


Abstract
Although approaches to the prediction of drug-drug interactions (DDIs) arising via time-dependent inactivation have recently been developed, such approaches do not account for simple competitive inhibition or induction. Accordingly, these approaches do not provide accurate predictions of DDIs arising from simple competitive inhibition (e.g., ketoconazole) or induction of cytochromes P450 (e.g., phenytoin). In addition, methods that focus upon a single interaction mechanism are likely to yield misleading predictions in the face of mixed mechanisms (e.g., ritonavir). As such, we have developed a more comprehensive mathematical model that accounts for the simultaneous influences of competitive inhibition, time-dependent inactivation, and induction of CYP3A in both the liver and intestine to provide a net drug-drug interaction prediction in terms of area under the concentration-time curve ratio. This model provides a framework by which readily obtained in vitro values for competitive inhibition, time-dependent inactivation and induction for the precipitant compound as well as literature values for fm and FG for the object drug can be used to provide quantitative predictions of DDIs. Using this model, DDIs arising via inactivation (e.g., erythromycin) continue to be well predicted, whereas those arising via competitive inhibition (e.g., ketoconazole), induction (e.g., phenytoin), and mixed mechanisms (e.g., ritonavir) are also predicted within the ranges reported in the clinic. This comprehensive model quantitatively predicts clinical observations with reasonable accuracy and can be a valuable tool to evaluate candidate drugs and rationalize clinical DDIs.
The cytochromes P450 superfamily of enzymes are the most important enzymes in the metabolism of a variety of compounds. Many drug-drug interactions (DDIs) result from altering the activities of these enzymes. A considerable effort in the area of drug metabolism is dedicated toward predicting clinical pharmacokinetics and DDIs from in vitro data (Ito et al., 1998, 2004; Mayhew et al., 2000; Wang et al., 2004; Venkatakrishnan and Obach, 2007). The prediction of DDIs based on an assumption of reversible P450 inhibition has been a standard part of preclinical programs in the pharmaceutical industry for many years. It is now well appreciated that this common practice will underpredict the magnitude of DDI when the underlying inhibitory mechanism includes time-dependent inactivation (e.g., erythromycin and verapamil). As such, recent efforts have yielded methods whereby in vitro data can be used to provide accurate predictions of DDI arising via a time-dependent inactivation mechanism (Mayhew et al., 2000; Wang et al., 2004; Obach et al., 2007). Although the magnitude of DDI predicted via these newer approaches is often close to that observed in the clinic for drugs known to inhibit CYP3A via time-dependent inactivation, it is important to acknowledge discrepancies that have been observed with some of the drugs tested, e.g., cyclosporin, erythromycin, ethinyl estradiol, indinavir, mibefradil, oleandomycin, rifampicin, and verapamil (Lamberg et al., 1998). Interestingly, some of these inactivators are also potent competitive inhibitors and inducers of CYP3A. As such, these discrepancies may represent the mixed nature of the underlying mechanisms by which the precipitants interact with CYP3A. In this work, we attempt to improve previously published mathematical models by simultaneously considering the relative contribution of competitive inhibition, inactivation, and induction in both the liver and intestine on the net DDI observed in the clinic. The validity of the derived approach was examined through a comparison of observed DDIs to those predicted using the mathematical model populated with competitive inhibition, inactivation, and induction parameters derived from in vitro systems. For this study, we have chosen the term “precipitant” for the drug causing the effect and “object” for the drug whose AUC is being affected. These terms are consistent with those used in the University of Washington database (http://www.druginteractioninfo.org).
Materials and Methods
Clinical Data Source. Literature compound clinical data were collected from the University of Washington Metabolism and Transport drug interaction database (http://www.druginteractioninfo.org/). The database contains in vitro and in vivo information on drug interactions in human from 5979 publications referenced in PubMed, 19 new drug applications, and 223 excerpts of product labels. Thirty-two drugs were chosen for this study, based on available data from clinical studies with midazolam, triazolam, simvastatin, and nifedipine and commercial availability. Also, efforts were made to choose precipitants that have exhibited net clinical inhibition as well as drugs that have exhibited net clinical induction as determined by the object (e.g., midazolam) AUC in the presence and absence of the precipitant. For rifampin, only the 15-mg midazolam dose studies were included to capture the maximum effect, along with all other object probes. For phenytoin and nifedipine, in vivo data were obtained from Backman et al. (1996) and Horsmans et al. (1991), respectively.
In Vitro Data. Data reflecting competitive inhibition, time-dependent inactivation, and induction of CYP3A were collected from the in vitro systems described below. Thirty-two drugs were included in this study. Of these, 16 drugs exhibited competitive inhibition, 18 drugs exhibited time-dependent inactivation, and 21 compounds exhibited induction. Thirteen drugs exhibited all three interaction mechanisms in vitro (troleandomycin, ethinyl estradiol, fluoxetine, mibefradil, pioglitazone, rosiglitazone, troglitazone, saquinavir, nelfinavir, ritonavir, ethinyl estradiol, mibefradil, and verapamil).
Competitive Inhibition Data. Competitive inhibition data were determined using previously described validated methods (Walsky and Obach, 2004) for CYP3A (midazolam 1′-hydroxylase). Human liver microsomes pooled from 53 individual donors were used as the source of enzyme activity. IC50 values were measured using the substrates at a concentration equal to previously determined KM values. Inhibitors were examined up to a maximum concentration of 300 μM.
Time-Dependent Inactivation Data. Data for the kinetic parameters determinations, Ki and kinact, were run in our laboratory using previously described methods (Walsky and Obach, 2004; Obach et al., 2006, 2007).
Induction Assay Data. CYP3A4 induction data were carried out as described by the manufacturer, using human cryopreserved hepatocytes (lot Hu4026; CellzDirect, Pittsboro, NC). Total RNA was extracted from cells using the RNeasy Mini kit according to instructions provided by the manufacturer (QIAGEN, Germantown, MD). Quantification of cytochrome CYP3A4 mRNA was performed using the TaqMan two-step reverse transcription-polymerase chain reaction method by using the ABI 7500 Fast Real Time polymerase chain reaction (Applied Biosystems, Foster City, CA). The relative quantity of the target CYP3A4 gene (Hs00604506_m1) compared with the endogenous control (glyceraldehyde-3-phosphate dehydrogenase) was determined by the ΔΔCT method. EC50 and Emax (Ψ) parameters were determined based on CYP3A4 mRNA, using a sigmoid three-parameter curve-fitting (Sigma Plot 9.0; Systat Software, Inc., Chicago, IL).
Mathematical Model. As reported previously, the ratio of area under the exposure-time curve in the presence (AUC′po) and absence (AUCpo) of a pharmacokinetic drug-drug interaction can be defined as stated in eq. 1 (Wang et al., 2004):  F′G and FG represent the fraction of drug escaping intestinal metabolism in the pharmacokinetically altered and unaltered state, respectively. CL′int,H and CLint,H represent the intrinsic hepatic clearance in the pharmacokinetically altered and unaltered state, respectively. The expanded forms of these ratios of intrinsic clearance and fraction escaping gut metabolism are derived separately in detail below.
F′G and FG represent the fraction of drug escaping intestinal metabolism in the pharmacokinetically altered and unaltered state, respectively. CL′int,H and CLint,H represent the intrinsic hepatic clearance in the pharmacokinetically altered and unaltered state, respectively. The expanded forms of these ratios of intrinsic clearance and fraction escaping gut metabolism are derived separately in detail below.
Alterations in hepatic intrinsic clearance. The intrinsic hepatic clearance of a theoretical drug can be defined as in eq. 2, where Vmax,H, Km, kcat,H, and Ess,H represent the maximum velocity, drug concentration associated with half-maximum velocity, catalysis rate, and the steady-state amount of clearing enzyme, respectively:  Since the steady-state amount of enzyme is determined by the ratio of the zero-order rate of synthesis (Ksyn,H) and first-order rate of degradation (kdeg,H), eq. 2 can be written as follows:
Since the steady-state amount of enzyme is determined by the ratio of the zero-order rate of synthesis (Ksyn,H) and first-order rate of degradation (kdeg,H), eq. 2 can be written as follows:  In the presence of competitive inhibition, the apparent Km (K′m) will also be determined by the concentration of inhibitor in the liver ([I]H) and its associated equilibrium dissociation constant (KI) as follows:
In the presence of competitive inhibition, the apparent Km (K′m) will also be determined by the concentration of inhibitor in the liver ([I]H) and its associated equilibrium dissociation constant (KI) as follows:  In the presence of inactivation, the overall rate of loss of the enzyme will also be determined by the pseudo-first-order apparent inactivation rate (kinact,app). This apparent inactivation rate is dependent upon [I]H, KI and the true first-order inactivation rate constant (kinact).
In the presence of inactivation, the overall rate of loss of the enzyme will also be determined by the pseudo-first-order apparent inactivation rate (kinact,app). This apparent inactivation rate is dependent upon [I]H, KI and the true first-order inactivation rate constant (kinact).  In the presence of induction, the rate of enzyme synthesis will also be determined by inducer concentrations in the liver [I]H, the maximum -fold induction (Ψ) and the concentration of inducer associated with half-maximum induction (EC50,I). The d parameter in eq. 6 represents an empirical calibration factor for the purposes of in vitro to in vivo induction scaling. As such, its value was estimated through correlation of predicted and observed AUC ratios:
In the presence of induction, the rate of enzyme synthesis will also be determined by inducer concentrations in the liver [I]H, the maximum -fold induction (Ψ) and the concentration of inducer associated with half-maximum induction (EC50,I). The d parameter in eq. 6 represents an empirical calibration factor for the purposes of in vitro to in vivo induction scaling. As such, its value was estimated through correlation of predicted and observed AUC ratios: 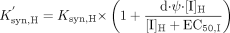 The expected net effect of simultaneous competitive inhibition, induction, and inactivation, can be illustrated by substituting eqs. 4, 5, and 6 into eq. 3:
The expected net effect of simultaneous competitive inhibition, induction, and inactivation, can be illustrated by substituting eqs. 4, 5, and 6 into eq. 3:  Assuming that the object drug is only fractionally metabolized by the altered pathway, the fm parameter represents the fraction of object drug cleared via the altered metabolic pathway (eq. 8):
Assuming that the object drug is only fractionally metabolized by the altered pathway, the fm parameter represents the fraction of object drug cleared via the altered metabolic pathway (eq. 8):  If one lets time-dependent inactivation term or
If one lets time-dependent inactivation term or  induction term or
induction term or  and reversible inhibition term or
and reversible inhibition term or  then the ratio of eqs. 3 and 8 will yield the expected ratio of object drug AUCpo due to the hepatic component of the drug-drug interaction arising from simultaneous inactivation (A), induction (B), and competitive inhibition (C):
then the ratio of eqs. 3 and 8 will yield the expected ratio of object drug AUCpo due to the hepatic component of the drug-drug interaction arising from simultaneous inactivation (A), induction (B), and competitive inhibition (C): 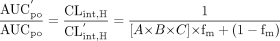
Alterations in intestinal intrinsic clearance. Assuming that absorption rate is unaffected, the ratio of the fraction of drug escaping intestinal extraction in the altered and unaltered state will equal the ratio of intrinsic clearance in the unaltered and altered state (eq. 10):  The intrinsic intestinal clearance of a theoretical drug in the altered and unaltered state can be derived in an identical manner to that depicted above for hepatic intrinsic clearance to provide eqs. 11 and 12:
The intrinsic intestinal clearance of a theoretical drug in the altered and unaltered state can be derived in an identical manner to that depicted above for hepatic intrinsic clearance to provide eqs. 11 and 12: 
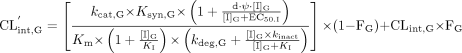 If one lets time-dependent inhibition term for the intestinal portion or
If one lets time-dependent inhibition term for the intestinal portion or  induction term for the intestinal portion or
induction term for the intestinal portion or  and reversible Inhibition term for the intestinal portion or
and reversible Inhibition term for the intestinal portion or  then the ratio of eq. 11 and 12 will yield the expected change in AUCpo due to the intestinal component of the DDIs arising from simultaneous inactivation (A), induction (B), and competitive inhibition (C):
then the ratio of eq. 11 and 12 will yield the expected change in AUCpo due to the intestinal component of the DDIs arising from simultaneous inactivation (A), induction (B), and competitive inhibition (C):  As reported previously (Obach et al., 2007), eq. 15 was used to estimate intestinal drug concentrations:
As reported previously (Obach et al., 2007), eq. 15 was used to estimate intestinal drug concentrations:  Dose, ka, fa, Qg, and freq represent, total daily dose of inhibitor given orally, first-order absorption rate constant, fraction of dose absorbed, enterocytic blood flow, and frequency of daily dose, respectively.
Dose, ka, fa, Qg, and freq represent, total daily dose of inhibitor given orally, first-order absorption rate constant, fraction of dose absorbed, enterocytic blood flow, and frequency of daily dose, respectively.
Net effect. Substituting eqs. 9 and 13 back into eq. 1, one obtains the following mathematical model for the net effect of competitive inhibition, inactivation and induction in both the intestine and liver: 
Prediction of AUC ratio.Equation 16 was used to make AUC ratio predictions against which comparisons were made to clinical AUC ratios obtained from the University of Washington Metabolism and Transport drug interaction database (http://www.druginteractioninfo.org). For purposes of prediction, fraction of precipitant drug absorbed (fa), and absorption rate (ka) were assumed to be 1.0 and 0.03 min-1. These values were combined with an assumed enterocytic blood flow (Qg) of 248 ml/min to calculate intestinal precipitant drug concentration using eq. 15. Average plasma concentrations and unbound fractions associated with the precipitant were also obtained from the literature as indicated under Results to calculate drug concentration available to interact at the level of the liver. Competitive inhibition, inactivation and induction parameters used in the predictions were estimated in vitro as described above (i.e., KI, kinact, EC50, and Ψ). Consistent with previous reports, the degradation rate for CYP3A4 (kdeg) was assumed to be 0.019 h-1 (t1/2 = 36 h) and 0.029 h-1 (t1/2 = 24 h) in the liver and intestine, respectively. The fraction of object drug metabolized by CYP3A4 was assumed to be the same as that reported previously for midazolam, nifedipine, simvastatin, and triazolam (fm = 0.93). The fraction of object drugs metabolized by CYP3A4 in the intestines was assumed to be 0.57, 0.66, 0.75, and 0.78 for midazolam, simvastatin, triazolam, and nifedipine, respectively, and as reported previously (Obach et al., 2007).
AUC ratio predictions were made under the following four conditions: 1) assuming a liver effect only and using total precipitant concentrations in the plasma, 2) assuming a gut and liver effect and using total precipitant concentrations in the plasma, 3) assuming a liver effect only and using unbound precipitant concentrations in the plasma, and 4) assuming a gut and liver effect and using unbound precipitant concentrations in the plasma.
The scaling parameter for induction (i.e., d) in each of the four sets of predictions was estimated through linear regression to a value that minimized the geometric mean -fold error (GMFE) of the prediction via linear weighted least-squares regression. Briefly, GMFE of the prediction set of interest was calculated as depicted in eq. 17. All parameters, with the exception of d, were fixed to those estimated in vitro. The value of d was then estimated via weighted least-squares regression to a value that provided the lowest GMFE. The derived GMFE was also subsequently used to assess the relative accuracy of the examined approaches: 
Results
The in vivo drug-drug interaction data gathered from the University of Washington database used in this study are summarized in Table 1. Clinical CYP3A4 probe substrate data were used from midazolam, triazolam, nifedipine, or simvastatin in vivo studies, where fm by CYP3A4 is hypothesized to be 0.93 (Obach et al., 2007).
Literature compounds dosing regimen information
Reversible Inhibition. The reversible inhibition of CYP3A4 for the test compounds was assessed, and the Ki data are shown in Table 2. About 16 compounds showed a Ki value less than 10 μM. The predicted clinical DDI predicted assuming a purely competitive mechanism is also shown, along with observed clinical DDI, expressed as AUC ratios in the presence and absence of inhibitor. Predictions depicted in Table 2 were made assuming competitive inhibition via total precipitant concentrations at the level of the liver via the C component of eq. 9. The results demonstrate that using the reversible inhibition data alone fail to predict the extent of drug-drug interaction within 2-fold for more than 50% of the examined cases. As one might expect, several compounds for which CYP3A4 interaction is known to be limited to a competitive inhibition mechanism are well predicted by this approach (e.g., cimetidine, fluonazole, itraconazole, and ketoconazole). As has been reported previously, this approach produced significant underpredictions of AUC ratio for compounds known to produce time-dependent inactivation of CYP3A4 (e.g., cyclosporin, diltazem, erythromycin, mibefradil, nelfinavir, ritonavir, saquinavir, troleandomycin, and verapamil).
Ki values obtained from reversible inhibition experiment and DDI-predicted AUC ratio (inhibition term used fromeq. 9) The values used for [I] in vivo were estimated total systemic plasma concentration.
Induction. As shown in Table 3, 21 compounds showed CYP3A4 induction in cryopreserved human hepatocytes. The scaling parameters for induction (i.e., d) in each of the two sets of predictions were estimated through linear least-squares regression to a value that minimized the GMFE. Using a composite of known strong inducers as described under Materials and Methods, the calibration constant, d, was set to 0.8. This gave a balance of slightly over predicting to slightly underpredicting AUC effects for the high inducers. Predictions made assuming induction via total precipitant concentrations at the level of the liver using the B component of eq. 9, provided AUC ratio predictions associated with greater than 2-fold error for the more than 50% of the examined cases. As one might expect, large underpredictions (exceeding an order of magnitude) of AUC ratio were associated with compounds possessing mixed mechanisms of CYP3A4 interaction (e.g., mibefradil, nelfinavir, ritonavir, saquinavir, troleandomycin, and verapamil).
Summary of induction data (induction term fromeq. 9) for the compounds tested and the DDI-predicted AUC ratios The values used for [I] in vivo were estimated total systemic plasma concentration.
Time-Dependent Inactivation. As shown in Table 4, 18 compounds showed time-dependent CYP3A4 inactivation. Predictions made assuming inactivation via total precipitant concentrations at the level of the liver using the A component of eq. 9 provided AUC ratio predictions associated with greater than 2-fold error for about 40% of the examined cases. As one might expect, many of the overpredictions of DDI were associated with drugs known to also possess the ability to induce CYP3A4 (e.g., fluoxetine, nelfinavir, pioglitazone, rosiglitazone, saquinavir, troglitazone, and verapamil). When these values are compared with estimates of AUC ratio changes obtained from clinical study reports, some compounds that show positive time-dependent inactivation (kinact > 0) are reasonably predicted.
Summary of the CYP3A4 inactivation data and the DDI-predicted values of AUC ratio based on time-dependent inactivation data only (TDI term ineq. 9) vs. observed values The values used for [I] in vivo were estimated total systemic plasma concentration.
Integrated Approach. To account for all known mechanisms affecting CYP3A4 activity, data from the three possible mechanisms: induction, inactivation, and competitive inhibition were used simultaneously to make a prediction on the AUC ratio change, as shown in Tables 5 and 6, and Fig. 1, A to D. Table 5 and Fig. 1, A and C, shows the AUC ratio predictions made by using total precipitant concentrations in the plasma, assuming a liver effect only and gut plus liver effect, by applying eqs. 9 and 16, respectively.
Predicted net DDI vs. that observed and associated geometric fold error assuming total plasma concentrations and interaction in the liver only ( eq. 9 ) or interaction in the liver plus intestine ( eq. 16 ), using all in vitro data (reversible inhibition, time-dependent inactivation, and induction)
The scaling parameter for induction (i.e., d) in each of the two sets of predictions was estimated through linear least-squares regression to a value that minimized the GMFE. The values for d were 0.8 and 0.3, and GMFE values were calculated as 1.8 and 2.5, for the liver and the gut plus liver analyses, respectively. Table 6 and Fig. 1, B and D, show the DDI predictions made by using unbound precipitant concentrations in the plasma, assuming a liver effect only and gut plus liver effect, by applying eqs. 9 and 16, respectively. The values for d were 1.0 and 0.4, and GMFE values were calculated as 1.9 and 2.0, for the liver and gut plus liver analyses, respectively.
Discussion
Pharmacokinetic DDIs via CYP3A pose a serious safety risk and attrition factor in drug development. As such, significant efforts are made in the pharmaceutical industry to design compounds that are devoid of this risk. These design efforts almost exclusively rely upon in vitro data. Frequently, mathematical models are also used that enable scaling of these and other pertinent information into a quantitative prediction of the expected magnitude of DDI (typically expressed as an AUC ratio of the object in the presence and absence of the precipitant). Prior work has demonstrated that this approach can indeed yield accurate DDI predictions (Ito et al., 1998, 2004; Mayhew et al., 2000; Wang et al., 2004; Venkatakrishnan and Obach, 2007). However, to date, such quantitative approaches have not been comprehensive in that they assume a singular mechanism of action. In cases where the mechanism of CYP3A interaction is mixed (e.g., competitive inhibition, time-dependent inactivation, and induction), such approaches may yield inaccurate predictions. A classic example of this is the underprediction of DDI for time-dependent inactivators when only a competitive mechanism is presumed (illustrated here in Table 2). Conversely, the presence of an induction mechanism may yield overpredictions of DDI in the event that only inhibition mechanisms are considered.
As such, we have developed and tested a comprehensive mathematical model that simultaneously accounts for competitive inhibition, time-dependent inhibition, and induction to yield a net DDI prediction from in vitro data. This model was combined with data from in vitro systems to provide 59 DDI predictions on 32 drugs. Of the 32 drugs examined in this study, 16 drugs exhibited in vitro reversible inhibition, 18 drugs exhibited time-dependent inactivation, and 21 drugs exhibited in vitro induction of CYP3A4. Thirteen drugs, namely; troleandomycin, ethinyl estradiol, fluoxetine, mibefradil, pioglitazone, rosiglitazone, troglitazone, saquinavir, nelfinavir, ritonavir, ethinyl estradiol, mibefradil, and verapamil, were positives in all three in vitro assays.
Application of this model to the data indicates that reasonably accurate predictions can be made with this approach (Fig. 1, A–D; Tables 5 and 6). In this particular case, the greatest predictive accuracy and least bias was observed when total plasma precipitant concentrations were used and the interaction at the level of the gut was excluded (Fig. 1A). Under this set of conditions, the geometric mean fold error of the prediction was 1.8-fold, with 66% of the predictions being within 2-fold (Table 5). Considering unbound precipitant concentrations (rather than total) yielded comparable accuracy, with a GMFE of 1.9, and 66% of predictions within 2-fold of actual (Fig. 1B; Table 5).
Assuming total precipitant concentrations and inclusion of the intestinal component (Fig. 1C) provided the least accuracy (GMFE = 2.5; 46% within 2-fold) and a clear bias at the extremes of induction and inhibition. Assuming unbound precipitant concentrations (Fig. 1D) and inclusion of the intestinal component reduced bias, but it provided less accuracy than those approaches using total precipitant concentrations (GMFE = 2.0; 64% within 2-fold).

Predicted versus observed AUC ratios and associated geometric fold error assuming total plasma concentration and hepatic interaction only (A), unbound plasma concentration and hepatic interaction only (B), total plasma concentration and interaction in the liver and intestine (C), and unbound plasma concentration and interaction in the liver and intestine (D). Solid black line represents unity. Dashed red lines represent 2-fold boundary around the line of unity.
Of particular interest in the examination of this approach are the subsets of precipitants that are CYP3A inducers. For this subset analysis, total drug concentrations and a hepatic only effect was assumed, and the relative predictive accuracy was assessed using in vitro data on competitive interaction only, TDI only or by including all data on competitive interaction, TDI and induction (Table 7). Overall, the least -fold error was observed using in vitro data on all the relevant mechanisms (GMFE = 3.2) for this subset of compounds. Importantly, drugs with a net induction effect in the clinic (e.g., avasimibe, carbamazepine, modafinil, nafcillin, phenytoin, and rifampin) were predicted with reasonable accuracy via this approach. This was achieved while maintaining reasonable predictive accuracy for compounds with a net inhibition in the clinic (e.g., mibefradil, nelfinavir, ritonavir saquinavir, troleandomycin, and verapamil) and for which the clinical DDI is negligible (e.g., ethinyl estradiol, nifedipine, pioglitazone and rosiglitazone).
Predicted net DDI vs. observed and associated geometric fold error assuming total plasma concentrations and interaction in the liver only (eq. 9) Predictions were made assuming an exclusively competitive mechanism, a TDI mechanism and a mixed mechanism.
Nevertheless, it is essential to note several important limitations of this work that represent areas for future improvement. Most of the limitations of this work relate to the mathematical model that is derived to address the extent of DDI under the equilibrium, steady-state condition. For example, this aspect of the mathematical model requires a singular value of precipitant concentrations in the intestine and liver for the prediction of DDI. Candidate measures of this exposure include maximum, minimum, or average systemic blood concentrations at steady-state. Calculated concentrations in the portal venous blood and intestine are also candidates for this singular exposure value. Last, each of these candidate exposure measures can be entered into the mathematical model as total or unbound exposure. As such, it is common practice with such approaches to empirically select surrogate measures of exposure in the liver and intestine that provide the best correlation of predicted and observed DDIs reported in the literature. Similar exercises have also been used herein to guide the incorporation or exclusion of the intestinal effect and the consideration of unbound fraction.
Although practically useful, such empirical oversimplifications surely limit predictive accuracy within the data set and may translate to lower accuracy when the method is applied outside the data set for which it was developed. In addition, caution should be used not to over interpret such empirical validation as mechanistic validation. For example, it would be inappropriate to conclude from this analysis that CYP3A4 interactions are fundamentally driven by total precipitant concentration or that the intestine is not a source of DDIs arising from alterations in CYP3A4 activity (only that, for whatever reason, the best correlation was observed when total steady-state average blood concentrations were used and intestinal interaction was ignored). Likewise, this empirical aspect makes it difficult to reasonably speculate as to the mechanistic source of observed prediction errors (e.g., troglitazone, pleconaril, and bosentan). Another empirical aspect of this work which imposes similar limitations relates to the estimation of the induction scaling parameter d. Because the value of d is estimated through correlation, its absolute value is shown to be dependent upon the assumptions regarding affected tissue (i.e., intestine, liver, or both) and exposure surrogate (total or unbound blood concentrations). Likewise, the value of this parameter is expected to depend upon the in vitro system used. For example, we have noted similar predictive accuracy associated with the need for a much higher value of d when in vitro induction data from immortalized hepatocytes, rather than cryopreserved hepatocytes, are used (data not shown).
All of these limitations indicate the need for more sophisticated approaches to the prediction of DDIs arising from interaction at the level of CYP3A4 metabolism. In this regard, approaches that link the time course of predicted precipitant and object liver and intestine exposure to the known CYP3A4 interaction mechanism will likely be required to address the aforementioned limitations. Ideally, such approaches should also provide the framework for mechanistic, physiologically based scaling of in vitro CYP3A4 interaction data such that interindividual variability in the interaction can also be predicted. Such approaches would also be much more appropriate for drawing fundamental mechanistic inferences through the comparison of predicted and observed DDIs.
Overall, this approach provides an accuracy comparable with previous methods designed to predict DDI arising from a singular CYP3A4 interaction mechanism (i.e., within 2-fold). The advantage of this approach is that it allows investigators to generate a DDI prediction that reflects the net influence of competitive inhibition, time-dependent inactivation, and induction. Use of this model requires six kinetic constants that are readily estimable from in vitro systems, namely, Ki and kinact for inhibitor binding and inactivation; d, Ψ, and EC50 for P450 induction; and fm for fraction of metabolism by the pathway being observed. As such, this method provides an attractive means to support early efforts to identify and mitigate DDI risk during drug design and development.
Acknowledgments
We thank Drs. Scott Obach, Michael Fisher, Sonia de Morais, Sharon Ripp, and Larry Tremaine for reviewing this manuscript.
Footnotes
-
This work was presented in part at the 2005 and 2006 Institute for Scientific Exchange Conferences on Drug-Drug Interactions and at the 2006 International Symposium on Microsomes and Drug Oxidations meeting.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.018663.
-
ABBREVIATIONS: P450, cytochrome P450; DDI, drug-drug interaction; AUC, area under the concentration-time curve; GMFE, geometric mean fold error; TDI, time-dependent inactivation; q.d., every day; GFE, geometric fold error.
- Received August 30, 2007.
- Accepted May 16, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}