


Abstract
Mouse CYP2C55 has been characterized as an enzyme that catalyzes synthesis of 19-hydroxyeicosatetraenoic acid (19-HETE), an arachidonic acid metabolite known to have important physiological functions such as regulation of renal vascular tone and ion transport. We have now found that CYP2C55 is induced by phenobarbital (PB) and pregnenolone 16α-carbonitrile (PCN) in both mouse kidney and liver. The nuclear xenobiotic receptors constitutive active/androstane receptor (CAR) and pregnane X receptor (PXR) regulate these drug inductions: CYP2C55 mRNA was increased 25-fold in PB-treated Car(+/+) but not in Car(−/−) mice and was induced in Pxr(+/+) but not Pxr(−/−) mice after PCN treatment. Cell-based promoter analysis and gel shift assays identified the DNA sequence −1679TGAACCCAGTTGAACT−1664 as a DR4 motif that regulates CAR- and PXR-mediated transcription of the Cyp2c55 gene. Chronic PB treatment increased hepatic microsomal CYP2C55 protein and serum 19-HETE levels. These findings indicate that CAR and PXR may play a role in regulation of drug-induced synthesis of 19-HETE in the mouse.
The human CYP2C subfamily of cytochrome P450 (P450) monooxygenases is responsible for metabolism of many therapeutically prescribed drugs, such as phenytoin, warfarin, tolbutamide, and numerous nonsteroidal anti-inflammatory drugs, as well as the metabolism of endogenous compounds such as arachidonic acid (AA) (Goldstein and de Morais, 1994; Miners and Birkett, 1998; Zeldin, 2001; Capdevila et al., 2007; Kaspera and Totah, 2009). The induction of members of the human CYP2C subfamily, such as CYP2C9, by drugs is regulated by the nuclear xenobiotic receptors constitutive active/androstane receptor (CAR) and pregnane X receptor (PXR) (Ferguson et al., 2002; Gerbal-Chaloin et al., 2002; Chen et al., 2004; Chen and Goldstein, 2009), leading to altered drug efficacies and causing drug-drug interactions (Honkakoski and Negishi, 2000). Once activated, these receptors bind response elements located within the 5′-flanking regions of target genes (Honkakoski et al., 1998; Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Tompkins and Wallace, 2007).
Mice are increasingly used as animal models for human disease and are an excellent system to investigate drug-induced regulation of P450 genes. However, the transcriptional regulation of murine Cyp2c genes is poorly understood at the present time. Fifteen murine Cyp2c genes have been identified (Luo et al., 1998; DeLozier et al., 2004; Nelson et al., 2004; Wang et al., 2004), including Cyp2c29, Cyp2c36, Cyp2c37, Cyp2c38, Cyp2C39, Cyp2c40, Cyp2c44, and Cyp2c55. The expression of the Cyp2c genes is regulated differentially (DeLozier et al., 2004; Jackson et al., 2004, 2006; Goetz et al., 2006). For example, the Cyp2c29 and Cyp2c37 genes were up-regulated by CAR activators but not the PXR activator pregnenolone 16α-carbonitrile (PCN) (Jackson et al., 2004, 2006). Cyp2c44, on the other hand, is not induced by either CAR or PXR activators (DeLozier et al., 2004), whereas Cyp2c40 is down-regulated by some triazoles (Goetz et al., 2006). CYP2C55 was characterized as an enzyme that catalyzes the biosynthesis of 19-hydroxyeicosatetraenoic acid (19-HETE) (Wang et al., 2004). A recent study showed that hepatic mRNAs for several murine CYP2C enzymes that are able to metabolize midazolam in recombinant studies were increased in Cyp3a knockout mice, particularly CYP2C55 (35-fold increase) (van Waterschoot et al., 2008). Although midazolam is thought to be metabolized primarily by the CYP3A enzyme in Cyp3a(+/+) mice, the CYP2C enzymes were found to be the major enzymes responsible for midazolam metabolism in Cyp3a(−/−) mice as a result of their increased expression. CYP2C55 is also induced by triazole fungicides, similar to Cyp3a11, thereby mediating triazole-induced hepatotoxicity (Goetz et al., 2006). However, the molecular mechanisms responsible for the induction of the Cyp2c55 gene by therapeutic drugs have not yet been investigated.
Murine CYP2C enzymes catalyze the metabolism of AA and produce various physiologically functional eicosanoids, including cis-epoxyeicosatrienoic acids (5,6-, 8,9-, 11,12-, and 14,15-EET), midchain hydroxyeicosatetraenoic acids (5-, 8-, 9-, 11-, 12-, and 15-HETE), and ω-terminal alcohols of AA (16-, 17-, 18-, 19-, and 20-HETE) (Capdevila et al., 2000; Zeldin, 2001; DeLozier et al., 2004). CYP2C55 showed high selectivity for 19-HETE production (Wang et al., 2004). 19-HETE has been reported to affect vascular tone and ion transport in the kidney and brain (Escalante et al., 1988; Carroll et al., 1996; Qu et al., 2001). Thus, induction of CYP2C55 by xenobiotics may have physiological effects as a result of changes in the biosynthesis of 19-HETE.
In this study, we examined whether hepatic and renal CYP2C55 mRNA was induced by the CAR agonist phenobarbital (PB) and the PXR ligand PCN in CAR- and PXR-null mice and wild-type controls. We performed cell-based promoter analyses and gel shift assays to delineate the mechanisms that regulate Cyp2c55 gene induction. Furthermore, we examined the levels of CYP2C55 protein in liver microsomes and of serum 19-HETE using Western blot analysis and liquid chromatography/tandem mass spectrometry, respectively. We found that both CAR and PXR play an essential role in regulation of the synthesis of 19-HETE by drugs.
Materials and Methods
Materials and Reagents.
Dimethyl sulfoxide (DMSO), PB sodium salt, PCN, and diethylnitrosamine (DEN) were purchased from Sigma-Aldrich (St. Louis, MO). The plasmid pGL3-basic was obtained from Promega (Madison, WI). Restriction endonucleases and DNA-modifying enzymes were purchased from New England Biolabs (Ipswich, MA). [32P]dATP was purchased from MP Biomedical (Solon, OH).
Animals.
C3H/HeNCrlBR (C3H) mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA). A CAR-null mouse (Ueda et al., 2002) was first crossbred with C3H to generate CAR-heterozygous offspring. Subsequently, CAR-heterozygous offspring were repeatedly backcrossed for at least five generations with C3H mice until the genetic background became more than 95% C3H. The resulting heterozygous mice were bred to produce the wild-type [Car(+/+)] and CAR-null [Car(−/−)] C3H mice. PXR-null 129S1/Sv*129 × 1/SvJ*C57BL/6 [Pxr(−/−)] and congenic wild-type mice [Pxr(+/+)] were obtained from Jeff L. Staudinger (University of Kansas, Lawrence, KS) (Staudinger et al., 2001) and maintained at the National Institute of Environmental Health Sciences (NIEHS). All the mice were housed in a room maintained at 22°C with a 12:12-h light/dark cycle (7:00 AM to 7:00 PM), and all the animal procedures were approved by the Animal Ethics Committee, NIEHS, National Institutes of Health (Research Triangle Park, NC). Chronic PB-treated mice were fed with Purina PicoChow 5058 (Ralston Purina Co., St. Louis, MO) and water ad libitum. Single treated mice were fed with NIH-31 open formula autoclavable diet (Zeigler Bros., Inc., Gardners, PA) and water ad libitum. The genotypes of offspring were determined by analyzing the mutant allele using polymerase chain reaction (PCR) with genomic DNA.
Single Animal Treatment.
Car(+/+), Car(−/−), Pxr(+/+), or Pxr(−/−) mice (7–8 weeks old) were randomly divided into two groups (three mice per group). Car(+/+) and Car(−/−) mice were given intravenous injections of saline or PB (100 mg/4 ml/kg); likewise, Pxr(+/+) and Pxr(−/−) mice were given intraperitoneal injections of DMSO or PCN (20 mg/4 ml/kg). The mice were sacrificed 12 or 24 h after a single injection, respectively.
Chronic Animal Treatment.
Car(+/+) and null [Car(−/−)] mice received a single dose of DEN and chronic treatment with PB as described previously in a liver tumor promotion model (Yamamoto et al., 2004). Car(+/+) and Car(−/−) mice were given a single intraperitoneal injection of DEN (90 mg/kg) at 5 weeks of age and were divided into four groups [groups 1 and 2, six Car(+/+) mice; groups 3 and 4, six Car(−/−) mice]. The mice in groups 2 and 4 were chronically treated with PB (500 ppm) in drinking water at 7 weeks of age until they were sacrificed after 6 or 32 weeks of PB treatment.
Expression Vectors and Cloning of the Cyp2c55 5′-Flanking Region.
For all the plasmids, m and h denote mouse and human, respectively. The following plasmids were constructed as described previously: pCMX/hRXR (Honkakoski et al., 1998), pcDNA3.1/mPXR (Squires et al., 2004), and pCR3/mCAR (Sueyoshi et al., 1999). To construct the reporter plasmid pGL3/Cyp2c55−2.5 kilobases (kb) (−2452/+38), amplified sequences from mouse genomic DNA were cloned into the XhoI and HindIII sites of pGL3 basic from Promega. Primers used for amplifications were 5′-CCGCTCGAGGACACTATTGTGGATGCCAAGAAGT-3′ and 5′-AAGAGAAAGCTGCCATGGATCCAGTAAGCTTGGG-3′. XhoI and HindIII sites are underlined. Reporter plasmid pGL3/Cyp2c55−1.6 kb (−1600/+38) was obtained by PCR (Pfu polymerase; Promega) using forward primer 5′-CTATCGATAGGTACCCAGTCTGTGTGACTC-3′, reverse primer 5′-GAGTCACACAGACTGGGTACCTATCGATAG-3′, and reporter plasmid pGL3/Cyp2c55−2.5 kb as a template. In the context of pGL3/Cyp2c55−2.5 kb, putative CAR and PXR binding sites, DR4 (16 bases) and DR5 (17 bases), were independently deleted using the Quick change site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) using the following primers: 5′-CATAGTTGATCCTGGGAAGCCTGAAGAGAA-3′ and 5′-TTCTCTTCAGGCTTCCCAGGATCAACTATG-3′ for the DR4 deletion, and 5′-GATTTTTGCACAAATGGGAAAATAGCTCAG-3′ and 5′-CTGAGCTATTTTCCCATTTGTGCAAAAATC-3′ for the DR5 deletion.
Cell-Based Transcription Assays.
Huh7 cells were cultured in minimum essential medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in an atmosphere of 5% CO2 at 37°C. The cells were plated on a 24-well plate at a density of 4.0 × 105 cells/well 24 h before plasmid transfection using FuGENE 6 (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions: the Cyp2c55 promoter firefly luciferase (50 ng), pRL-CMV (5 ng; Promega), and a given gene expression plasmid (10 ng) while the total mount of transfected plasmids was equalized by adding the empty vector pcDNA3-V5-His (Invitrogen, Carlsbad, CA). After being transfected for 24 h, the cells were treated with drug in fetal bovine serum-free medium for an additional 24 h before harvesting to prepare lysates for luciferase assays using Dual-Luciferase Reporter Assay System (Promega).
Quantitative Reverse Transcription-PCR.
Total RNA from cells and tissue was extracted using TRIzol reagent (Invitrogen), from which cDNA was synthesized using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). Real-time PCR was performed with an ABI prism 7700 sequence detection system (Applied Biosystems) using 2× SYBR Green Master Mix (Applied Biosystems) and the following primers: forward 5′-GAACAGAAACCACAAACATTACTCTAAGA-3′ and reverse 5′-TGATTGGCAGACACAGGAGC-3′ for Cyp2c55. The TaqMan rodent glyceraldehyde-3-phosphate dehydrogenase control reagent (Applied Biosystems) was used as an internal control.
Gel Shift Assays.
Double-stranded DNA containing a CAR/PXR putative binding site (5′-GATCCTGGTGAACCCAGTTGAACTGAAGGATC-3′, −1683 to −1660) was labeled with [α-32P]dATP and DNA polymerase Klenow fragment (New England Biolabs). The underlines indicate additional sequences used to fill in with by Klenow fragment. PXR, CAR, and retinoid X receptor proteins were produced using the in vitro transcription/translation system (TNT T7 quick-coupled system; Promega) and were incubated with the 32P-labeled probe (100,000 cpm) in 10 μl of binding buffer containing 50 mM NaCl, 10 mM HEPES, pH 7.5, containing 0.05% Nonidet P40, 6% glycerol, and 1.5 μg of poly(dI-dC). The proteins were separated on a 5% acrylamide gel in Tris/acetate/EDTA running buffer at 150 V for 2 h, and the gel was dried under vacuum and subjected to autoradiography at −70°C.
Western Blot Analysis.
Mice were given a single intraperitoneal injection of DEN (90 mg/kg) and treated chronically with PB (500 ppm) for 6 weeks. Livers were removed, and hepatic microsomes were prepared as described previously (Sueyoshi et al., 1995). Immunoblotting of the microsomes was performed as described previously (Honkakoski et al., 1998) using a specific polyclonal antibody for CYP2C55 (Wang et al., 2004). Purified recombinant CYP2C55 was prepared as described previously (Wang et al., 2004). The microsomes (6 μg/lane for CYP2B10, 20 μg/lane for CYP2C55) and recombinant CYP2C55 (0.5 pmol of P450/lane) were electrophoresed in 12% Tris glycine gels, and the resolved proteins were transferred onto polyvinylidene difluoride membranes. Membranes were immunoblotted using the rabbit anti-CYP2C55 antibodies (1:1500 dilution), rabbit anti-CYP2B10 antibodies (1:20,000 dilution), goat anti-rabbit IgG (1:5000 dilution) conjugated to horseradish peroxidase (GE Healthcare Bio-Sciences, Little Chalfont, Buckinghamshire, UK), and the ECL Plus reagent (GE Healthcare Bio-Sciences). Protein determinations were performed using reagents from Bio-Rad Laboratories (Hercules, CA).
19-HETE in Mouse Sera.
Quantification of 19-HETE was performed using a liquid chromatography/tandem mass spectrometry method adapted from a published method (Newman et al., 2002). On-line liquid chromatography of extracted samples was performed with Agilent Technologies 1100 Series capillary high-performance liquid chromatography. Separations were achieved using a Phenomenex (Torrance, CA) Luna C18(2) column (5 μm, 150 × 2 mm), which was held at 40°C. The flow rate was 350 μl/min. Mobile phase A was 0.1% acetic acid in water. Mobile phase B was 0.1% acetic acid in 85:15 acetonitrile/methanol. Gradient elution was used, and the mobile phase percentage B was varied as follows: 15% B at 0 min, ramp to 2 min to 30% B, ramp from 2 to 5 min to 55% B, and ramp from 5 to 25.5 min to 75.5% B. Samples were spiked with 30 ng of 10,11-dihydroxynonadecanoic acid (10,11-DiHN) in 10 μl of ethanol as an internal standard before extraction. 10,11-DiHN was supplied by John Newman (University of California, Davis, CA). 19-HETE for external calibration was supplied by John R. Falck (University of Texas Southwestern, Dallas, TX). Dried sample extracts were reconstituted in 100 μl of 50% ethanol. Triplicate injections of 20 μl were analyzed.
Electrospray ionization/tandem mass spectrometry was used for detection. Analyses were performed on an Applied Biosystems/MDS Sciex (Foster City, CA) API 3000 equipped with a TurboIonSpray source. Turbo desolvation gas was heated to 350°C at a flow rate of 7 l/min. All the analytes were monitored as negative ions with the instrument in multiple-reaction monitoring mode. Analytes were monitored at the following parent ion–product ion mass/charge ratio pairs and retention times (tR): 19-HETE, 319.2–275.0 (tR = 16.8 ± 0.1 min); and 10,11-DiHN, 329.2–311.2 (tR = 18.7 ± 0.1 min).
Statistics.
Statistical analysis was performed using Student's t test for the drug responses in Car(+/+), Car(−/−), Pxr(+/+), or Pxr(−/−) mice.
Results
Induction of CYP2C55 mRNA in Livers and Kidneys.
Car(+/+) and Car(−/−) mice were given intravenous injections of saline or PB (100 mg/kg) and sacrificed 24 h later. The mRNA levels in these mouse livers were determined by real-time reverse transcription (RT)-PCR. The PB treatment increased hepatic CYP2C55 mRNA 140-fold in Car(+/+) but not in Car(−/−) mice (Fig. 1A). Likewise, the hepatic CYP2C55 mRNA was examined in Pxr(+/+) and Pxr(−/−) mice intraperitoneally treated with DMSO or PCN (20 mg/kg). The hepatic CYP2C55 mRNA was induced more than 15-fold in Pxr(+/+) mice treated with PCN but not in Pxr(−/−) mice (Fig. 1B). Next we examined the effect of chronic PB treatment on the hepatic CYP2C55 mRNA. Car(+/+) and Car(−/−) mice were given a single intraperitoneal dose of DEN (90 mg/kg) and treated chronically with PB (500 ppm) for 6 and 32 weeks. The hepatic CYP2C55 mRNA was induced approximately 30-fold in Car(+/+) mice treated with PB for 6 and 32 weeks but not in Car(−/−) mice (Fig. 1, C and D).

CAR- and PXR-dependent induction of hepatic CYP2C55 mRNA in mice. A and B, mice were given intravenous injections of saline, PB (100 mg/kg, i.v.), intraperitoneal DMSO, or PCN (20 mg/kg, i.p.) and sacrificed 12 and 24 h after injection, respectively. Relative mRNA levels in these mouse livers were determined by real-time RT-PCR and were expressed by taking those in the Car(+/+) or Pxr(+/+) mice treated with vehicle as equal to one. Values express mean ± S.D. (n = 3). **, p < 0.01 for vehicle-injected group versus drug-injected group in the Car(+/+) and Pxr(+/+) mice. C and D, mice were given a single intraperitoneal dose of DEN (90 mg/kg) and chronically treated with PB (500 ppm) for 6 and 32 weeks as described under Materials and Methods. These mice were sacrificed, and relative hepatic mRNA levels were determined by real-time RT-PCR and were expressed by taking those in the Car(+/+) mice treated with only DEN as equal to one. Values express mean ± S.D. (n = 6). ***, p < 0.005 for DEN versus DEN + PB in the Car(+/+) mice.
Car(+/+) and Car(−/−) mice received intravenous injections of saline or PB (100 mg/kg) and were sacrificed 12 h after the injection. Renal CYP2C55 mRNA levels were induced 50-fold in Car(+/+) mice treated with PB but not in Car(−/−) mice (Fig. 2A). Likewise, renal CYP2C55 mRNA levels were examined in Pxr(+/+) and Pxr(−/−) mice treated with DMSO or PCN (20 mg/kg) at 24 h after the injection. Renal CYP2C55 mRNA was induced more than 4-fold only in the Pxr(+/+) mice (Fig. 2B).

CAR- and PXR-dependent induction of renal CYP2C55 mRNA in mice. A, mice were given intravenous injections of saline or PB (100 mg/kg) and sacrificed 12 h after the injection. B, mice were given intraperitoneal injections of DMSO or PCN (20 mg/kg) and sacrificed 24 h after the injection. Relative mRNA levels in these mouse kidneys were determined by real-time RT-PCR and were expressed by taking those in the Car(+/+) or Pxr(+/+) mice treated with vehicle as equal to one. Values express mean ± S.D. (n = 3). **, p < 0.01 for vehicle-injected group versus drug-injected group in the Car(+/+) and Pxr(+/+) mice.
Microsomal CYP2C55 in Mouse Livers.
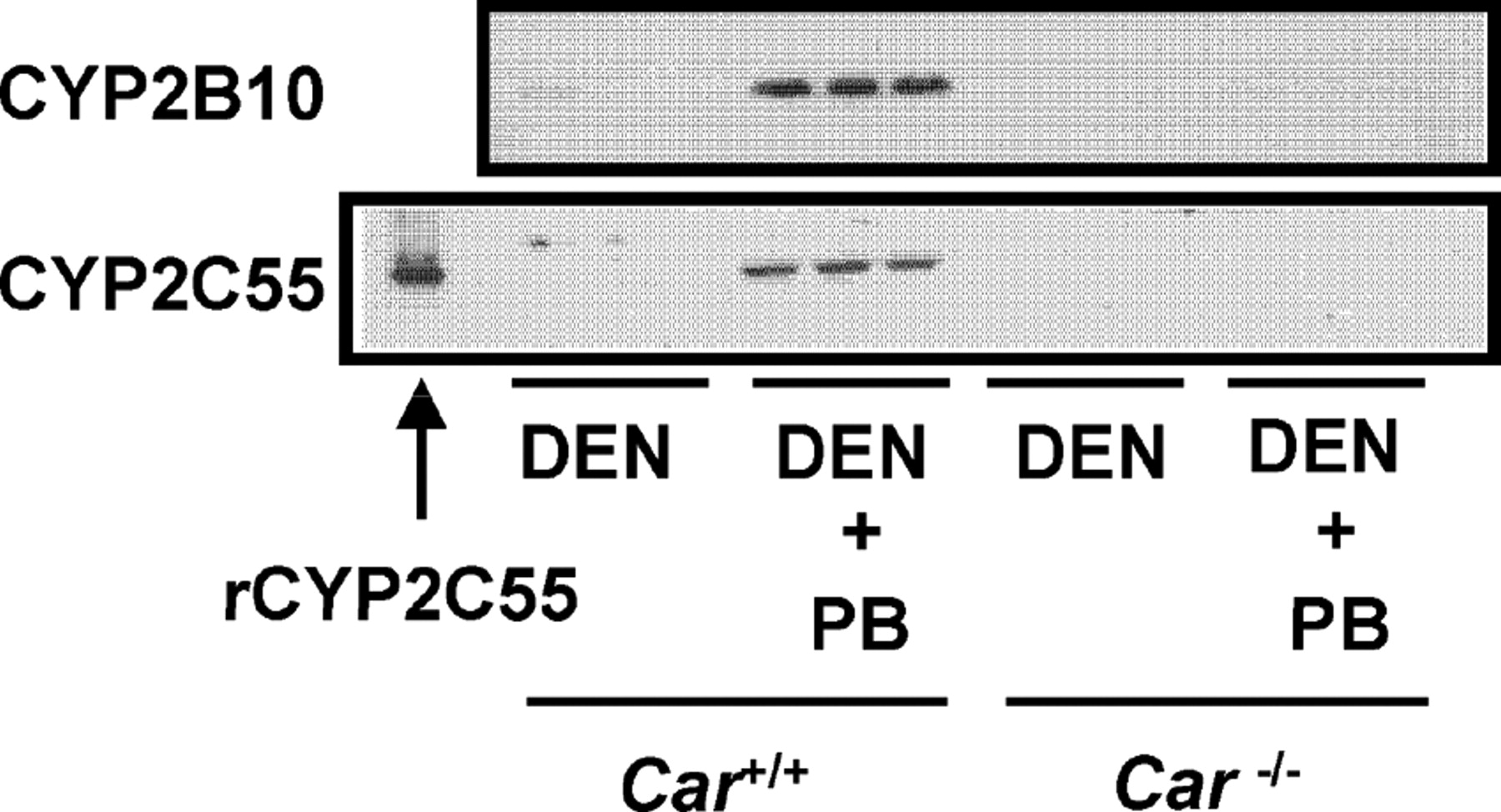
Car(+/+) and Car(−/−) mice were given a single intraperitoneal dose of DEN (90 mg/kg) and treated chronically with PB (500 ppm) for 6 weeks. Western blot analysis was performed with liver microsomes prepared from these mice. Hepatic CYP2C55 protein was clearly increased in Car(+/+) mice by PB treatment but not in Car(−/−) mice (Fig. 3). In addition, we confirmed that the hepatic CYP2B10 protein was also only induced in the Car(+/+) mice by PB.
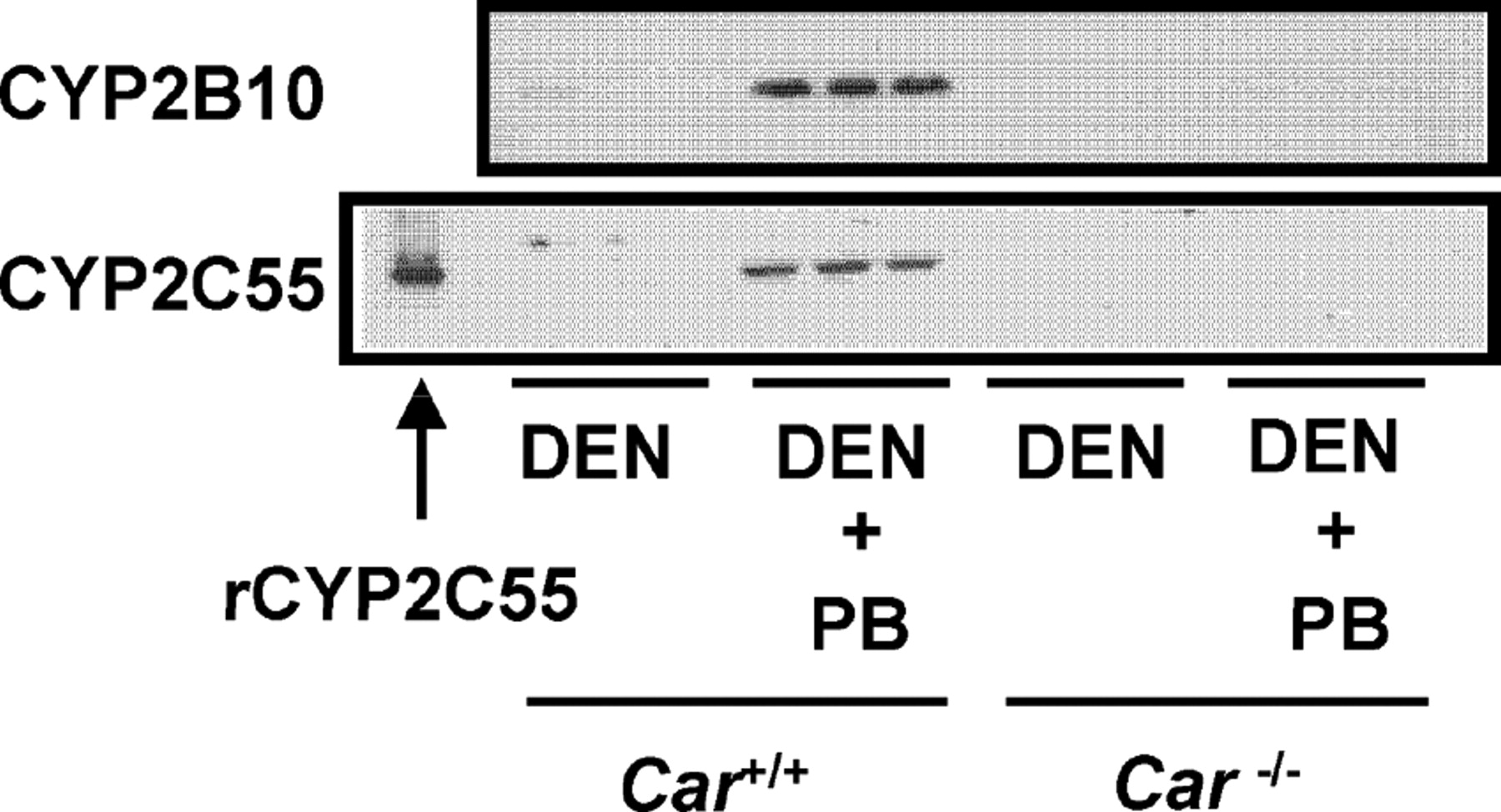
CAR-dependent induction of hepatic CYP2C55 protein in mice. Mice (three per group) were given a single intraperitoneal dose of DEN (90 mg/kg) and chronically treated with PB (500 ppm) for 6 weeks. Recombinant CYP2C55 (0.5 pmol of P450/lane) and microsomes (6 μg/lane for CYP2B10, 20 μg/lane for CYP2C55) prepared from the mice livers were analyzed by Western blot analysis as described under Materials and Methods.
Serum 19-HETE Level in Mice.
Car(+/+) mice (three mice for each group) were given a single intraperitoneal dose of DEN (90 mg/kg) and treated chronically with PB (500 ppm) for 6 weeks. Serum 19-HETE levels in these mice were measured by liquid chromatography/tandem mass spectrometry analysis. The serum 19-HETE level was significantly induced 2-fold by PB treatment: 2.5 ± 0.5 and 5.5 ± 1.5 for DEN + PB treatment versus DEN treatment, respectively, p < 0.05.
A CAR/PXR Response Element within the Cyp2c55 Promoter.
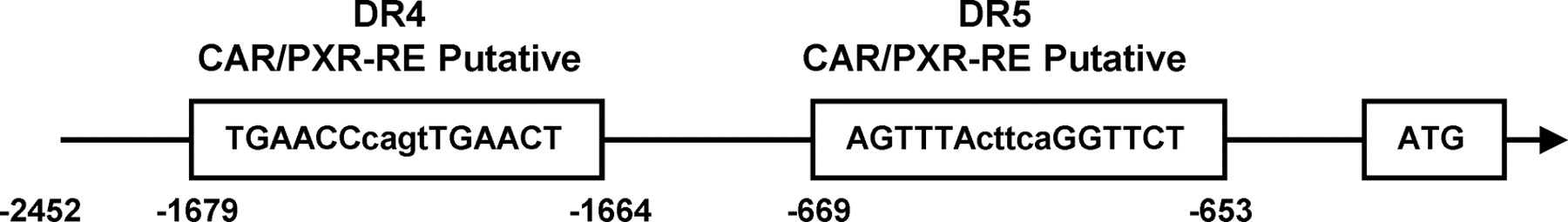
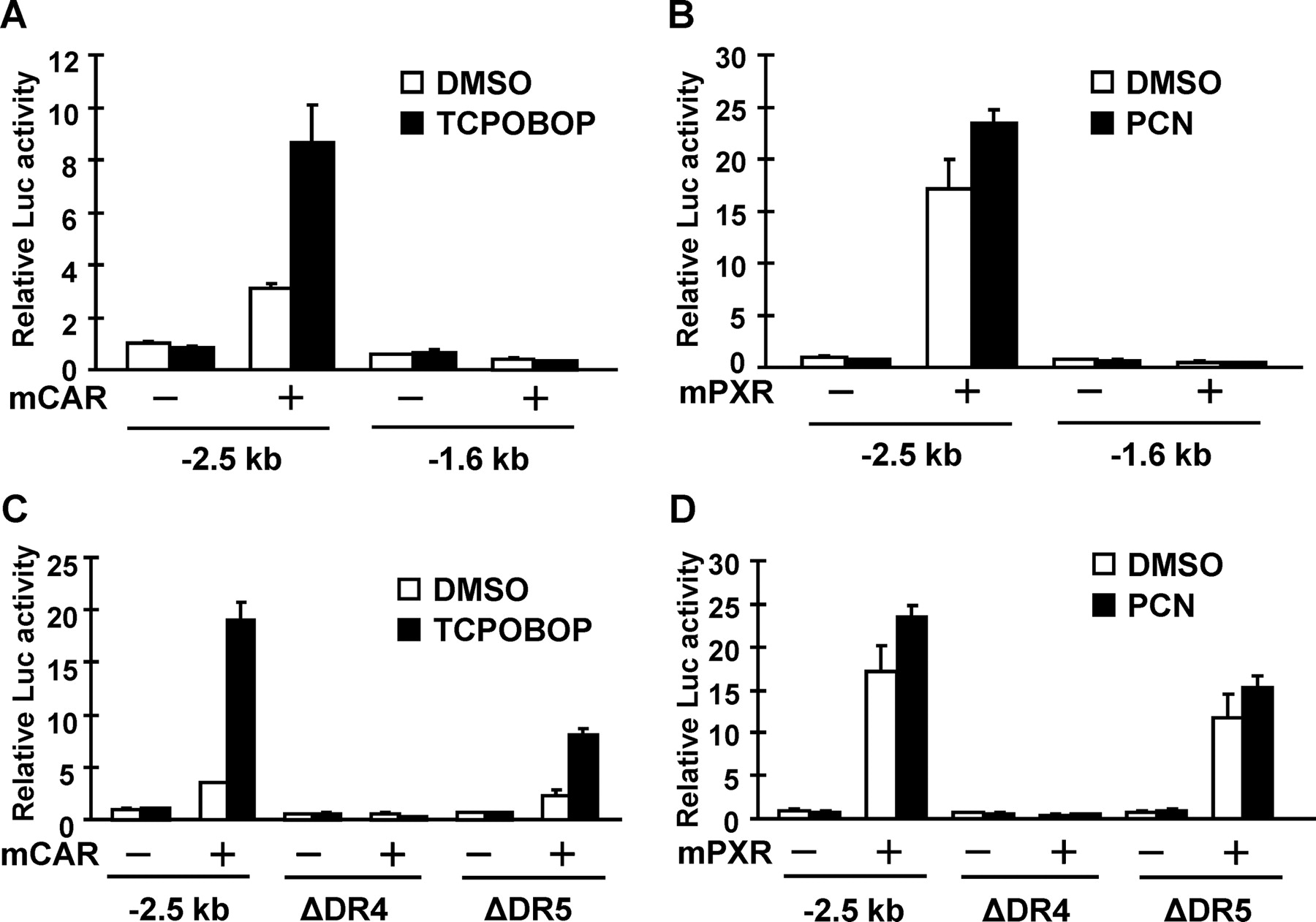
SeqLab GcG (Accelrys, San Diego, CA) was used to search a 10-kb DNA sequence of the Cyp2c55 5′-flanking region for a CAR/PXR response element, an imperfect direct repeat of AGGTCA spaced by three to five nucleotides (DR-n). Two putative response elements were found upstream of the Cyp2c55 5′-flanking region, including a DR4 motif (−1679/−1664) and a DR5 motif (−669/−653) (Fig. 4). Luciferase reporters containing various lengths of the Cyp2c55 5′-flanking region were constructed and cotransfected with CAR or PXR into Huh7 cells for transient transfection assays. Whereas the Cyp2c55−1.6 kb luciferase reporter was not activated by the CAR ligand 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), the Cyp2c55−2.5 kb luciferase reporter was activated 8-fold (Fig. 5A). This Cyp2c55−2.5 kb luciferase reporter was also activated 17- and 25-fold by PCN when PXR was cotransfected (Fig. 5B). To determine the role of the DR4 site (−1679/−1664) in this activation, this site was internally deleted from the Cyp2c55−2.5 kb luciferase reporter. Neither TCPOBOP nor PCN activated the DR4-deleted promoter (Fig. 5, C and D). Furthermore, gel shift assays confirmed specific binding of CAR and PXR to the DR4 sequence (data not shown).

Alignment of CYP2C55 5′-flanking region. A schematic representation of the Cyp2c55 5′-flanking region provides the putative CAR and PXR binding sites.

Transcriptional activation analysis of the Cyp2c55 promoter by mCAR and mPXR in Huh7 cells. A and B, reporter plasmid, pGL3/Cyp2c55−2.5 kb and pGL3/Cyp2c55−1.6 kb, were cotransfected with or without pCR3/mCAR and pcDNA3.1/mPXR as indicated. At 24 h after transfection, cells were treated with DMSO, TCPOBOP (250 nM), and PCN (10 μM) and incubated for an additional 24 h. Relative luciferase activities were expressed by taking the activity of the DMSO-treated cells transfected with the −2.5-kb reporter plasmid alone as equal to one. C and D, reporter plasmid, pGL3/Cyp2c55−2.5 kb, and the internal deletion mutants of the putative binding sites, DR4 and DR5, were transfected with or without pCR3/mCAR and pcDNA3.1/mPXR as indicated. At 24 h after transfection, cells were treated with DMSO, TCPOBOP (250 nM), and PCN (10 μM) and incubated for an additional 24 h. Relative luciferase activities were expressed by taking the activity of the DMSO-treated cells transfected with the −2.5-kb reporter plasmid alone as one.
Discussion
CYP2C subfamily enzymes are known to metabolize xenochemicals and endogenous compounds such as AA. Mouse CYP2C55 was characterized as an enzyme that preferentially catalyzes the biosynthesis of 19-HETE (Wang et al., 2004), an AA metabolite known to have potent physiological functions such as effects on renal vascular tone and ion transport (Escalante et al., 1988; Ma et al., 1993; Carroll et al., 1996). Herein, we found that CAR and PXR regulate drug induction of CYP2C55 mRNA in mouse liver and kidney by using Car(+/+), Car(−/−), Pxr(+/+), and Pxr(−/−) mice. Furthermore, we also found that chronic PB treatment increased serum 19-HETE levels. Because other P450 isoforms (e.g., CYP2E1, CYP4A, CYP2C, and CYP2J9) (Laethem et al., 1993; Luo et al., 1998; Nguyen et al., 1999; Qu et al., 2001; Poloyac et al., 2004; Wang et al., 2004; Cowpland et al., 2006) are known to synthesize 19-HETE, the actual contribution of CYP2C55 to the PB-increased 19-HETE remains to be determined in future investigations. 19-HETE has been suggested to vasodilate renal arcuate arteries (Ma et al., 1993) and stimulate the renal cortical Na+/K+ ATPase (Escalante et al., 1988) and proximal tubule transporters (Quigley et al., 2000). Although there have been no reports that chronic usage of CAR and PXR activators such as PB and rifampicin resulted in alteration of renal function, this might be an area of future research. Moreover, it is of interest that among the human CYP2C enzymes, CYP2C19 is unique in that it produces primarily 19-HETE from AA (Bylund et al., 1998). Moreover, CYP2C19 is inducible by CAR and PXR (Chen et al., 2003). It is noteworthy that in the studies in Cyp3a-null mice that CYP2C55 was markedly induced and that the authors also suggested that food-derived xenobiotics might up-regulate CYP2C55 (van Waterschoot et al., 2008). Additional studies by the same group (van Watershoot et al., 2009) showed that the CAR ligand TCBPOBOP induced CYP2C55. Dexamethasone induced CYP2C55 in wild-type mice but not PXR knockout mice. There were differences in CYP2C55 expression in mice being fed with semisynthetic versus commercial chow, for which the mechanism was not investigated further.
Drug-induced transcriptional regulation of the human CYP2C subfamily genes, such as CYP2C9, by CAR and PXR has been characterized (Ferguson et al., 2002; Gerbal-Chaloin et al., 2002; Chen et al., 2004; Ferguson et al., 2005). Within the mouse CYP2C subfamily, the Cyp2c29 and Cyp2c37 genes were reported to be up-regulated by CAR but not by PXR (Jackson et al., 2004, 2006). Herein, we identified a functional CAR and PXR responsive element (−1679/−1664) within the Cyp2c55 promoter. Cyp2c55 is the first murine Cyp2c gene that has been shown to be regulated by PXR. Both CAR and PXR bind to the same responsive element DR4 (−1679/−1664) and activate the Cyp2c55 gene in liver and kidney. Induction of Cyp2c55 may be responsible for the increase of 19-HETE levels in serum of DEN-PB-treated mice.
Acknowledgments.
We thank the NIEHS for the sequencing core.
Footnotes
This work was supported in part by the Intramural Research Program of the National Institutes of Health National Institute of Environmental Health Sciences [Grants Z01-ES7100501, Z01-ES025034, Z01-ES050167, Z01-ES02124] (to M.N., D.C.Z., K.B.T., and J.A.G., respectively).
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032334.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- AA
- arachidonic acid
- CAR
- constitutive active/androstane receptor
- PXR
- pregnane X receptor
- PCN
- pregnenolone 16α-carbonitrile
- HETE
- hydroxyeicosatetraenoic acid
- EET
- epoxyeicosatrienoic acid
- PB
- phenobarbital
- DMSO
- dimethyl sulfoxide
- DEN
- diethylnitrosamine
- C3H
- C3H/HeNCrlBR (mice)
- NIEHS
- National Institute of Environmental Health Sciences
- PCR
- polymerase chain reaction
- kb
- kilobase(s)
- 10,11-DiHN
- 10,11-dihydroxynonadecanoic acid
- tR
- retention time
- RT
- reverse transcription
- DR-n
- direct repeat spaced by n nucleotides
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene.
- Received January 19, 2010.
- Accepted April 6, 2010.
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}