


Abstract
Ombitasvir (also known as ABT-267) is a potent inhibitor of hepatitis C virus (HCV) nonstructural protein 5A (NS5A), which has been developed in combination with paritaprevir/ritonavir and dasabuvir in a three direct-acting antiviral oral regimens for the treatment of patients infected with HCV genotype 1. This article describes the mass balance, metabolism, and disposition of ombitasvir in humans without coadministration of paritaprevir/ritonavir and dasabuvir. Following the administration of a single 25-mg oral dose of [14C]ombitasvir to four healthy male volunteers, the mean total percentage of the administered radioactive dose recovered was 92.1% over the 192-hour sample collection in the study. The recovery from the individual subjects ranged from 91.4 to 93.1%. Ombitasvir and corresponding metabolites were primarily eliminated in feces (90.2% of dose), mainly as unchanged parent drug (87.8% of dose), but minimally through renal excretion (1.9% of dose). Biotransformation of ombitasvir in human involves enzymatic amide hydrolysis to form M23 (dianiline), which is further metabolized through cytochrome P450–mediated oxidative metabolism (primarily by CYP2C8) at the tert-butyl group to generate oxidative and/or C-desmethyl metabolites. [14C]Ombitasvir, M23, M29, M36, and M37 are the main components in plasma, representing about 93% of total plasma radioactivity. The steady-state concentration measurement of ombitasvir metabolites by liquid chromatography–mass spectrometry analysis in human plasma following multiple doses of ombitasvir, in combination with paritaprevir/ritonavir and dasabuvir, confirmed that ombitasvir is the main component (51.9% of all measured drug-related components), whereas M29 (19.9%) and M36 (13.1%) are the major circulating metabolites. In summary, the study characterized ombitasvir metabolites in circulation, the metabolic pathways, and the elimination routes of the drug.
Introduction
Hepatitis C virus (HCV) infection affects approximately 170 million individuals worldwide (World Health Organization, 2011). Untreated chronic HCV infection can result in cirrhosis or hepatocellular carcinoma, both of which are leading causes of liver transplantation (Pawlotsky, 2004; Lavanchy, 2011; Mohd Hanafiah et al., 2013). Recently, several interferon-free combinations of direct-acting antivirals (DAAs) have been developed to cure chronic HCV infection with high success rates (Shah et al., 2013; Zeuzem, 2014). Ombitasvir has been developed for the HCV genotype-1 infection in combination with an NS3 protease inhibitor, paritaprevir with ritonavir, and/or a nonstructural protein 5B (NS5B) non-nucleoside polymerase inhibitor (dasabuvir) with or without ribavirin (Feld et al., 2014; Ferenci et al., 2014; Kowdley et al., 2014; Poordad et al., 2014). Ombitasvir is an inhibitor of HCV NS5A (DeGoey et al., 2014; Krishnan et al., 2015). Ombitasvir exhibited picomolar activities against HCV genotype 1a and 1b subgenomic replicons in vitro, with EC50 values of 14 and 5 pM, respectively. Ombitasvir also demonstrated robust in vivo responses with mean maximum decreases in HCV RNA up to 3.10 log10 IU/ml following 3-day monotherapy in treatment-naïve HCV genotype-1–infected subjects (Lawitz et al., 2012).
Clinically, ombitasvir has favorable safety, tolerability, and pharmacokinetic profiles when given as a monotherapy or combination therapy at doses administered to date (Dumas et al., 2011; Menon et al., 2013). Ombitasvir shows linear pharmacokinetics with dose-proportional increases in exposure over the range of 5–100 mg after once-daily multiple-dose administration. Ombitasvir has a half-life (t1/2) of approximately 24 hours when administered once daily. The mean Cmax and area under the curve (0–24-hour) (AUC0-24h) values of ombitasvir were 27 and 62% higher, respectively, on day 10 compared with day 1 following 5- to 200-mg once-daily (QD) multiple doses, suggesting minimal accumulation (Dumas et al., 2011).
This report describes the metabolism and disposition of a single 25-mg oral dose of [14C]ombitasvir in four healthy human subjects. The purpose of this study was to assess the mass balance, elucidate the routes and rates of excretion, identify and quantify the exposure of circulating metabolites in human plasma, elucidate the metabolite structures, and determine the metabolite profiles in excreta and the metabolic pathway of ombitasvir in humans. In addition, the circulating metabolites of ombitasvir at steady state in the 3DAA regimen (paritaprevir/ritonavir, dasabuvir, and ombitasvir) were assessed, and mechanisms of metabolite formation are also described.
Materials and Methods
Drugs and Reagents.
The following reference standards were supplied by Process Chemistry, AbbVie, Inc. (North Chicago, IL) and were used as high-performance liquid chromatography (HPLC) and mass spectrometric standards: ombitasvir [dimethyl((2S,2'S)-((2S,2'S)-2,2'-(((((2S,5S)-1-(4-(tert-butyl)phenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl))bis(carbonyl))bispyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl))dicarbamate], M23 [4,4'-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline], M29 [1-(4-((2S,5S)-2,5-bis(4-aminophenyl)pyrrolidin-1-yl)phenyl)ethanone], and M36 [1-(4-((2S,5S)-2,5-bis(4-aminophenyl)pyrrolidin-1-yl)phenyl)-2-hydroxyethanone. [14C]Ombitasvir was supplied by Process Chemistry, AbbVie, Inc. The chemical structure of [14C]ombitasvir is shown in Fig. 1; * denotes the [14C]label position. The radiochemical synthesis of [14C]ombitasvir started from para-nitroacetophenone[carbonyl-14C] ([14C]4'-NAP) and was completed in six steps. Purification of the compound by crystallization provided >99% radiochemical purity by HPLC.

Structure of [14C]ombitasvir. *Position of [14C] radiolabel.
Clinical Study.
The clinical study was conducted at Covance Laboratories Inc., in conjunction with the Covance Clinical Research Unit (Madison, WI). In this open-label study, a total of four adult male subjects (N = 4) in general good health were selected to participate in the study according to the selection criteria. On the morning of study day 1, subjects received a single oral dose of [14C]ombitasvir under nonfasting conditions. The study drug, ombitasvir (25 mg active, 100 μCi), was administered as a single liquid-filled capsule. The total amount of liquid taken was approximately 240 ml, 30 minutes after starting a standardized breakfast. Subjects were confined to the study site for a minimum of 192 hours postdose, or up to a maximum of 360 hours postdose. Subjects were released from the study site at any time after 192 hours postdose if the preset release criteria were met.
Blood samples were collected by venipuncture into vacutainer collection tubes containing potassium (K2) EDTA at the following times: 0 (predose), 1, 2, 4, 6, 8, 10, 12, 24, 48, 72, 96, 120, 144, 168, and 192 hours after dosing of [14C]ombitasvir on day 1. Plasma was separated via centrifugation and stored at –70°C.
Urine samples were collected over the following intervals: 0–12, 12–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, and 168–192 hours after dosing of [14C]ombitasvir on study day 1. Urine samples were collected into a collection jar containing approximately 1.2 g of dodecylbenzenesulfonic acid sodium salt to minimize nonspecific binding with the container. Aliquots of the urine were frozen and maintained at –20°C prior to metabolite profiling.
Fecal samples were collected predose (upon check-in before dosing) and over the following intervals after dosing: 0–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, and 168–192 hours. All feces collected during a collection interval were kept frozen at −20°C.
Total Radioactivity Measurement by Liquid Scintillation Counting.
All sample combustion was performed using a Model 307 Sample Oxidizer (Packard Instrument Company), and the resulting 14CO2 was trapped in a mixture of PermaFluor and Carbo-Sorb (Perkin Elmer). The oxidation efficiency was evaluated each day of sample combustion by analyzing a commercial radiolabeled standard both directly in scintillation cocktail and by oxidation. Acceptance criteria were defined as combustion recoveries of 95–105%. Ultima Gold XR scintillation cocktail (Perkin Elmer, Waltham, MA) was used for samples analyzed directly. All samples were analyzed for radioactivity in Model 2900TR liquid scintillation counters (Packard Instrument Company, Downers Grove, IL) for at least 5 minutes or 100,000 counts. Each sample was homogenized, and an aliquot was mixed with scintillation cocktail before radioanalysis. All samples were analyzed in duplicate if the sample size allowed, unless the entire sample was used for analysis. If the results from sample replicates (calculated as 14C dpm/g sample) differed by more than 10% from the mean value, and sample aliquots had radioactivity greater than 200 dpm, the sample was rehomogenized and reanalyzed.
After mixing, duplicate blood samples were weighed (approximately 0.2 g), combusted, and analyzed by liquid scintillation counting (LSC). The representative lower limit of quantitation for blood was 13.1 ng equivalents/g. Plasma samples were mixed, and duplicate weighed aliquots (approximately 0.2 g) were analyzed directly by LSC. The representative lower limit of quantitation for plasma was 11.9 ng equivalents/g. The urine samples were mixed and duplicate weighed aliquots (approximately 0.2 g) were analyzed directly by LSC. The representative lower limit of quantitation for urine was 11.3 ng equivalents/g. Fecal samples were combined by subject at 24-hour intervals, and the weight of each combined sample was recorded. A weighed amount of water was added, and the sample was mixed. The sample was removed from the freezer and homogenized, or immediately homogenized using a probe-type homogenizer. Duplicate weighed aliquots (approximately 0.2 g) were combusted and analyzed by LSC.
Sample Preparation for Metabolite Profiling.
Plasma samples were thawed at room temperature and pooled across subjects at selected time points in addition to AUC plasma pooling utilizing the Hamilton method (Hamilton et al., 1981). Plasma samples were processed using a solvent extraction method. In brief, pooled plasma was extracted with a 4-fold volume of acetonitrile/methanol mixture (3:1, v/v), followed by vortexing and sonication. The sample was then centrifuged at 3000 rpm (2465 × g) for 15 minutes at 4°C. The supernatant was transferred to a glass tube. The protein pellets were extracted sequentially four times, each using 2-fold the original sample volume of acetonitrile/methanol mixture (3:1, v/v), with vortexing and sonication (15 minutes). After combining the supernatants, 100 µl of formamide was added, and the solution was concentrated to ∼100 µl under a stream of nitrogen. The residues were diluted with 75 µl of acetonitrile/methanol mixture (3:1, v/v) and 150 µl of water before HPLC–mass spectrometry (MS)–radiochemical detection analysis. An aliquot of the reconstituted sample was subjected to LSC analysis to determine total radioactivity recovery. Another aliquot of the reconstituted sample was transferred to an HPLC autosampler vial and was injected for HPLC–MS–radiochemical detection analysis. The mean radioactivity recovery in the processed plasma samples was about 93.1 ± 12.7% (S.D.).
Equal volumes of urine were pooled across subjects at each time point before processing. The pooled urine was centrifuged at 3220 × g for 15 minutes at 4°C. Aliquots were dried under a stream of nitrogen at room temperature and reconstituted for metabolite profiling using HPLC–MS–radiochemical detection analysis. Aliquots were also cleaned up using a Strata SAX solid-phase extraction (SPE) cartridge (Phenomenex, Torrance, CA) to remove the detergent in the urine sample. In brief, an SPE cartridge (1 g/12 ml) was conditioned with 15 ml of methanol and 15 ml of deionized water. Aliquots of pooled urine were loaded to the preconditioned column, followed by washing with 10 ml of water. The elution was achieved by using 4 × 5 ml of acetonitrile/methanol mixture (3:1, v/v). The eluate was dried under the nitrogen stream at room temperature. The residue was reconstituted in the initial mobile phase for HPLC–MS–radiochemical detection analysis. The overall extraction recovery was about 59.3 ± 13.1% (S.D.).
The feces samples were processed using multiple solvent extractions with acetonitrile/methanol mixture (3:1, v/v) using a 1:3 sample:solvent ratio, followed by centrifugation at 3220 × g for 20 minutes at 4°C. The repeated extraction was stopped either when 80% of the radioactivity had been recovered or until less than 2% of the radioactivity was extracted. Aliquots of extracted samples were subjected to LSC for total radioactivity. The extract was dried under a nitrogen stream at room temperature. The final residues were reconstituted in acetonitrile/methanol (3:1; v/v) and further diluted with 30% water for HPLC–MS–radiochemical detection analysis. An aliquot of the reconstituted solution was subjected to LSC analysis for extraction recovery calculation. The overall extraction recovery for the fecal sample was 98.6 ± 24.1% (S.D.).
Method for Metabolite Profiles and Identification.
HPLC separation of ombitasvir and the corresponding metabolites was achieved using a Thermo Accela HPLC system (Thermo Fisher, San Jose, CA), which consisted of Accela autosampler, 1250 Series binary pump, and Accela PDA detector. The elution of metabolites in plasma and urine was achieved at room temperature on an Agilent Eclipse XDB C18, 5-µm, 4.6 × 250–mm HPLC column (Agilent Technologies, Santa Clara, CA). Mobile phases were as follows: 50 mM ammonium acetate aqueous solution (A), and acetonitrile/methanol mixture (1:1, v/v) (B); the flow rate was maintained at 1.0 ml/min. The gradient was as follows: 0–3 minutes: 5% B; 3–10 minutes: 5–40% B; 10–53 minutes: 40–75% B; 53–72 minutes: 75–95% B; 72–75 minutes: 95% B; 75–76 minutes: 95–5% B; 76–80 minutes: 5% B. The HPLC system was interfaced with a mass spectrometer. The high-resolution MS and MSn acquisitions were performed with a Thermo Fisher Orbitrap Discovery mass spectrometer (Thermo Fisher, San Jose, CA) fitted with an electrospray ionization source (typical source parameter: sheath gas 25.0; auxiliary gas 10; electrospray ionization source +4500 volts; capillary temperature 300°C; capillary voltage 43 V; tube lens 80 V). The instrument was calibrated daily using external calibration reference compounds, with mass resolution set at 30,000 for full scan and 7500 for MSn scan. Typical mass errors of analytes relative to theoretical masses are less than ±5 ppm in daily operations. MS data were processed using Thermo Xcalibur 2.10 and MetWorks 1.2 utilizing a multiple mass defect filtering algorithm.
For metabolite analysis in fecal samples, separation was accomplished on an Agilent Eclipse XDB C18, 5-µm, 4.6 × 250–mm HPLC column; mobile phases were as follows: 0.1% formic acid in water (A), and acetonitrile (B); the flow rate was maintained at 1.0 ml/min. The gradient was as follows: 0–2 minutes: 15–20% B; 2–5 minutes: 20–30% B; 5–25 minutes: 30–45% B; 25–50 minutes: 45–65% B; 50–53 minutes: 65–75% B; 53–60 minutes: 75–95% B; 60–61 minutes: 95% B.
Radioactive components in plasma, urine, or feces samples were collected in a TopCount 96 Deep Well Luma Plate (PerkinElmer, Waltham, MA) and counted by using a PerkinElmer TopCount NXT system. The HPLC eluent was split postcolumn between the mass spectrometer and Agilent 1100 fraction collector at a ratio of 20:80. The Agilent 1100 fraction collector was set to collect fractions at intervals of 0.3 minute/well.
Pharmacokinetic Calculations.
Plasma concentration-time radioactivity data were analyzed with SAS software (version 9.2; SAS Institute Inc., Cary, NC). Maximum plasma concentration (Cmax), time at which Cmax was achieved (Tmax), area under the concentration-time curve from time zero to the last measurable time point for total radioactivity, [14C]ombitasvir, and its metabolites in plasma were estimated. Area under the concentration-time curve from time zero to infinity and t1/2 for total radioactivity, and [14C]ombitasvir in plasma were also calculated.
Metabolism of M23 by Recombinant CYP2C8.
Synthetic reference material of M23 was incubated with recombinant CYP2C8 enzyme (BD Gentest, Bedford, MA) in the presence of NADPH. The incubation mixture (225 μl) included the substrate (10 µM final concentration), 0.1 mM phoshphate buffer (pH 7.4), and recombinant CYP2C8 protein (final concentration 100 pmol/ml). After a 5-minute prewarm in a 37°C water bath, 25 µl of 10 mM NADPH was added to initiate the reaction (NADPH final concentration 1 mM). The samples were incubated in a 37°C water bath for 60 minutes. The reaction was stopped by adding 1 volume of quenching solution (acetonitrile/methanol; 1:1, v/v) to the incubation mixture. The mixture was centrifuged at 3220 × g for 20 minutes at 4°C. Aliquots of the supernatant were subjected to HPLC-MS analysis.
Human Plasma for Quantitative Analysis by Liquid Chromatography–MS.
Human plasma samples were obtained from 12 subjects in a phase 1 open-label, pharmacokinetics, safety and tolerability study. Healthy subjects were orally administered ombitasvir (25-mg tablet, QD), paritaprevir/ritonavir (150/100-mg tablet, QD), and dasabuvir (400-mg tablet, twice a day) for 14 days. Blood samples for plasma concentration analysis were collected on day 14 at 0, 1, 2, 3, 4, 6, 9, 12, 16, and 24 hours after dosing. The plasma samples were pooled using an equal 100-µl volume across subjects at each time point for HPLC–tandem mass spectrometry (MS/MS) quantitation.
Liquid Chromatography–MS Quantitation Method for Ombitasvir and Metabolites in Plasma.
A bioanalytical method was developed for the simultaneous quantitation of parent drug and the metabolites M23, M29, M36, and M37 in human plasma. In brief, the method separated the components of interest and internal standards from a plasma aliquot using protein precipitation with a mixture of acetonitrile and methanol (9:1, v/v). Spiked plasma standards were analyzed simultaneously with the samples. Parent drug, the selected metabolites and the internal standards (D13-ombitasvir and [6-(4-fluoro-benzoyl)-1H-benzoimidazol-2-yl]-carbamic acid methyl ester) were separated at room temperature on an Ascentis Express C18, 2.7-µm, 30 × 3–mm HPLC column (Sigma-Aldrich, St. Louis, MO). Mobile phases were as follows: acetonitrile (A), and 20 mM ammonium acetate, pH 7.6 (B). The flow rate was maintained at 0.8 ml/min. The gradient was as follows: 0–0.2 minute: 20% A; 0.2–0.22 minute: 20–30% A; 0.22–0.35 minute: 30–45% A; 0.35–0.9 minute: 45%; 0.9–0.92 minute: 45–20% A; 0.92–1.1 minutes: 20% A. Analysis was performed on an API5500 Mass Spectrometer (AB Sciex, Framingham, MA) with a turbo-ionspray interface, with ionization of the analytes in the positive-ion mode; detection was in the multiple reaction monitoring mode. The peak areas of all components of interest were determined using Sciex Analyst software. The concentration of each sample was calculated by least-squares linear regression analysis of the peak area ratio (compound/internal standard) of the spiked human plasma standards versus concentration.
Results
Excretion of Radioactivity
Following a single oral dose of [14C]ombitasvir (25 mg, 100 µCi) to four healthy, male volunteers, the excretion of radioactivity in urine and feces from all subjects was measured over a period of up to 192 hours postdose. Figure 2 presents the mean cumulative recovery of total radioactivity in excreta, expressed as a percentage of dose. The overall mean recovery of radioactivity in urine and feces samples was 92.2% (± 0.82%, S.D.) over the 192-hour collection period, with recovery in individual subjects ranging from 91.4 to 93.1%. The radioactivity was excreted primarily through fecal elimination (mean, 90.2% of dose). Renal excretion was relatively minor (mean, 1.9% of dose).

Mean cumulative percentage of radioactive dose recovered in urine and feces at specified intervals after a single 25-mg (100-µCi) oral dose of [14C]ombitasvir to healthy male subjects.
Pharmacokinetic Data Analysis
The mean concentration-time profiles of ombitasvir and total radioactivity in human plasma after oral administration of [14C]ombitasvir are graphically depicted in Fig. 3. The pharmacokinetic parameters for ombitasvir and total radioactivity are summarized in Table 1. The concentration of total radioactivity was measured by LSC, expressed as ng equivalent/g. The concentrations of ombitasvir were determined using a validated liquid chromatography (LC)–MS/MS bioanalytical method, and expressed as ng/ml. Cmax for the parent drug and total radioactivity were 27.8 ng/ml and 142.5 ng equivalent/g, respectively. The AUC0-192h for the parent drug and total radioactivity were 359 ng⋅h/ml and 17,411 ng equivalent⋅h/g, respectively.

Mean (standard deviation) plasma concentration-time curves for ombitasvir (ng/ml) and total radioactivity (ng equivalent/g) in male subjects administered a single oral dose of [14C]ombitasvir (25 mg; n = 4).
Mean ± S.D. pharmacokinetic parameters of total radioactivity and ombitasvir
Metabolite Profiles of [14C]Ombitasvir in Circulation and Excreta
Plasma.
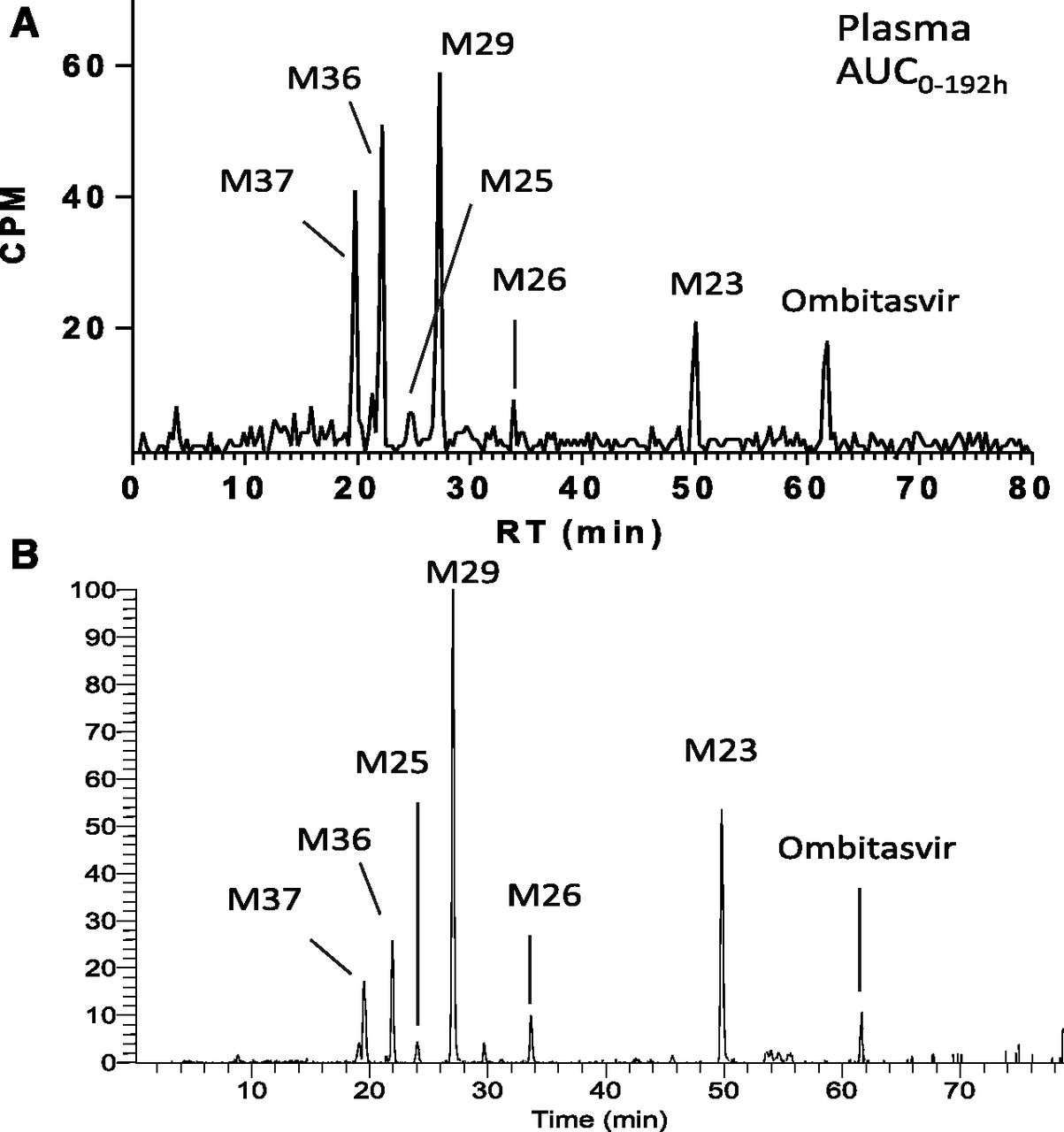
A representative HPLC radiochromatogram of [14C]ombitasvir and its metabolites in pooled human plasma using the Hamilton method (t = 0–192 hours postdose) is shown in Fig. 4. [14C]Ombitasvir and metabolites M29, M36, M37, and M23 were the main components in plasma. In addition, M25 and M26 were also detected at low levels. The relative amounts of ombitasvir and metabolites in human plasma, expressed as percentage of radioactivity in plasma, are summarized in Table 2. Metabolites M29, M36, M37, and M23 accounted for 32.9, 25.7, 16.3, and 10.0% of drug-related material in plasma, respectively. The unchanged drug represented approximately 8.5% of radioactivity. The concentration-time profile of ombitasvir and its metabolites is summarized in Table 3. Concentrations of metabolites are generally low, approximately in the nanomolar range.

Representative HPLC radiochromatogram (A) and HPLC-extracted ion chromatogram (B) of ombitasvir and its metabolites in AUC0-192h pooled human plasma after a single 25-mg oral dose of [14C]ombitasvir. CPM, counts per minute; RT, retention time.
Percentages of radioactivity for ombitasvir and its metabolites in pooled human plasma (n = 4, AUC0-192h) following administration of a single 25-mg oral dose of [14C]ombitasvir
Concentration-time profile of [14C]ombitasvir metabolites in pooled plasma at selected time point across subjects (n = 4)
Urine and Feces.
The recovered radioactivity of administered [14C]ombitasvir in urine was relatively low. The mean cumulative recovery of the dose in the entire sample collection (0–192 hours postdose) is only 1.91% (±0.36%). Chromatographic evaluation of selected pooled urine samples showed several small poorly separable radiochemical peaks (unknown metabolites in urine [Mu1–Mu5]) in the HPLC retention time between 8 and 20 minutes. The representative HPLC radiochromatogram of pooled human urine (48–72 hours postdose) is shown in Fig. 5A. Due to the detergent interference in the urine and low radioactive concentration of analytes, LC-MS analysis of SPE-enriched urine samples failed to provide molecular identities for these peaks. Only trace levels of [14C]ombitasvir were observed in 12- to 24-hour pooled urine (0.03% of dose; Table 4) but were not observed in late time points.

Representative HPLC radiochromatograms of ombitasvir and its metabolites in human excreta, urine (A) and feces (B), after a single 25-mg oral dose of [14C]ombitasvir. CPM, counts per minute; RT, retention time.
Percentages of excretory metabolites of ombitasvir in humans following administration of a single 25-mg oral dose of [14C]ombitasvir (n = 4)
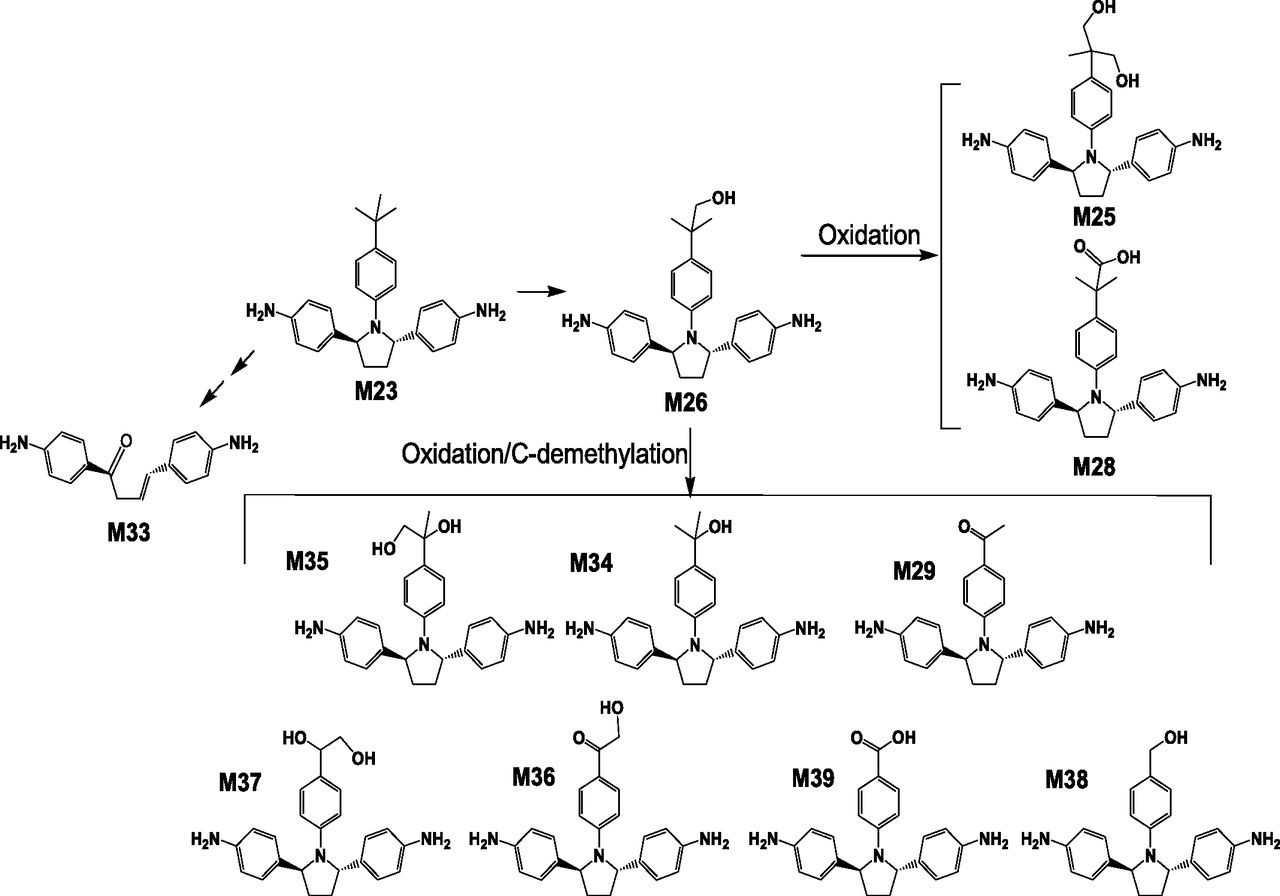
The representative HPLC radiochromatogram of pooled human feces is shown in Fig. 5B. Unchanged parent drug was the most abundant radiochemical component in feces throughout the sample collection periods from 0 to 192 hours postdose. The amount of ombitasvir and corresponding metabolites in urine and feces, expressed as the mean percentage of the administered radioactive dose, is tabulated in Table 4. The unchanged parent drug represents about 87.8% of the dose, indicating both absorbed and unabsorbed [14C]ombitasvir was mainly eliminated as unchanged parent drug in feces. Metabolites detected in feces are very minor (≤1% of total dose), including M9 (0.7% of dose), M3 (0.6%), M2 (0.2%), M5 (0.2%), and M6 (0.2%). The proposed metabolic scheme for ombitasvir in humans is shown in Fig. 6.

Proposed metabolic pathways of ombitasvir in humans.
LC-MS/MS Characterization of Ombitasvir and Metabolites
As described under Method for Metabolite Profiles and Identification, metabolites of ombitasvir were characterized using a combination of positive-ionization high-resolution full-scan MS and product ion scan (MS/MS) analyses. The structures of metabolites M5, M23, M29, M36, and M37 were confirmed against the synthesized materials, whereas the structures of other metabolites were proposed based on the high-resolution MS/MS fragmentation pattern analysis. The measured accurate masses and characteristic fragment ions are listed in Table 5.
Molecular ions and characteristic fragment ions of ombitasvir and metabolites in human plasma, urine, or feces
Ombitasvir yielded a protonated molecular ion at m/z 894.5113 (calculated mass m/z 894.5124, chemical formula C50H68N7O8+) in the positive-ion mode. The key MS/MS fragment ions were m/z 737.4377, 640.3844, 588.3170, 547.3270, 431.2433, and 334.1914 (Table 5). The collision-induced dissociation spectrum and detailed assignment of the fragments are provided in the Supplemental Materials.
Metabolite M23.
M23 was detected in human plasma with a protonated molecular ion at m/z 386.2587 (calculated mass m/z 386.2591, C26H32N3+). The characteristic MS/MS fragments of M23 include m/z 369.1820 (−NH3), 293.2005 (loss of aniline), 237.1379 (loss of tert-butyl aniline), and 144.0802 (loss of aniline and tert-butyl aniline). Metabolite M23 was further confirmed by comparing the MS/MS fragmentation pattern and HPLC coinjection analysis using the synthetic reference standard.
Metabolite M29.
M29 was detected in human plasma by LC-MS with a protonated molecular ion at m/z 372.2068, indicating a loss of two methyl groups and two hydrogens, plus the addition of one oxygen atom to M23. The predicted molecular formula was C24H26N3O+ (calculated mass m/z 372.2070). The characteristic fragment ions of M29 are m/z 355.2841 (loss of NH3), 279.1485 (loss of aniline), and common fragment ions at m/z 237.1379 and 144.0802 as in M23. M29 was assigned as 1-(4-((2S,5S)-2,5-bis(4-aminophenyl)pyrrolidin-1-yl)phenyl)ethanone, and was further confirmed by comparing the MS/MS fragmentation pattern and HPLC coinjection analysis using the synthetic reference standard.
Metabolite M36.
M36 was detected in human plasma by LC-MS with a protonated molecular ion at m/z 388.2015. The measured accurate mass data suggested the molecular formula of C24H26N3O2+ (calculated mass m/z 388.202). The major fragment ions of M36 included m/z 371.2258 (loss of NH3), 295.1435 (loss of aniline), and common fragment ions at m/z 237.1380 and 144.0802 as in M23. M36 was assigned as a hydroxylated metabolite of M29; hydroxylation occurred at the 1-(4-aminophenyl)ethanone moiety. The structure of M36 was further confirmed by comparing the MS/MS fragmentation pattern and HPLC coinjection analysis using the synthetic reference standard.
Metabolite M37.
M37 was detected in human plasma by LC-MS with a protonated ion at m/z 390.2171, suggesting a molecular formula of C24H28N3O2+ (calculated mass: 390.2176). The collision-induced dissociation of M37 produced major fragment ions at m/z 297.1611 (loss of aniline), m/z 237.1380, and 144.0803. M37 is dihydroxylated 4-ethylphenyl-pyrrolidine-2,5-diyl-dianiline. The structure of M37 was further confirmed by comparing the MS/MS fragmentation pattern and HPLC coinjection analysis using the synthetic reference standard.
Metabolite M25.
M25 was detected at low levels in plasma, and yielded a protonated molecular ion at m/z 418.2490. The predicted molecular formula was C26H32N3O2+ with calculated mass of m/z 418.2489. The collision-induced dissociation of M25 produced major fragment ions at m/z 237.1378 and 144.0804. M25 was tentatively assigned as tert-butyl dihydroxyl metabolite of M23.
Metabolite M26.
M26 was detected at low levels in plasma and yielded a protonated molecular ion at m/z 402.2538 in LC-MS, indicating the addition of 16amu (+O) to M23. The predicted molecular formula is C26H32N3O+ with calculated mass of m/z 402.2540. The collision-induced dissociation of M26 produced major fragment ion at m/z 309.1947, 237.1377, and 144.0803. The presence of m/z 237.1379 indicated the hydroxylation occurred at the tert-butylaniline moiety. Therefore, M26 was assigned as a tert-butyl hydroxyl metabolite of M23.
Metabolite M28.
M28 was observed as a trace metabolite in plasma; it gave a protonated molecular ion at m/z 416.2338. The predicted molecular formula is C26H30N3O2+ (calculated mass: 416.2333). The collision-induced dissociation of M28 produced major fragment ions at m/z 237.1382 and 144.0805. Metabolite M28 was tentatively assigned as the tert-butyl carboxylic acid metabolite of M23.
Metabolite M34.
M34 was detected as a trace metabolite in plasma; it gave a protonated molecular ion at m/z 388.2383, suggesting a molecular formula of C25H30N3O+ (calculated mass: 388.2383). The collision-induced dissociation of M34 generated major fragment ions at m/z 371.2265 (loss of NH3), 237.1379, and 144.0801. M34 was tentatively assigned as the tert-butyl demethylation and hydroxylation metabolite of M23.
Metabolite M35.
M35 was detected as a trace metabolite in plasma; it produced a protonated molecular ion at m/z 404.2330, suggesting a molecular formula of C25H30N3O2+ (calculated mass: 404.2333). The collision-induced dissociation of M35 generated major fragment ions at m/z 237.1382 and 144.0800. M35 was tentatively assigned as the tert-butyl demethylation and dihydroxylation metabolite of M23.
Metabolite M5.
M5 was present at a low level in both plasma and feces; it gave a protonated molecular ion at m/z 910.5056, suggesting an addition of oxygen to the parent drug. Due to low ion intensity, no MS/MS spectrum was obtained. M5 was tentatively assigned as a hydroxylation metabolite of parent drug.
Metabolite M6.
Metabolite M6 was present at a trace level in both plasma and feces, and gave a protonated molecular ion at m/z 640.3856, suggesting a molecular formula of C38H50N5O4+ (calculated mass: 640.3857). The collision-induced dissociation of M6 generated major fragment ions at m/z 547.3279 (loss of aniline), 491.2649 (loss of tert-butyl aniline), 334.1912 (further cleavage of aniline amide from 491), and 255.1336 (aniline amide cleavage). M6 was assigned as a monohydrolysis product of the parent drug.
Metabolite M7.
M7 was only detected by LC-MS and gave a protonated molecular ion at m/z 273.1441, suggesting a molecular formula of C12H21N2O5+ (calculated mass: 273.1445). The collision-induced dissociation of M7 generated major fragment ions at m/z 255.1337, 227.1380, and 116.0703. M7 was confirmed by coinjection HPLC-MS analysis using a reference standard of (S)-1-((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid.
Metabolite M9.
M9 was a trace metabolite present in both plasma and feces; it gave a protonated molecular ion at m/z 912.5224, suggesting the addition of water to the parent drug, with a predicted molecular formula of C50H70N7O9+ (calculated mass: 912.5230). The collision-induced dissociation of M9 generated major fragment ions at m/z 755.4494, 738.4403, 640.3842, 547.3271, 491.2643, and 334.1904. M9 was tentatively assigned as a hydration metabolite of the parent drug.
Trace levels of M2 (measured m/z 910.5075) and M3 (measured m/z 910.5075) were also detected in fecal samples. Due to their overall low abundance in feces (each <1% of dose; Table 4), no further characterization was performed. M2 and M3 were assigned as hydroxylated metabolites of the parent drug.
Quantification of Ombitasvir Metabolites following Multiple Oral Dosing in Human
As ombitasvir is not intended for use as a single agent, the metabolic profile of ombitasvir in human plasma was further investigated at steady state, following administration of 3DAA combination. Plasma samples obtained from 12 healthy subjects, following 14 days of dosing with ombitasvir (25 mg, QD), administered in combination with paritaprevir/ritonavir (150/100 mg, QD) and dasabuvir (400 mg, twice a day), were pooled. Concentrations of M23, M29, M36, and M37 were measured against synthetic reference standards. The measured concentrations and AUCs of these metabolites are listed in Table 6. Unchanged parent drug was the major component in plasma, with a Cmax of 125 ng/ml and an AUC0-24h of 1745 ng⋅h/ml. Two downstream metabolites, M29 and M36, provided Cmax values more than 4-fold lower (31.4 and 23.0 ng/ml, respectively), with correspondingly lower AUC values (669 and 442 ng⋅h/ml, respectively). Plasma concentrations of M37 (Cmax 16.1 ng/ml; AUC 312 ng⋅h/ml) and M23 (Cmax 8.6 ng/ml; AUC 194 ng⋅h/ml) were even lower. Parent drug accounted for 51.9% of the drug-related material, followed by M29 (19.9%), M36 (13.1%), M37 (9.3%), and M23 (5.8%).The same set of plasma samples was also pooled using the Hamilton method, extracted, and analyzed using a high-resolution mass spectrometer for metabolite profiling (Fig. 7). Qualitatively similar metabolites were detected in the plasma following multiple doses of ombitasvir with paritaprevir/ritonavir and dasabuvir, and no new metabolites were identified.
Estimated relative amounts (%AUCt) of ombitasvir and its metabolites in human plasma following multiple doses of ombitasvir

HPLC-MS–extracted ion chromatogram of metabolites generated from pooled human plasma following oral administration of ombitasvir (25 mg, once daily), paritaprevir/ritonavir (150/100 mg, once daily), and dasabuvir (400 mg, twice daily) for 14 days. RT, retention time; SM, smoothing function using 3-point boxcar function.
Metabolism of M23 by Recombinant CYP2C8
Since the enzymatic amide hydrolysis product M23 is a precursor to M29 and M36, the metabolic pathway of M23 was characterized using an in vitro hepatic system. In vitro cytochrome P450 reaction phenotyping indicated that CYP2C8 is the primary enzyme to metabolize M23. Figure 8 showed the extracted ion chromatogram of downstream metabolites of M23 following in vitro incubation of M23 in recombinant CYP2C8 enzyme. At least 11 downstream metabolites of M23 were identified. M28 (tert-butyl acid), M25 (tert-butyl dihydroxyl), and M29 are the most abundant components in HPLC-MS analysis. The proposed metabolic pathway of M23 by CYP2C8 was illustrated in Fig. 9. M23 undergoes a tert-butyl hydroxylation (Rodrigues et al., 1995; Weber et al., 1999; Polsky-Fisher et al., 2006; Prakash et al., 2008) to form M26, followed by further oxidation to generate oxidative and/or desmethyl metabolites, such as M25, M29, M34, and M35. The uncommon metabolic pathway involving oxidation at the tert-butyl group followed by C-demethylation has been reported previously (Prakash et al., 2008; Yoo et al., 2008). Although the exact chemical mechanism for the carbon-cleavage reaction from M23 to M29, M36, or other demethylated metabolites is yet to be further characterized, it is likely that it involves a similar reaction mechanism postulated by Prakash et al. (2008), who showed that C-demethylation may involve the oxidation of the tert-butyl alcohol to form an aldehyde metabolite, followed by P450-mediated deformylation to lose formic acid, to produce an unstable carbon-centered radical which reacts with water to form downstream demethylated metabolites.

HPLC-MS–extracted ion chromatogram of metabolites generated from in vitro incubation of M23 in human recombinant CYP2C8 enzymes. RT, retention time.

Proposed metabolic pathways of M23 mediated by human CYP2C8.
Discussion
The mass balance, disposition, and metabolism of ombitasvir were evaluated in four healthy human subjects. Following the administration of a single 25-mg oral dose of [14C]ombitasvir, the mean total recovery of the administered radioactive dose was 92.2% (± 0.82%, S.D.), with recovery in individual subjects ranging from 91.4 to 93.1%. Nearly all of the administered radioactive dose (90.2% of dose) was recovered in feces, whereas a very limited amount of radioactivity (1.9%) was recovered in urine through the last collection interval, indicating that ombitasvir and metabolites were predominantly eliminated in humans through feces and minimally through renal clearance.
Metabolites of ombitasvir in plasma, urine, and feces were profiled using HPLC-radioactivity detection, and structures of metabolites were characterized using HPLC–high-resolution mass spectrometry. Of the total radioactivity excreted in human feces, unchanged parent drug constituted 87.8% of the total dose, and metabolites M2, M3, M5, M6, and M9 accounted only for <1% of the total dose each. In urine, only trace levels of parent drug and several small polar components (Mu1–Mu5) were present. In human plasma, [14C]ombitasvir, M23, M29, M36, and M37 were the main components after a single 25-mg dose of [14C]ombitasvir alone, representing about 93% of total plasma radioactivity, with at least nine additional metabolites (M5, M6, M7, M9, M25, M26, M28, M34, and M35) observed at either minor or trace levels. Similar to preclinical toxicology species, biotransformation of ombitasvir in humans primarily involves enzymatic amide hydrolysis at the aniline amide linker to generate metabolites M6 (monoaniline), M7 (pyrrolidine acid), and M23. It is not clear what enzymes are involved in the amide hydrolysis or where the process occurs. Only trace levels of metabolites M6 and M7 were produced in in vitro hepatocytes or liver microsomes across species (Abbvie, unpublished data). In humans, M23 further undergoes CYP2C8-mediated oxidative metabolism at the tert-butyl group to generate oxidative and/or C-desmethyl metabolites, such as M26 (hydroxy tert-butyl dianiline), M25 (dihydroxy tert-butyl dianiline), M34 (tert-butyl desmethyl hydroxy dianiline), M35 (tert-butyl desmethyl dihydroxy dianiline), M36 (hydroxyacetophenone dianiline), M37 (tert-butyl desmethyl dihydroxy dianiline), and M29 (acetophenone dianiline).
Based on the assessment of metabolite exposures at steady state, M29 and M36 were defined as major circulating metabolites, representing 19.9 and 13.1%, respectively, of the total drug-related material in plasma, whereas the parent drug accounted for 51.9% of the total drug-related material. M29 and M36 are downstream metabolites of M23, which is present in preclinical species at higher levels than in humans, providing safety coverage in all toxicology species. M29 and M36 have not been observed in studies using animal- and human-derived hepatic in vitro systems, in plasma, or excreta of in vivo preclinical animals used in absorption, distribution, metabolism, and excretion or toxicology studies. These two metabolites were further characterized independently (i.e., not combined) and proved to be negative in in vitro Ames tests and a chromosomal aberration assay. There were no adverse findings in independent 4-week repeat-dose and embryo-fetal developmental toxicity studies in mice with M29 and M36.
Whereas the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use M3 R2 (http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm) and Food and Drug Administration Guidance on Safety Testing of Metabolites (http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm) focus on the relative abundance of metabolites, there has been considerable emphasis on the fact that absolute exposures (as circulating concentrations or total body burden) of metabolites need to be taken into consideration, especially for drugs at low doses (Smith et al., 2009). These disproportionate metabolites, M29 and M36, are present at low nanomolar plasma concentrations (average concentration 17–31 ng/ml) in humans receiving a 25-mg dose of ombitasvir as a part of the 3DAA treatment regimen. They are highly bound to plasma proteins, not active against HCV replicons in vitro, and are not expected to have clinically relevant on-target or off-target pharmacologic activity.
Clinical drug-drug interactions of the complete 3DAA regimen including ombitasvir have been extensively characterized and summarized elsewhere (Menon et al., 2015). The disproportional metabolites M29 and M36 were de facto tested as part of ombitasvir administration. No safety findings were attributed to ombitasvir and its metabolites. Detailed in vitro studies to profile cytochrome P450 enzymes and drug transporters for ombitasvir and other DAA components have been conducted (M. Shebley, D. Bow, J. Liu, O. Kavetskaia, J. Sydor, S. M. de Morais, V. Fischer, and M. Nijsen, manuscript in preparation), and physiologically based pharmacokinetic models were established to provide a mechanistic understanding of potential drug-drug interactions. In summary, the overall disposition and metabolism of 25-mg [14C]ombitasvir in healthy volunteers was investigated. The overall study objectives were met, with good recovery of radioactivity dose from all subjects. The mass balance results confirm that orally administered ombitasvir is primarily eliminated by the biliary-fecal route. The metabolite structures of the main circulating metabolites were elucidated, with a proposed metabolic pathway of enzymatic amide hydrolysis of ombitasvir followed by tert-butyl hydroxylation of M23 to generate secondary oxidative or C-demethylation metabolites.
Acknowledgments
Special thanks to Anthony R. Haight, Benoit Cardinal-David, Shashank Shekhar, and Brian Kotecki for preparation of M29 and M36 reference materials.
Authorship Contributions
Participated in research design: Shen, Menon, Kavetskaia, Fischer.
Conducted experiments: Serby, Ma.
Contributed new reagents or analytic tools: Serby, Surber.
Performed data analysis: Shen, Serby, Ma, Badri, Menon.
Wrote or contributed to the writing of the manuscript: Shen, Serby, Surber, Lee, Ma, Badri, Menon, Kavetskaia, de Morais, Sydor, Fischer.
Footnotes
- Received October 1, 2015.
- Accepted May 11, 2016.
↵1 Current affiliation: Global Clinical Pharmacology, Pfizer, Groton, Connecticut.
Disclosure Statement: The design, study conduct, and financial support for this study were provided by AbbVie. AbbVie participated in the interpretation of data, writing, review, and approval of the publication. All authors are current employees of AbbVie, except Olga Kavetskaia, who was an AbbVie employee at the time the manuscript was developed.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ABT-267
- ombitasvir
- AUC
- area under the curve
- Cmax
- maximum plasma concentration
- DAA
- direct-acting antiviral agent
- HCV
- hepatitis C virus
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- LSC
- liquid scintillation counting
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- Mu
- unknown metabolites in urine
- NS
- nonstructural protein
- QD
- once daily
- SPE
- solid-phase extraction
- t1/2
- half-life
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}