differs between hCE1 and hCE2 (Satoh et al., 2002). For hCE1, the liver-dominant form, extrahepatic mRNA expression observed in decreasing order are the stomach, testis, kidney, spleen, and colon. The intestinal-dominant form, hCE2, has extrahepatic mRNA expression in decreasing order in the colon, small intestine, and heart. In addition to the studies examining mRNA expression, the relative activity of the two enzymes has been recently shown in the liver and small intestine (Imai et al., 2006). Using tissue preparations, hCE1 and hCE2 were separated in nondenaturing gels, and the hydrolysis of 1-naphthylbutyrate was used to determine the relative activity of the two enzymes. Both enzymes were detected in the liver, but hCE1 was clearly the dominant form. Unlike the liver, the small intestine showed that hCE2 was the dominant form with a minor contribution by hCE1. This pattern of tissue distribution has been further demonstrated (Taketani et al., 2007).

The metabolic pathway of prasugrel leading to its active metabolite.
Although hCE1 and hCE2 have overlapping substrate recognition, clear evidence of ester-based substrate specificity has been observed (Satoh et al., 2002). Two products result from ester hydrolysis, an alcohol and an acyl moiety. In general, hCE1 prefers substrates with a large acyl moiety, whereas hCE2 prefers substrates with a large alcohol substituent. Based on this substrate-activity relationship, prasugrel would be predicted to be a preferred substrate for hCE2.
This study aims to investigate the role of hCE1 and hCE2, the dominant forms in the liver and intestinal tract, respectively, in the bioactivation of prasugrel. To accomplish this endeavor, hCE1 and hCE2 enzymes were expressed and purified and used to determine the formation kinetics of R-95913 from prasugrel. Additional experiments were conducted with Caco-2 monolayers to assess the conversion of prasugrel to R-95913 and their relative transit across this intestinal model.
Materials and Methods
In Vitro Hydrolysis Assays. For the hydrolysis assays, each reaction tube contained buffer, enzyme, prasugrel, and acetonitrile in a total volume of 600 μl. The buffer used was Dulbecco's phosphate buffered saline (14040-117, Invitrogen, Carlsbad, CA). A 5 mM stock of prasugrel was prepared in acetonitrile. The final reaction volume contained a total of 2% acetonitrile. Prasugrel concentrations used with hCE1 were 0.855, 1.71, 3.42, 6.84, 13.7, 27.4, 54.7, and 109 μM. In addition to the concentrations used with hCE1, the hCE2 studies also used 21.9, 40.5, 71.2, and 87.6 μM. The expression and purification of hCE1 and hCE2 were previously described (Williams et al., 2008). All the protein preparations are homogenous based on analysis of an SDS-polyacrylamide gel electrophoresis gel stained with SimplyBlue Safe-Stain (Invitrogen; data not shown). The tubes containing the various prasugrel concentrations were preincubated in a water bath at 37°C for approximately 1 min. The enzyme, either 1 μg/ml hCE1 or 0.25 μg/ml hCE2, was preincubated with buffer for approximately 5 min at 37°C and added to the tubes with prasugrel to start the reaction. A 100-μl aliquot was removed from the reaction tube at 1, 2, 3, and 6 min after reaction initiation, and then 100 μl of acetonitrile containing 2 μg/ml of a d4-labeled R-95913 as the internal standard was added to each aliquot to terminate the reaction. All the studies were conducted in triplicate.
For the standards, a stock solution of R-95913 was prepared in acetonitrile at 2 mg/ml. The concentrations ranged from 4.88 ng/ml to 40 μg/ml in 2% acetonitrile in Dulbecco's phosphate buffered saline. Each standard (100 μl) was added to 100 μl of acetonitrile with internal standard. In addition to the standards, two blanks were also included. One blank was lacking R-95913 but included the internal standard. The second blank was lacking both R-95913 and internal standard. Samples and standards had two dilution schemes for analysis, 1:25 and 1:275 dilution, with the diluent being methanol/water (1:1, v/v). The appropriate dilution was used for analysis to ensure samples did not saturate the detector response of the mass spectrometer.
Study samples were analyzed by liquid chromatography/tandem mass spectrometry using a Sciex API 4000 triple quadrupole mass spectrometer (Applied Biosystems/MDS, Foster City, CA) equipped with a TurboIonSpray interface and operated in positive ion mode. The analytes were chromatographically separated using a Betasil C18 2.1 × 20 mm, 5 μm, Javelin high-performance liquid chromatography column (Thermo Fisher Scientific, Inc., Waltham, MA), with a gradient liquid chromatography system composed of water/1 M ammonium bicarbonate (200:1, v/v) (mobile phase A) and methanol/1 M ammonium bicarbonate (200:1, v/v) (mobile phase B). The pumps were a Shimadzu (Kyoto, Japan) LC-10AD with an SCL-10A controller. Also, a Gilson (Middleton, WI) 215 liquid handler was used. The gradient profile changed from 30% B at 0 min, 42% B at 0.01 to 0.10 min, 75% B at 0.20 to 0.30 min, and 98% at 0.31 to 0.76 min at a flow rate of 1.5 ml/min. Chromatography was performed at ambient temperature, with 1 ml/min directed to the mass spectrometer between 0.18 and 0.5 min (0.5 ml/min split to waste). Selected reaction monitoring (M + H)+ transitions m/z 332.2 > 177.1 and 336.2 > 149.2 were monitored for R-95913 and the internal standard, respectively. The TurboIonSpray temperature was maintained at 725°C, with collision, curtain, nebulizing, and desolvation gas (nitrogen) settings of 8, 10, 50, and 70, respectively. The ionspray voltage was set to 4000 V, and the respective declustering, entrance, collision, and exit potentials were 55, 10, 27, and 4 for R-95913 and 55, 10, 33, and 8 for the internal standard. The mass spectrometer quadrupoles were tuned to achieve unit resolution (0.7 Dalton at 50% full width at half maximum). Data were acquired and processed with Analyst 1.4.1 (Applied Biosystems).
For the assays with hCE1, the standards for the 1:25 dilution ranged from 19.5 to 2500 ng/ml. A correlation coefficient of 0.999 was achieved with less than a 7% deviation from the relative mean using a quadratic curve with a weighting of 1/x2. The standards for the 1:275 dilution ranged from 156 to 20,000 ng/ml. A correlation coefficient of 0.998 was achieved with less than a 12% deviation from the relative mean using a quadratic curve with a weighting of 1/x2. For the assays with hCE2, the standards for the 1:25 dilution ranged from 78.1 to 2500 ng/ml. A correlation coefficient of 0.998 was achieved with less than a 9% deviation from the relative mean using a quadratic curve with a weighting of 1/x2. The standards for the 1:275 dilution ranged from 625 to 10,000 ng/ml. A correlation coefficient of 0.999 was achieved with less than a 6% deviation from the relative mean using a quadratic curve with a weighting of 1/x2.
Enzyme Kinetic Modeling. Hydrolysis reaction rates (nanomole of product per minute per microgram of protein) were calculated from the linear portion of the product concentration versus time curve. Fitting the rate data to standard kinetic models (Copeland, 1996) was accomplished using WinNonlin (Pharsight Corp., Mountain View, CA). Michaelis-Menten, substrate inhibition, and Hill kinetic models with differing weighting were used. For the results with hCE1, Michaelis-Menten kinetics using a weighting of 1/y2 provided the best fit. However, the unusual kinetics of the formation of R-95913 by hCE2 could not be fit to the models described above as shown by the standard errors of the fit being large as compared with the kinetic values. Therefore, the product formation rate data obtained with incubation with hCE2 between 0.855 and 40.5 μM of prasugrel were found to best fit the Hill model using a weighting of 1/ŷ2. The rate data with hCE2 and prasugrel concentrations between 27.4 and 109 μM were fit to an IC50 model using Prism (GraphPad Software Inc., San Diego, CA). The various estimated parameters are reported as the value ± S.E. of the estimate.
Intestinal Transport and Metabolism Assays. The Caco-2 cell line was obtained from the American Type Culture Collection (Manassas, VA) and grown under standard culture conditions of 37°C in a humidified atmosphere containing 5% CO2. Caco-2 cell monolayers were cultured in 175-cm2 flasks in Dulbecco's modified Eagle's medium (12430-054; Invitrogen) supplemented with 10% fetal bovine serum (FB-01; Omega Scientific, Inc., Tarzana, CA), 1 mM sodium pyruvate (25-000-CI; Mediatech, Inc., Herndon, VA), 100 mM nonessential amino acids (25-025-CI), 2 mM l-glutamine, 100 U/ml penicillin (30-002-CI), and 100 μg/ml streptomycin. Cells were seeded at a density of 60,000 cell/cm2 onto collagen-coated 12-well Transwell polycarbonate membranes (0.4-μm pore size, 1.13-cm2 surface area) (Corning Life Sciences, Acton, MA) and used between 21 and 28 days after seeding. The culture medium was changed every other day for 10 days after seeding onto Transwell filters, and daily afterward. Cells of passage number 61 were used for these studies.
Studies were performed with Hanks' balanced salt solution (HBSS) (14065-056; Invitrogen) containing 10 mM HEPES (15630-080) and 15 mM glucose (G-5400; Sigma-Aldrich, St. Louis, MO), pH 7.4 (HBSS+), at 37°C with 5% CO2 in a humidified incubator. At the start of the experiment, monolayers were rinsed twice with HBSS+, and the transepithelial electrical resistance of each cell monolayer was measured using an Endohm-12 resistance meter. Monolayers having transepithelial electrical resistance values outside the range of 450 to 650 Ω · cm2 were discarded. Volumes in the apical and basolateral chambers were 0.5 and 1.5 ml, respectively. All the studies were conducted in triplicate.
The apical-to-basolateral studies used an apical dosing solution of 5 μM prasugrel along with reference compounds, 100 μM atenolol (A-7655; Sigma-Aldrich), and 10 μM pindolol (P-0778; Sigma-Aldrich). At each sampling time (0, 15, 30, 60, 90, and 120 min), a 200-μl aliquot of drug solution was removed from the basolateral receiver chamber and immediately replaced with an equal volume of drug-free buffer. Similarly, a 20-μl aliquot was removed from the apical donor chamber without replenishing the donor solution.
Monolayers were dosed with 0.5 mM Lucifer Yellow (L-453; Invitrogen) to determine postexperimental monolayer integrity. Lucifer Yellow was measured with a BMG FLUOstar 403 microplate reader using an excitation wavelength of 485 nm and an emission wavelength of 538 nm. Each determination was performed in triplicate and was not significantly greater than baseline.
The basolateral-to-apical studies used a basolateral dosing solution of 5 μM prasugrel along with reference compounds, 100 μM atenolol, and 10 μM pindolol. At each sampling time (0, 15, 30, 60, 90, and 120 min), a 200-μl aliquot of drug solution was removed from the apical receiver chamber and immediately replaced with an equal volume of drug-free buffer. Similarly, a 20-μl aliquot was removed from the basolateral donor chamber without replenishing the donor solution. A postexperimental monolayer integrity check was conducted with Lucifer Yellow, as described above.
A 10 mM stock solution of prasugrel and R-95913 in dimethyl sulfoxide was prepared. Further dilution in 1:1 acetonitrile/water gave a 100 μM stock. Atenolol stocks (10 mM in water and 100 μM in 1:1 acetonitrile/water) and pindolol stocks (10 mM in dimethyl sulfoxide and 10 μM in 1:1 acetonitrile/water) were prepared previously. Standard curves were obtained by serial dilution in 1% formic acid in acetonitrile/Hanks buffer, pH 7.4 (1:1).
Caco-2 study samples were diluted in 1% formic acid in acetonitrile/Hanks buffer, pH 7.4 (1:1), then analyzed by liquid chromatography/tandem mass spectrometry using a Sciex API 3000 triple quadrupole mass spectrometer (Applied Biosystems/MDS) equipped with a TurboIonSpray interface, and operated in positive ion mode. The analytes were chromatographically separated using a BDS Hypersil C18 30 × 2.1-mm, 3-μm high-performance liquid chromatography column (Thermo Fisher Scientific, Inc.). The buffer consisted of 25 mM NH4OH, adjusted to pH 3.5 with 88% formic acid. The gradient liquid chromatography system composed of 10% buffer in water (mobile phase A) and 10% buffer in acetonitrile (mobile phase B). Rheos 2000 micropumps (Thermo Fisher Scientific, Inc.) and CTC Analytics HTC PAL autosampler (Zwingen, Switzerland) were used. The mobile phase composition changed from 0% B at 0 min, 100% B at 0.02 to 1.00 min, 100% B at 1.00 to 2.50 min, 0% at 2.50 to 2.60 min, and 0% from 2.60 to 4.00 min, at a flow rate of 0.3 ml/min. Chromatography was performed at ambient temperature. Selected reaction monitoring (M + H)+ transitions m/z 374.0 > 206.1, 332.2 > 109.3, 267.2 > 145.0, and 249.0 > 116.2 were monitored for prasugrel, R-95913, atenolol, and pindolol, respectively. The TurboIonSpray temperature was maintained at 450°C, with collision, curtain, and nebulizer gas settings of 10, 10, and 8, respectively. The ionspray voltage was set to 5000 V, whereas the respective declustering, focusing, entrance, collision, and exit potentials were 46, 200, 10, 35, and 12 for prasugrel; 31, 150, 10, 50, and 10 for R-95913; 36, 170, 10, 35, and 8 for atenolol; and 36, 170, 10, 35, and 6 for pindolol.
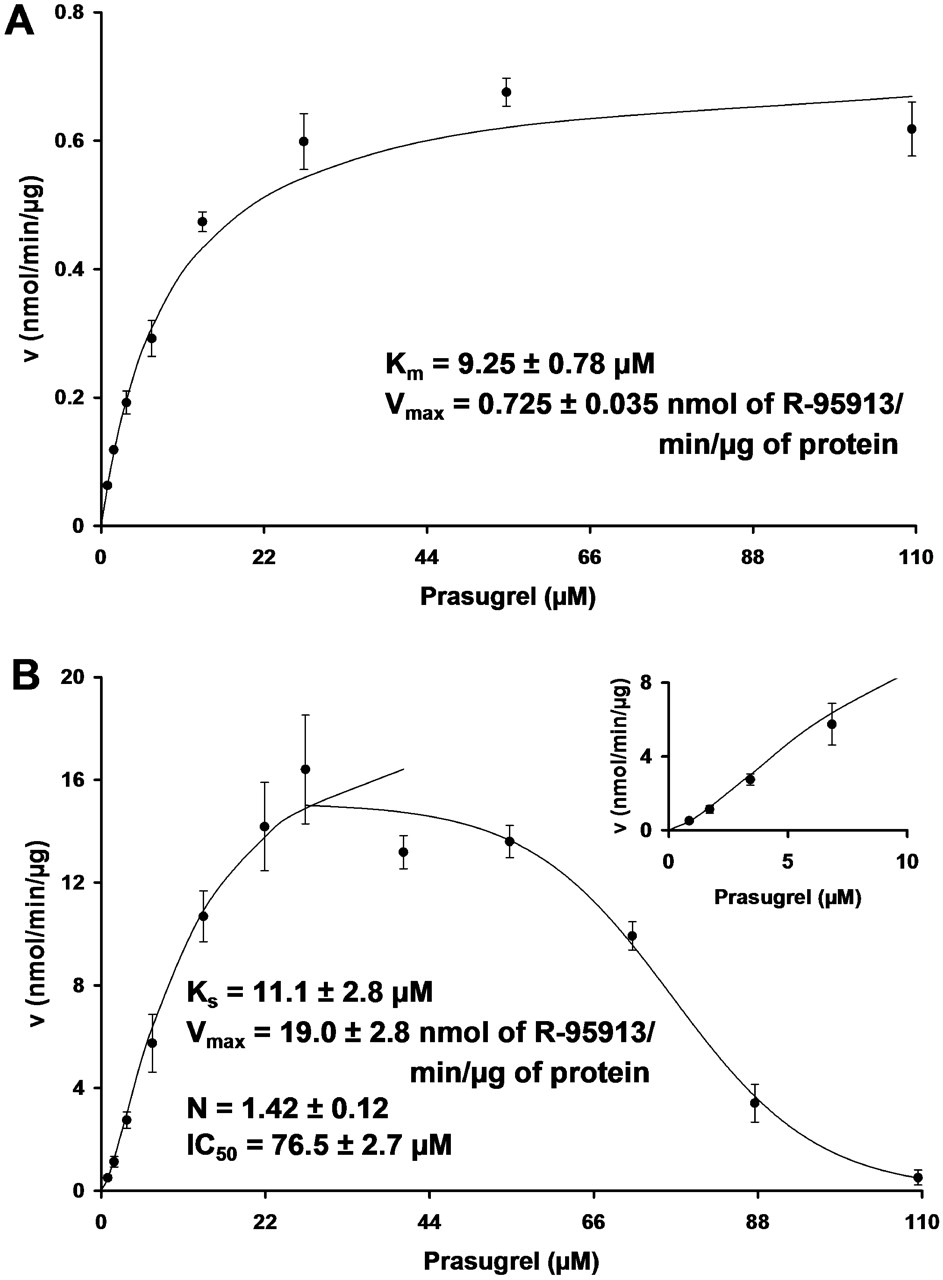
The kinetics plots for the hydrolysis of prasugrel by hCE1 (A) and hCE2 (B). The points with the error bars represent the average and S.E., whereas the lines represent the best model fit of their respective kinetic models. A, for hCE1, the curve is standard Michaelis-Menten kinetics. B, for hCE2, data up to 40.5 μM were fit to the Hill equation, whereas the data 27.4 μM and above were fit to a standard IC50 model. The inset shows the sigmoidicity of the Hill kinetics.
The standards for prasugrel, R-95913, pindolol, and atenolol ranged from 10 to 1000 nM. For prasugrel and R-95913, the correlation coefficients were 1.00 and 1.00, respectively, with a maximum deviation of 3 and 4%, respectively, from the relative mean using a quadratic curve with a weighting of 1/x2. For pindolol and atenolol, the correlation coefficients were 0.998 and 0.999, respectively, with a maximum deviation of 8.8 and 10%, respectively, from the relative mean using a linear curve with a weighting of 1/x2.
The formation and disappearance rates of prasugrel and R-95913 were calculated using Microsoft Excel (Redmond, WA). The rates (nanomole per hour) are the result of using the SLOPE function with data points in the linear range, as described under Results.
Results
The human carboxylesterases hCE1 and hCE2 efficiently hydrolyzed prasugrel to R-95913, as shown in Fig. 2. Figure 2A depicts the fit of standard Michaelis-Menten kinetics to the rate data for hCE1, which yields an apparent Km of 9.25 ± 0.78 μM and an apparent Vmax of 0.725 ± 0.035 nmol product/min/μg protein. However, the results obtained using hCE2 (Fig. 2B) were determined to fit poorly to all the standard enzyme kinetic models, including that for substrate inhibition. Therefore, the results obtained with hCE2 were divided into two datasets for modeling. The first dataset consisted of the results between prasugrel concentrations of 0.855 and 40.5 μM to model for standard enzyme kinetics. The best fit with this dataset was produced using the Hill equation, which gave an apparent Ks of 11.1 ± 2.8 μM, an apparent Vmax of 19.0 ± 2.8 nmol product/min/μg protein, and an apparent Hill coefficient (N) of 1.42 ± 0.12. The second portion of the curve resembles an inhibition plot, and as such the data between 27.4 and 109 μM were modeled for inhibition to yield an apparent IC50 of 76.5 ± 2.7 μM.

Cumulative amounts of prasugrel (solid line) and R-95913 (dotted line) in the donor and receiver wells for apical to basolateral (A and B, respectively) and basolateral to apical (C and D, respectively) experiments using a Caco-2 monolayer. Graphs are displayed as the average with the error bars representing the S.E.
Because of the different models needed to fully describe the diverse kinetics observed, the clearance near the therapeutic concentration range (Williams et al., 2002) was also compared. The clearance by each enzyme was calculated using the substrate concentrations between 0 and 14 μM, which resulted in a linear slope of 32 and 798 μl/min/μg for hCE1 and hCE2, respectively, yielding R2 values of 0.955 and 0.938, respectively. This comparison indicates that the rate of hydrolysis of prasugrel at low substrate concentrations is about 25 times greater for hCE2 than hCE1.
The Caco-2 monolayer transport and metabolism study showed the conversion of prasugrel to R-95913 (Fig. 3). The active metabolite of prasugrel, R-138727, was not monitored because CYP3A4, which is the primary P450 involved in its formation from the thiolactone, R-95913 (Rehmel et al., 2006), is not routinely expressed in Caco-2 monolayers (Cummins et al., 2004). Figure 3, A and B, shows the apical to basolateral (representing lumen to blood) conversion and transport of the cumulative amounts of prasugrel and R-95913 in the donor and receiver compartments, respectively. As shown in Fig. 3A, the appearance rate of R-95913 in the donor (apical) buffer is 1.65 ± 0.40 nmol/h between 0 and 30 min, and the loss of prasugrel between 0 and 30 min occurs at a rate of 2.00 ± 0.34 nmol/h. For Fig. 3B, the appearance rate of R-95913 in the receiver (basolateral) buffer is 0.730 ± 0.028 nmol/h between 0 and 90 min, and prasugrel was not detected. Similarly, Fig. 3, C and D, shows the basolateral to apical (representing blood to lumen) prasugrel conversion and transport. Figure 3C shows a loss of prasugrel in the donor (basolateral) buffer at a rate of 1.98 ± 0.10 nmol/h between 30 and 120 min, and the appearance rate of R-95913 between 0 and 90 min is 1.41 ± 0.08 nmol/h. In Fig. 3D, the appearance rate for R-95913 in the receiver (apical) buffer is 0.510 ± 0.043 nmol/h between 0 and 90 min, and prasugrel was not detected.
Discussion
These studies show that both hCE1 and hCE2 have similar Km and Ks values for the ester hydrolysis of prasugrel, suggesting they bind prasugrel with similar affinity. However, the rate of hydrolysis (Vmax) by hCE2 appears to be 26 times higher than that of hCE1. Because the pattern of kinetics for the hydrolysis of prasugrel by hCE1 and hCE2 was found to be quite different, the hydrolysis rates at low substrate concentrations, as previously described (Williams et al., 2002), were used as a potentially more meaningful comparison. This comparison indicates the hydrolysis of prasugrel by hCE2 at low substrate concentrations is 25 times greater than that for hCE1. This pattern of substrate selectivity by carboxylesterases is consistent with published data (Satoh et al., 2002). On hydrolysis of the ester, thioester, or amide bond, an alcohol and acyl moiety are released as metabolites. The relative sizes of these two moieties have been shown to predict which enzyme will have preferential recognition of the substrate (Satoh et al., 2002). Substrates yielding a smaller alcohol moiety, such as clopidogrel (Tang et al., 2006), are preferred by hCE1. However, hCE2 has a preference for substrates that yield a smaller acyl moiety on hydrolysis, like prasugrel.
Prasugrel appears to have a higher binding affinity (lower Km value) for the hCEs as compared with other characterized carboxylesterase substrates. When compared with irinotecan (CPT-11), the binding affinity of prasugrel is about 4 times greater for hCE1 but 10 times lower for hCE2 (Sanghani et al., 2004). Compared with heroin, the binding of prasugrel to hCE1 and hCE2 is substantially greater, by about 600 times (Kamendulis et al., 1996). Similar to heroin, the binding affinities of cocaine and 4-methylumbelliferyl acetate (Pindel et al., 1997) are lower by at least 10 times for prasugrel with both enzymes. Also, when compared with 4-nitrophenyl butyrate the binding affinity of prasugrel is about 10 times greater (Williams et al., 2008). Therefore, prasugrel appears to have a higher binding affinity for the hCEs than most carboxylesterase probe substrates.
For the hydrolysis of prasugrel by hCE2, the kinetic terms determined, Ks, Vmax, and IC50, are estimates of these descriptors because of the unusual shape of the kinetic curve. Because of the highly significant substrate inhibition observed, the reported Vmax is likely underestimated. Thus, it is best described in this context as the maximum achievable velocity. Because the Vmax is likely underestimated, the determined Ks and IC50 values may also be inaccurate. However, the observed values are relevant in the context of this system because the values reflect the maximal values possible using purified enzymes without other metabolic or clearance pathways involved.
The most unique feature of the prasugrel hydrolysis curve obtained with hCE2 is the steep inhibition phase that becomes apparent around the substrate concentration of 50 μM and results in nearly complete inhibition by 109 μM. Fitting the entire kinetic curve to the classical substrate inhibition model resulted in an extremely poor fit of the data at high substrate concentrations because the model does not accommodate nearly 100% inhibition. However, an enzyme kinetic model has been proposed to describe such results (Kühl, 1994). Kühl proposed that before the completion of the first catalytic cycle, an extended recovery period for the enzyme's active site could permit a side interaction with a second substrate molecule. This interaction would prevent the enzyme from returning to its initial state for a new reaction cycle until the second substrate molecule dissociates from the active site. Interestingly, this concept is consistent with the carboxylesterase reaction cycle, which involves the covalent binding of the acyl moiety of the substrate to the carboxylesterase active site, followed by cleavage of the ester bond and the release of the alcohol moiety, then regeneration of the active enzyme by the release of the acyl moiety from the enzyme (Satoh and Hosokawa, 1998). In the case of hCE2, which has a preference for a small acyl moiety, the initial substrate molecule would bind and cleavage would occur, allowing the bulky alcohol moiety to vacate the active site. Before the regeneration of the enzyme active site by the removal of the small acyl moiety, a second substrate molecule could bind and prevent the enzyme from completing the catalytic cycle. This would prevent the new molecule from being hydrolyzed and result in the inhibition observed for hCE2. This hypothesis is consistent with the model proposed by Kühl and is the subject of future work.
Additional possible explanations for the inhibition observed for hCE2 at high substrate concentrations involve the potential for multiple substrate binding sites. The active site of carboxylesterases contains two binding pockets (Satoh et al., 2002), and the entrance to the active site of hCE2 is less restrictive than hCE1 (Wadkins et al., 2001). Thus, a potential explanation is that multiple molecules may interact with the active site of hCE2 resulting in the unproductive binding of substrates. Other than the active site itself, hCE2 may have an external binding site similar to the Z-site found with hCE1 that could affect catalysis. It is known that substrate binding to the Z-site of hCE1 results in conformational changes that alter enzyme activity (Bencharit et al., 2003). Because a crystal structure does not yet exist for hCE2, it is unknown whether it has a similar Z-site or can bind multiple substrate molecules.
Despite the apparent in vitro inhibition of hCE2 at high prasugrel concentrations, there is no evidence that this occurs in vivo. A linear relationship has been shown between the prasugrel dose (2.5–75 mg) and the plasma exposure (area under the curve) of R-95913 (Asai et al., 2006). The micromolar equivalents of the 2.5-, 10-, and 75-mg doses of prasugrel dissolved in a standard glass of water (250 ml) would result in concentrations of 26.8, 107, and 803 μM, respectively. If the in vitro inhibition results were combined with these calculated intestinal lumen prasugrel concentrations and used to predict the in vivo inhibition, then hCE2 would not be substantially inhibited with the 2.5-mg dose but might be inhibited at the other doses. If such inhibition were to occur, this would result in a disproportionately higher exposure to R-95913 after a 2.5-mg dose compared with the higher doses, which has not been observed. Therefore, the observed in vitro hCE2 inhibition does not translate to in vivo relevance. The lack of observed in vivo inhibition could be caused by the large intestinal surface area; thus, intracellular hCE2 is not exposed to the high calculated intestinal concentrations of prasugrel. Also, hCE1 or other esterases may have sufficient capacity to compensate for a loss in hCE2 activity, if any.
The Caco-2 monolayer transport and metabolism study showed efficient conversion of prasugrel to R-95913 (Fig. 3). For both apical and basolateral administration, prasugrel is only found in the donor buffer and not the receiver buffer, which indicates that prasugrel does not pass through this intestinal model unmodified. Also, the loss of prasugrel, R-95913 formation, and R-95913 transit across the monolayer occur slightly faster when dosed on the apical versus the basolateral surface. Whereas Caco-2 cells are not a perfect intestinal model system to study carboxylesterases because of the relatively high levels of hCE1 versus hCE2 (Imai et al., 2005), the results obtained from these studies with prasugrel are consistent with the ideas presented when temocapril and 4-nitrophenyl acetate were used as substrates. In the study conducted by Imai et al. (2005), the brush-border membrane vesicles (BBMV) of the Caco-2 cells were isolated from the remaining cellular S9 fraction. Temocapril, a substrate for hCE1, was hydrolyzed in the S9 fraction but not as well in the BBMV; the generic esterase substrate 4-nitrophenyl acetate had much higher hydrolysis in the BBMV. Despite the presence of esterase activity in the BBMV, hCE1 is predominantly in the S9 fraction. Because the formation rate of R-95913 was similar whether dosed apically or basolaterally, S9 hydrolysis is suggested. The slightly higher hydrolysis rate when prasugrel was dosed on the apical membrane could be because of the additional hydrolysis occurring in the BBMV.
The results of the Caco-2 studies are in agreement with the data obtained in humans where prasugrel was found to be rapidly absorbed and metabolized and was not detected in plasma (Farid et al., 2007b). In addition, prasugrel and its hydrolysis product, R-95913, were not detected in human feces, but only metabolites of these two compounds were found in the feces. The above data suggest that after a p.o. dose, prasugrel was essentially fully absorbed and metabolized before excretion (Farid et al., 2007b). However, a role for other enzymes or intestinal microflora in the metabolism of prasugrel cannot be precluded.
Conclusion
Both hCE1 and hCE2 appear to have similar binding affinities (Km/s ≈ 10 μM) for prasugrel, but hCE2 has about a 26 times greater maximal hydrolysis rate (Vmax = 19.0 nmol R-95913/min/μg protein) than hCE1 (Vmax = 0.725 nmol R-95913/min/μg protein). Furthermore, the initial formation of R-95913 at low prasugrel concentrations is about 25 times higher with hCE2 (798 μl/min/μg) than hCE1 (32 μl/min/μg). Thus, although both hCE1 and hCE2 are capable of hydrolyzing prasugrel to R-95913, hCE2 appears to be the more efficient carboxylesterase for this conversion. Based on these results, it is proposed that the biotransformation of prasugrel to R-95913 is mediated efficiently by hCE2, the dominant carboxylesterase in the intestine, before reaching the portal vein. Any remaining parent compound would likely be hydrolyzed by hepatic esterases. The results of the hCE in vitro metabolism assays and the Caco-2 monolayer metabolism and transport studies, coupled with the fact that CYP3A accounts for ∼80% of the total P450 content in the human small intestine (Paine et al., 2006), help explain the rapid formation of the active metabolite of prasugrel, R-138727, in humans, in whom maximum concentrations of R-138727 are observed 30 min after a prasugrel p.o. dose, and also the rapid onset of the inhibition of platelet aggregation by prasugrel (Brandt et al., 2007; Farid et al., 2007b).
Acknowledgments
We thank Barbara Ring, Eli Lilly and Company, for her thoughtful discussion of the work.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.020248.
-
ABBREVIATIONS: prasugrel, 2-acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine; hCE, human carboxylesterase; P450, cytochrome P450; R-95913, 2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone; HBSS, Hanks' balanced salt solution; R-138727, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid; BBMV, brush-border membrane vesicle.
- Received December 19, 2007.
- Accepted March 26, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}