


Abstract
Dasabuvir [also known as ABT-333 or N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide] is a potent non-nucleoside NS protein 5B polymerase inhibitor of the hepatitis C virus (HCV) and is being developed in combination with paritaprevir/ritonavir and ombitasvir in an oral regimen with three direct-acting antivirals for the treatment of patients infected with HCV genotype 1. This article describes the mass balance, metabolism, and disposition of dasabuvir in humans. After administration of a single oral dose of 400-mg [14C]dasabuvir (without coadministration of paritaprevir/ritonavir and ombitasvir) to four healthy male volunteers, the mean total percentage of the administered radioactive dose recovered was 96.6%. The recovery from the individual subjects ranged from 90.8% to 103%. Dasabuvir and corresponding metabolites were predominantly eliminated in feces (94.4% of the dose) and minimally through renal excretion (2.2% of the dose). The biotransformation of dasabuvir primarily involves hydroxylation of the tert-butyl group to form active metabolite M1 [N-(6-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(1-hydroxy-2-methylpropan-2-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide], followed by glucuronidation and sulfation of M1 and subsequent secondary oxidation. Dasabuvir was the major circulating component (58% of total radioactivity) in plasma, followed by metabolite M1 (21%). Other minor metabolites represented < 10% each of total circulating radioactivity. Dasabuvir was cleared mainly through cytochrome P450–mediated oxidation metabolism to M1. M1 and its glucuronide and sulfate conjugates were primarily eliminated in feces. Subsequent oxidation of M1 to the tert-butyl acid, followed by formation of the corresponding glucuronide conjugate, plays a secondary role in elimination. Cytochrome P450 profiling indicated that dasabuvir was mainly metabolized by CYP2C8, followed by CYP3A4. In summary, the biotransformation pathway and clearance routes of dasabuvir were characterized, and the structures of metabolites in circulation and excreta were elucidated.
Introduction
The hepatitis C virus (HCV) affects approximately 150 million people worldwide (http://www.who.int/mediacentre/factsheets/fs164/en/). The virus, which replicates predominantly in the cytoplasm of hepatocytes, can lead to acute or chronic liver infections that introduce the likelihood of developing liver cirrhosis or cancer. Dasabuvir, also known as ABT-333 [N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide], is a nonstructural protein 5B (NS5B) inhibitor that has been developed for the HCV genotype 1 infection in combination with an NS3 protease inhibitor paritaprevir with ritonavir and/or an NS5A non-nucleoside polymerase inhibitor (ombitasvir) with or without ribavirin (Maring et al., 2009; Feld et al., 2014; Kowdley et al., 2014; Zeuzem et al., 2014). RNA polymerase (NS5B) is a key element in the replication of HCV utilizing an atypical ability of initiating RNA synthesis without using an RNA primer (Moradpour et al., 2007; Beaulieu, 2009; Rigat et al., 2010). Non-nucleoside inhibitors commonly interrupt the start of the RNA synthesis phase (Legrand-Abravanel et al., 2010). Dasabuvir exhibits potent inhibition against genotype 1a and 1b HCV polymerases (IC50 = 2.2–10.7 nM in a biochemical enzymatic assay) and against genotype 1a and 1b HCV replicons (EC50 = 7.7 nM and 1.8 nM, respectively) (Maring et al., 2009).
Clinically, dasabuvir has favorable safety, tolerability, and pharmacokinetic profiles as a monotherapy or combination therapy at doses administered to date. Minimal to no accumulation was observed from dasabuvir at doses ranging from 200 mg to 600 mg twice daily. A modest accumulation of 65% was observed after a 1000 mg twice-daily dose regimen of dasabuvir, and approximately 2-fold accumulation was observed when dasabuvir was dosed above 1000 mg twice daily. The absolute bioavailability of the dasabuvir 400-mg tablet was 46% compared with an intravenous microdose of approximately 85 μg [14C]dasabuvir administered at the same time as the oral dose. According to dasabuvir phase 1 studies, when the nonradiolabeled dasabuvir was administered as a single agent, the dasabuvir M1 metabolite-to-parent ratio was around 0.35.
This report describes the absorption, metabolism, and excretion of a single 400-mg oral dose of [14C]dasabuvir in four healthy human subjects. The purposes of the study are to assess the mass balance, elucidate the routes and rates of excretion, identify and quantify the exposure of circulating metabolites in human plasma, elucidate the metabolite structures, determine the metabolite profiles in excreta, and understand the metabolic pathway of dasabuvir in humans. In addition, in vitro characterization of the major drug metabolism enzymes that are responsible for the metabolism of dasabuvir is also described.
Materials and Methods
Drugs and Reagents
Dasabuvir, its tert-butyl hydroxyl metabolite (M1) and tert-butyl carboxylate metabolite (M5), [14C]dasabuvir, and [3H]dasabuvir were supplied by Process Chemistry, AbbVie Inc. (North Chicago, IL). The structures of dasabuvir and its metabolite standards are shown in Fig. 1. The radiochemical synthesis of [14C]dasabuvir was conducted in two steps using (2-[14C]) uracil as the radiolabeled starting material. Purification of the compound by crystallization provided > 99% radiochemical purity by high-performance liquid chromatography (HPLC). [3H]Dasabuvir was prepared via reduction of 5-bromouracil dasabuvir with tritium for in vitro assays. The radiochemical purity after HPLC purification was > 99%. Metabolite standards were as follows: N-(6-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(1-hydroxy-2-methylpropan-2-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (M1) and 2-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxy-3-(6-(methylsulfonamido)naphthalen-2-yl)phenyl)-2-methylpropanoic acid (M5). These reference standards were used as HPLC and mass spectrometric standards.

Structure of [14C]dasabuvir and synthetic metabolites.
Clinical Study
The clinical study was conducted at Covance Laboratories Inc., in conjunction with the Covance Clinical Research Unit (Madison, WI). In this open-label study, a total of four adult male subjects in general good health were selected to participate in the study, according to the selection criteria. On the morning of study day 1, subjects received a single oral dose of [14C]dasabuvir under nonfasting conditions. The study drug, [14C]dasabuvir (400 mg active, 100 μCi [14C]), was administered as a liquid suspension. Subjects were confined to the study site for a minimum of 120 hours postdose or up to a maximum of 312 hours postdose. Subjects were released from the study site at any time after 120 hours postdose if the preset release criteria were met.
Blood samples were collected by venipuncture into vacutainer collection tubes containing potassium EDTA at the following times: 0 (predose), 1, 2, 4, 6, 8, 10, 12, 24, 48, 72, 96, 120, 144, 168, 192, 216, and 240 hours after dosing of [14C]dasabuvir on day 1 of the study. Plasma was separated via centrifugation and stored at –70°C.
Urine samples were collected over the following intervals: 0–12, 12–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, and 216–240 hours after dosing of [14C]dasabuvir on study day 1. Urine samples were spiked with dodecylbenzenesulfonic acid sodium salt at a concentration of 0.6 mg/ml prior to sample aliquoting. Aliquots of urine were frozen and maintained at –20°C prior to metabolite profiling.
Fecal samples were collected predose (upon check in before dosing) and over the following intervals after dosing: 0–24, 24–48, 48–72, 72–96, 96–120, 120–144, 144–168, 168–192, 192–216, and 216–240 hours. All feces collected during a collection interval were kept frozen at –20°C prior to metabolite profiling.
Total Radioactivity Measurement by Liquid Scintillation Counting
All sample combustion was performed using a Model 307 Sample Oxidizer (Packard Instrument Company, Meriden, CT) and the resulting 14CO2 was trapped in a mixture of Perma Fluor (Perkin Elmer, Waltham, MA) and Carbo Sorb (Perkin Elmer). The efficiency of the oxidizer was evaluated each day of sample combustion by analyzing a commercial radiolabeled standard both directly in the scintillation cocktail and by oxidation. Acceptance criteria were defined as combustion recoveries of 95%–105%. Ultima Gold XR scintillation cocktail was used for samples analyzed directly. All samples were analyzed for radioactivity in Model 2900TR liquid scintillation counters (Packard Instrument Company) for at least 5 minutes or 100,000 counts. Each sample was homogenized and an aliquot was mixed with scintillation cocktail before radioanalysis. All samples were analyzed in duplicate if the sample size allowed unless the entire sample was used for analysis. If results from sample replicates (calculated as 14C dpm/g sample) differed by > 10% from the mean value and sample aliquots had radioactivity > 200 dpm, the sample was rehomogenized and reanalyzed.
After mixing, duplicate blood samples were weighed (approximately 0.2 g), combusted, and analyzed by liquid scintillation counting (LSC). The representative lower limit of quantitation for blood was 195 ngEq/g. Plasma samples were mixed and duplicate weighed aliquots (approximately 0.2 g) were analyzed directly by LSC. The representative lower limit of quantitation for plasma was 173 ngEq/g. The urine samples were mixed and duplicate weighed aliquots (approximately 0.2 g) were analyzed directly by LSC. The representative lower limit of quantitation for urine was 160 ngEq/g. Fecal samples were combined by subject at 24-hour intervals and the weight of each combined sample was recorded. A weighed amount of water was added and the sample was mixed. The sample was removed from the freezer and homogenized, or it was immediately homogenized using a probe-type homogenizer. Duplicate weighed aliquots (approximately 0.2 g) were combusted and analyzed by LSC.
Sample Preparation for Metabolite Profiling
Plasma samples were thawed at room temperature and pooled across subjects at selected time points in addition to area under the curve (AUC) plasma pooling utilizing the Hamilton method (Hamilton et al., 1981) for each subject. The pooled plasma was processed using the following protein precipitation method. In brief, a 6-fold volume of acetonitrile/methanol [3:1 (v/v)] was added to each sample, followed by vortexing and 5-minute sonication. The sample was then centrifuged at 3500 rpm (2465g) at 4°C. The supernatant was transferred a glass tube. The protein pellets were further washed with 3–5 ml acetonitrile-methanol [4:1 (v/v)], followed by centrifugation at 3500 rpm (2465g). After combining the supernatants, 50 µl dimethylsulfoxide (DMSO) was added and the solution was concentrated to approximately 50–100 µl volume under a stream of nitrogen. The remaining material was diluted with 150 µl 0.1% formic acid in water. An aliquot of the reconstituted sample was subjected to LSC counting to determine total radioactivity recovery. Another aliquot of the reconstituted sample was transferred to an HPLC autosampler vial and 75 µl of the reconstituted sample was injected for liquid chromatography (LC)/mass spectrometry (MS) and radiochromatographic analysis.
Urine samples were pooled at selected time points across subjects. Pooled samples were processed using solid-phase extraction (SPE) (Agilent Accu-Bond II Octyl SPE cartridge, 1000 mg/6 ml, PN188-0360; Agilent Technologies, Santa Clara, CA). In brief, the SPE cartridges were conditioned with acetonitrile (2× bed volume), methanol (2×), and 0.1% formic acid/water (4×) before use. Aliquots of urine were loaded to preconditioned columns, washed with water (4× bed volume), and eluted with 3 × 4 ml acetonitrile/methanol [3:1 (v/v)]. The eluents were combined, followed by the addition of 100 µl DMSO. The combined eluents were then concentrated under a stream of nitrogen to about 100–200 µl, followed by dilution of the sample with 250 µl 0.1% aqueous formic acid before LC-MS analysis.
Fecal homogenate samples were pooled at each time point across subjects before processing. The fecal samples were processed using multiple solvent extractions with acetonitrile-methanol [3:1 (v/v)] using a 1:3 sample/solvent ratio, followed by centrifugation at 4000 rpm for 20 minutes at 4°C. The extraction was repeated until either 80% of the radioactivity had been recovered or < 1% of the radioactivity was extracted. Aliquots of extracted samples were subjected to LSC for total radioactivity. A 50-µl aliquot of DMSO was added to the combined supernatants before concentrating the sample under a nitrogen flow at room temperature. The final residues were reconstituted with 0.1% formic acid/acetonitrile (1:1) before HPLC/MS/radio analysis.
Method for Metabolite Profiles and Identification
HPLC separation of dasabuvir and the corresponding metabolites was conducted using a Thermo Accela HPLC system (Thermo Fisher Scientific, San Jose, CA), which consisted of a Thermo Accela autosampler, a 1250 series binary pump, and an Accela PDA detector. Separation was achieved on an Agilent Eclipse XDB-C18 (3.5 µm, 4.6 × 150 mm column; Agilent Technologies). The HPLC mobile phase consisted of 0.1% formic acid in water (solvent A) and 100% acetonitrile (solvent B). The HPLC flow rate was 1.0 ml/min. The gradient was as follows: 0–3 minutes: 20% B; 3–10 minutes: 20%–40% B; 10–50 minutes: 40%–63% B; 50–52 minutes: 63%–95% B; 52–58 minutes: 95% B; 58–59 minutes: 95% to 20% B; and 59–65 minutes: 20% B. The HPLC system was interfaced with a Thermo Fisher Orbitrap Discovery mass spectrometer (Thermo Fisher Scientific). The mass spectrometric analyses were conducted using electrospray ionization operating in negative ionization mode. The MS settings were as follows: electrospray ionization voltage, −2.6 kV; capillary temperature, 275°C; capillary voltage, 35 V; and tube lens, 110 V. The unchanged parent drug and its metabolites were detected using data-dependent multiple-stage mass analysis with mass isolation of 2 Da, with normalized collision energy of 35% for both the MS2 and MS3 scans. The mass resolution was set at 30,000 for the full scan and 7500 for the MS2 and MS3 scans. Accurate mass measurements were obtained after performing a daily external calibration. Data acquisition and processing were carried out using Xcalibur 2.2 software (Thermo Fisher Scientific).
Radiolabeled components in plasma, urine, or feces samples were detected by the PerkinElmer TopCount 96-Deep-Well Luma Plate (Perkin Elmer, Waltham, MA). The HPLC eluent was split postcolumn between the mass spectrometer and the Agilent 1100 fraction collector at a ratio of 20:80. The Agilent 1100 fraction collector was set to collect fractions at intervals of 0.3 minutes per well. Radioactivity counting was conducted using a PerkinElmer TopCount NXT system.
Quantitation of Dasabuvir and M1 in Plasma
Plasma concentrations of dasabuvir and metabolite M1 were determined using a validated LC–tandem mass spectrometry (MS/MS) bioanalytical method. A common protein precipitation procedure, followed by online SPE using a small aliquot of plasma combined with an aliquot of stable isotope–labeled internal standard solution in acetonitrile, was used in all validated methods. After centrifugation, an aliquot of the supernatant was injected onto the online SPE LC-MS/MS system. The chromatographic separation of dasabuvir, M1, and internal standards from the coextracted matrix components was achieved with a reverse-phase analytical column. Analysis was performed on an AB Sciex triple quadrupole mass spectrometer with a TurboIonSpray interface (AB Sciex, Framingham, MA). Detection was performed in the multiple reaction monitoring mode at mass-to-charge ratios (m/z) of 494.2 → 359.2 for dasabuvir, 498.2 → 363.2 for 13CD3-dasabuvir (the internal standard of dasabuvir), 510.4 → 412.4 for M1, and 516.4 → 418.4 for the stable isotope–labeled internal standard of M1.
Pharmacokinetic Calculations
Plasma concentration-time radioactivity data were analyzed with SAS software (version 9.2; SAS Institute Inc., Cary, NC). Maximum plasma concentration (Cmax), time at which Cmax was achieved (Tmax), area under the concentration time curve from time zero to the last measurable time point (AUC0–t) for total radioactivity, [14C]dasabuvir, and its metabolites in plasma were estimated. The area under the concentration time curve from time zero to infinity (AUC0–∞) and half-life for total radioactivity, [14C]dasabuvir, and the M1 metabolite in plasma were also calculated.
In Vitro Metabolism and Cytochrome P450 Phenotyping
Studies to identify the cytochrome P450 (P450) isoform(s) responsible for formation of M1 were conducted by incubation of the substrate in the presence of recombinant P450 isoforms. Chemical inhibition was studied using selected inhibitors in the human liver microsomal preparations. Studies with recombinant human P450 isoforms were conducted using P450 isoforms CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 and flavin-containing monooxygenase (FMO) isoforms FMO1, FMO3, and FMO5, all of which were obtained from BD Bioscience (San Jose, CA). Standard 200-µl incubations were performed in duplicate with NADPH (1 mM), 0.25 µM [3H]dasabuvir, 50 mM phosphate buffer (pH 7.4), and recombinant human P450 (20 pmol). Reactions were initiated with the addition of an NADPH solution to the incubation mixture and the incubation was conducted at 37°C. At 0 and 60 minutes, incubations were quenched with a 2× volume of the acetonitrile/methanol mixture [1:1 (v/v)]. Quenched samples were centrifuged at 1900g for 5 minutes at 4°C. An aliquot of the supernatant was analyzed using HPLC with radiochemical detection.
Chemical inhibition studies were conducted using human liver microsomes with the following inhibitors of specific P450 isoforms: ketoconazole (CYP3A4/5), quercetin (CYP2C8), quinidine (CYP2D6), and 2-phenyl-2-(1-piperidinyl)propane (CYP2B6). Inhibitor concentrations were selected to bracket the inhibitor Ki. The inhibitor Ki used was 0.0037 µM, 0.027 µM, 1.1 µM, and 5.6 µM for ketoconazole, quinidine, quercetin, and 2-phenyl-2-(1-piperidinyl)propane, respectively. A standard 0.2-ml incubation contained 0.5 mg/ml human liver microsomes (In Vitro Technologies, Noble Park North, VIC, Australia), 0.25 µM [3H]dasabuvir, and selected concentrations of chemical inhibitors [ketoconazole, 0.03–1 µM; quinidine, 0.01–2 µM; quercetin, 0.5–45 µM, and 2-phenyl-2-(1-piperidinyl)propane, 10–150 µM] in 50 mM phosphate buffer (pH 7.4). After a 5-minute preincubation at 37°C, the reaction was initiated with the addition of NADPH for a final concentration of 1 mM and incubated in a water bath at 37°C. After 0 and 60 minutes, the incubations were quenched with an equal volume of acetonitrile/methanol [1:1 (v/v)]. The samples were then centrifuged at 3500g for 30 minutes at 4°C. The supernatants were analyzed by HPLC with radiochemical detection.
Results
Excretion of Radioactivity
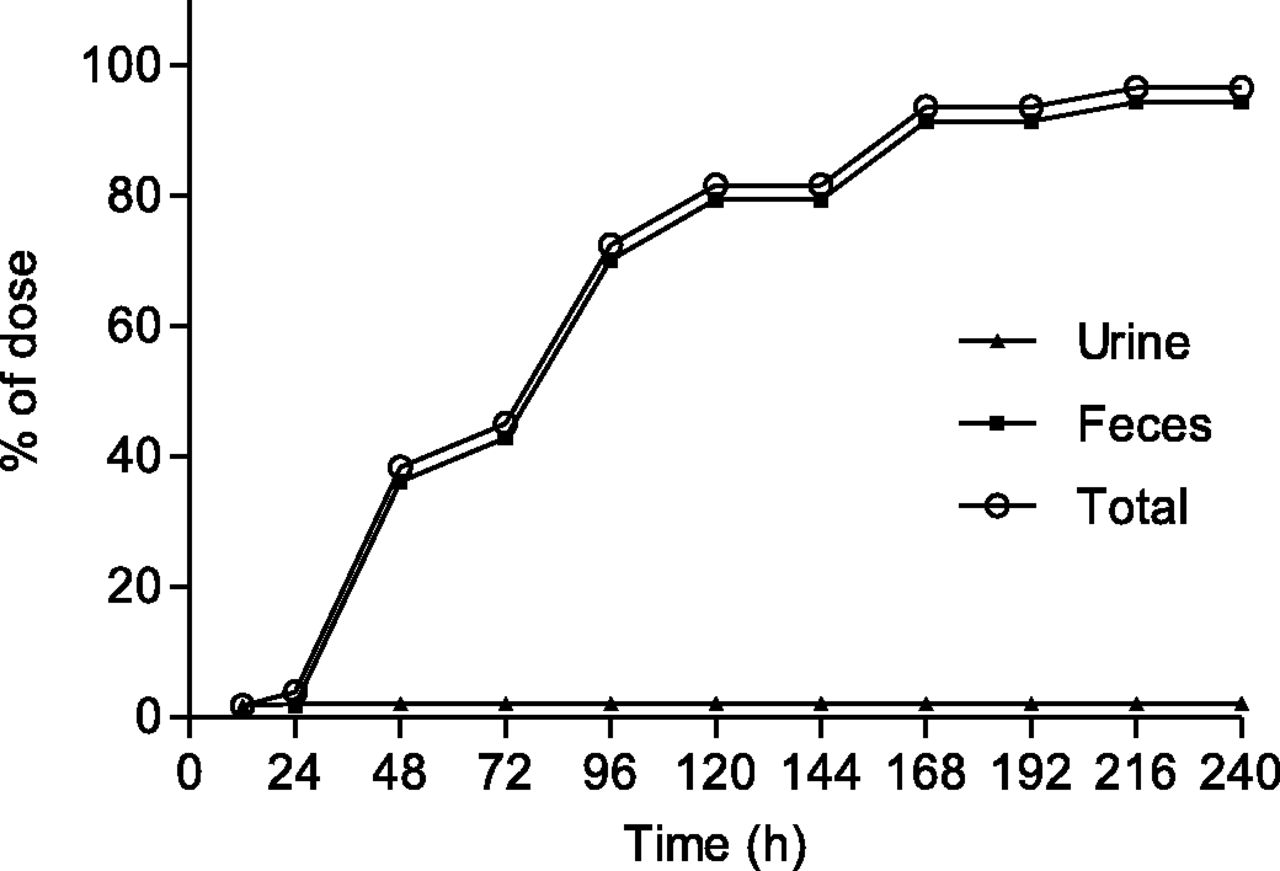
After a single oral dose of [14C]dasabuvir (400 mg, 100 µCi) to four healthy, male volunteers, the excretion of radioactivity in urine and feces from all of the subjects was measured over a period of up to 240 hours postdose. Figure 2 presents the mean cumulative recovery of total radioactivity in excreta expressed as the percentage of dose. The overall mean recovery of radioactivity in urine and feces samples was 96.6% ± 5.1% over the 240-hour collection period, with recovery in individual subjects ranging from 90.8% to 103%. The radioactivity was excreted primarily through fecal elimination (mean 94.4% of the dose). Renal excretion was relatively minor (mean 2.2% of the dose).

Mean cumulative percent of radioactive dose recovered in urine and feces at specified intervals after a single 400-mg (100 µCi) oral dose of [14C]dasabuvir to healthy male subjects.
Pharmacokinetic Data Analysis
The pharmacokinetic parameters for dasabuvir, M1, and total radioactivity are summarized in Table 1. The concentration of total radioactivity was measured by LSC, expressed as nanogram equivalents per gram. The concentrations of dasabuvir and metabolite M1 were determined using a validated LC-MS/MS bioanalytical method, expressed as nanograms per milliliter. The Tmax for dasabuvir, M1, and total radioactivity occurred approximately 3 to 4 hours after a single oral dose. Mean peak plasma concentrations (Cmax) for the parent drug, M1, and total radioactivity were 658 ng/ml, 267 ng/ml, and 1300 ngEq/g, respectively. The concentrations of dasabuvir and total radioactivity apparently declined in parallel after reaching the peak concentration (Fig. 3). The AUC0–last values for the parent drug, M1, and total radioactivity were 6260 ng⋅h/ml, 2230 ng⋅h/ml, and 8251 ngEq⋅h/g, respectively. The sum of dasabuvir and M1 exposures measured by the LC-MS bioanalytical method matches reasonably well with the total plasma radioactivity exposure, indicating that dasabuvir and M1 are the primary species in plasma.
Pharmacokinetic parameters of total radioactivity, dasabuvir, and metabolite M1
Data are presented as means ± S.D.

Mean (S.D.) plasma concentration-time curves for dasabuvir (in nanograms per milliliter) and total radioactivity (in nanogram equivalents per gram) in male subjects administered a single 400-mg oral dose of [14C]dasabuvir (N = 4).
Metabolite Profiles of [14C]Dasabuvir in Excreta and Circulation
Plasma.
A representative HPLC radiochromatogram of [14C]dasabuvir and its metabolites in pooled human plasma using the Hamilton method (t = 0–12 hours postdose) is shown in Fig. 4. The relative amounts of dasabuvir and metabolites in human plasma, expressed as the percentage of radioactivity in plasma, are summarized in Table 2. [14C]Dasabuvir is the predominant component in human plasma, representing 58.1% of total radioactivity in circulation. M1 is the most significant metabolite, accounting for 21.4% of radioactivity, followed by minor metabolites M2, M3, M4, M5, and M6.

Representative HPLC radiochromatograms of dasabuvir and its metabolites in human plasma after a single 400-mg oral dose of [14C]dasabuvir. CPM, counts per minute.
Percentages of radioactivity for dasabuvir and corresponding metabolites in human plasma (N = 4) after administration of a single 400-mg oral dose of [14C]dasabuvir
Data are presented as means ± S.D.
Urine and Feces.
The representative HPLC radiochromatogram of pooled human urine is shown in Fig. 5A. The unchanged parent drug and a total of nine metabolites, including M1–M7, M11, and U1, were detected in human urine. All of these metabolites were present at trace or low levels with respect to the administered dose; M1 was the most significant component in urine, accounting for 0.85% of the dose.

Representative HPLC radiochromatograms of dasabuvir and its metabolites in human excreta, urine (A) and feces (B), after a single 400-mg oral dose of [14C]dasabuvir. CPM, counts per minute; RT, retention time.
The representative HPLC radiochromatogram of pooled human feces is shown in Fig. 5B. M1 was the most abundant radiochemical component in feces, accounting for 31.5% of the total dose, followed by unchanged parent drug dasabuvir (26.2%), M2 (15.2%), and M5 (11.1%). Minor metabolites M8, M9, and M10 were also detected in feces, each representing < 5% of the dose. The mean quantification results for dasabuvir and corresponding metabolites in urine and feces, expressed as the percentages of the administered radioactive dose, are tabulated in Table 3. The proposed metabolic scheme for dasabuvir in humans is shown in Fig. 6.
Percentages of excretory metabolites of dasabuvir in humans (N = 4) after administration of a single 400-mg oral dose of [14C]dasabuvir

Proposed metabolic pathways of dasabuvir in humans.
LC-MS/MS Characterization of the Metabolites
As described in the Materials and Methods (see the section on method for metabolite profiles and identification), metabolites of dasabuvir were characterized using a combination of negative ionization high-resolution full-scan MS and product ion scan (MS/MS) analyses. The structures of metabolites M1 and M5 were confirmed against the synthesized materials, and the structures of other metabolites were proposed based on the high-resolution MS/MS fragmentation pattern analysis. Table 4 lists the approximate retention time and key mass spectral fragmentation of dasabuvir and metabolites. The collisional induced dissociation (CID) spectrum and detailed assignment of the fragments of dasabuvir and metabolites are provided in the Supplemental Material.
Retention time, molecular ions, and characteristic fragment ions of dasabuvir and metabolites in human plasma, urine, or feces
Dasabuvir yielded a deprotonated molecular ion at m/z 492.1601 (calculated mass m/z 492.1599) ([M-H]−) in negative ion mode. The CID of the m/z 492 ion gave the characteristic MS/MS fragment ions at m/z 477.1372 (base peak), 462.1137, 449.1543, 435.0897, 414.1825, and 399.1590 (Table 4). The fragment ions at m/z 477.1372 and 462.1137 were the results of loss of one and two methyl groups, respectively. The fragment ion at m/z 435.0897 was derived from a loss of a tert-butyl group. Minor fragment ions were also observed at m/z 414.1825 (loss of CH2SO2) and m/z 399.1590 (loss of CH3 and CH2SO2).
Metabolite M1.
Metabolite M1 gave a deprotonated molecular ion at m/z 508.1550, which is 16 atomic mass units (amu) higher than that of parent drug and is consistent with the chemical formula C26H26N3O6S− ([M-H]−) (calculated mass m/z 508.1548, parent + O). The CID of M1 produced major fragment ions including the base peak at m/z 493.1316 (loss of CH3) and other ions at m/z 476.1289 (loss of CH3OH), 463.1213 (loss of CH3 and CH2O), 435.0897 (loss of C4H9O), and 415.1543 (loss of CH3 and CH2SO2). The presence of an ion at m/z 435.0897 suggests that hydroxylation occurred at the tert-butyl moiety. Comparison of HPLC retention times using coinjection analysis and characteristic MS/MS fragmentation patterns of M1 with an authentic reference standard confirmed the M1 structure to be a tert-butyl hydroxylated form of dasabuvir.
Metabolite M2.
The deprotonated molecular ion of M2 was observed at m/z 588.1111, which is consistent with the proposed chemical formula C26H26N3O9S2− ([M-H]−) (calculated mass m/z 588.1116). The CID of M1 gave a major fragment ion at m/z 509.1267 (loss of CH3SO2 group). Other fragment ions included m/z 545.1053 (loss of NCO), 494.1024 (loss of CH3SO2 and CH3), 466.1202 (loss of NCO and CH3SO2), and 465.1490 (loss of NCO and SO3). The structure of M2 was proposed to be the sulfate conjugates of M1.
Metabolite M3.
The deprotonated molecular ion of M3 was observed at m/z 684.1861, which is 192 amu higher than that of the parent drug, and was consistent with the proposed chemical formula C32H34N3O12S− ([M-H]−) (calculated mass m/z 684.1869). The CID of M3 gave the main product ion at m/z 476.1284, a result of the loss of a glucuronide and CH3OH. Other product ions included m/z 641.1804 (loss of NCO), 597.1913 (loss of NCO and CO2), and 433.1223 (loss of C7H12O7 and NCO). M3 was proposed to be the glucuronide conjugate of M1. M3 was produced by both in vitro human and rat liver microsomal incubations of M1 in the presence of uridine diphosphoglucuronic acid and alamethicin (Fisher et al., 2000), confirming that M3 was formed through glucuronidation of M1. M3 isolated from rat bile was readily hydrolyzed by β-glucuronidase (Zenser et al., 1999), indicating that M3 is an O-glucuronide of M1 (data not shown).
Metabolite M4.
The deprotonated molecular ion of M4 was observed at m/z 506.1381, which is 14 amu higher than that of the parent drug and is consistent with the chemical formula C26H24N3O6S−([M-H]−) (calculated mass m/z 506.1391). The CID of M4 produced a major fragment ion at m/z 463.1208, a result of loss of CH3 and CO. The presence of the fragment ion at m/z 435.0901 (loss of C4H7O) also suggested oxidation occurred on tert-butyl group. M4 was proposed to be the tert-butyl aldehyde metabolite.
Metabolite M5.
The deprotonated molecular ion of M5 was observed at m/z 522.1337, which is 30 amu higher than that of parent drug and is consistent with the proposed chemical formula C26H24N3O7S− ([M-H]−) (calculated mass m/z 522.1340). The characteristic fragment ions included base peak m/z 478.1435 (loss of CO2) and 463.1212, which was a result of loss of a methyl group and CO2. The presence of ion at m/z 435.0895 (loss of C4H7O2) suggested that carboxylation occurred on the tert-butyl group. The structure of M5 was further confirmed with an authentic synthetic reference by comparison of LC retention times and characteristic MS/MS fragmentation patterns.
Metabolite M6.
The deprotonated molecular ion of M6 was m/z 698.1657, which is consistent with the chemical formula C32H32N3O13S− ([M-H]−) (calculated mass m/z 698.1661), a glucuronide conjugate of M5 (expected mass 698.1661, molecular formula C32H32N3O13S−). The CID of M6 produced the main product ion at m/z 478.1434, a result of loss of glucuronide and CO2, and other product ions at m/z 522.1334 (loss of C6H8O6), 463.1204, and 383.1267. M5 was also produced by both in vitro human and rat liver microsomal incubations of M5 in the presence of uridine diphosphoglucuronic acid and the pore-forming peptide alamethicin. Based on this information, M5 was identified as the glucuronide conjugate of M5.
Metabolite M7.
The deprotonated molecular ion of M7 was m/z 700.1801, which is consistent with the chemical formula of C32H34N3O13S− ([M-H]−) (calculated mass m/z 700.1818). The CID of ion at m/z 700 yielded the fragment ion of m/z 524.1493, indicating a loss of glucuronide. Further fragmentation of m/z 524 by MS3 produced a fragment of m/z 444.1568 (loss of CH4SO2). M7 is assigned as a dihydro metabolite of M6, with the addition of two protons at the pyrimidine-2,4(1H,3H)-dione moiety.
Metabolite M8.
Metabolite M8 was only observed in feces. The deprotonated molecular ion of M8 was m/z 524.1487, which is consistent with the chemical formula C26H26N3O7S− ([M-H]−) (calculated mass m/z 524.1497). The CID of M8 gave the main product ion at m/z 465.1367, which corresponds to the loss of CO2 and a methyl group. Other product ions included m/z 480.1601 (loss of CO2), 401.1745 (loss of CO2 and CH3SO2), and 387.1593 (loss of CO2, CH2SO2, and CH3). Based on these data, M8 was assigned as a dihydro metabolite M5, with the addition of two protons at the pyrimidine-2,4(1H,3H)-dione moiety.
Metabolite M9.
Metabolite M9 was only observed in feces. The deprotonated molecular ion of M9 was m/z 510.1694, which is consistent with the chemical formula C26H28N3O6S− ([M-H]−) (calculated mass m/z 510.1704). The CID of M9 yielded product ions at m/z 495.1474 (loss of CH3), 478.1445 (loss of CH3OH), 465.1366 (loss of C2H5O), and 437.1054 (loss of C4H9O). The presence of the ion at m/z 437.1054 suggested a loss of tert-butyl hydroxyl. M9 was assigned as a dihydro metabolite of M1, with the addition of two protons at the pyrimidine-2,4(1H,3H)-dione moiety.
Metabolite M10.
Metabolite M10 was only observed in feces. The deprotonated molecular ion of M10 was m/z 590.1260, which is consistent with the chemical formula C26H28N3O9S2− ([M-H]−) (calculated mass m/z 590.1272). Because of low ion intensity, the CID spectrum of M10 was not obtained. M10 was assigned as a dihydro metabolite of M2, possibly with the addition of two protons at the pyrimidine-2,4(1H,3H)-dione moiety.
Metabolite M11.
The deprotonated molecular ion of M11 was m/z 494.1380, which is consistent with the chemical formula C25H24N3O6S− ([M-H]−) (calculated mass m/z 494.1391) but is not consistent with the dihydro dasabuvir (C26H28N3O5S−, calculated mass m/z 494.1755). The observed mass has a –76 ppm mass error compared with the latter proposed structure. The CID of M11 gave product ions at 479.1155 (loss of CH3), 464.0918 (loss of two CH3), 421.0724 (loss of tert-butyl hydroxyl), 416.1612 (loss of SO2CH2), and 401.1380 (loss of SO2CH2 and CH3). M11 was assigned as a desmethyl metabolite of M1.
Unknown U1.
U1 eluted at the solvent front by HPLC and represented < 5% of the radioactivity in plasma and trace amount in urine. Two potential structures are proposed for the compound (Fig. 6), each of the structures formed from the cleavage of the pyrimidine-2,4(1H,3H)-dione ring. After utilizing multiple methods to identify the compound, a definitive m/z was not obtained.
Reaction Phenotyping of M1 Formation
Identification of enzymes involved in the metabolism of dasabuvir using recombinant human P450 enzymes and FMO enzymes in vitro suggests that it is predominantly metabolized by CYP2C8, followed by CYP3A4 and to a minor extent by CYP2B6 and CYP2D6. Figure 7 shows the representative HPLC radiochromagrams of [3H]dasabuvir and the corresponding metabolite in recombinant CYP2C8, CYP3A4, CYP2D6, and CYP2B6 incubations. M1 was the only metabolite radiochemically detected in these incubations. P450 enzyme involvement in the metabolism of dasabuvir was further confirmed by chemical inhibition assays. Incubation of dasabuvir in human liver microsomes in the presence of selective chemical inhibitors revealed that CYP2C8 is the major contributor to dasabuvir metabolism, followed by CYP3A4 and CYP2D6 (approximately 60%, 30%, and 10% of dasabuvir metabolism was inhibited by quercetin, ketoconazole, and quinidine, respectively). The contribution of CYP2B6 was negligible [<1% inhibition of dasabuvir metabolism was observed with 2-phenyl-2-(1-piperidinyl)propane)].

Representative HPLC radiochromatograms of dasabuvir and its metabolites in recombinant human CYP2C8 (A), CYP3A4 (B), CYP2D6 (C) and CYP2B6 (D) isoforms.
Discussion
The mass balance, disposition, and metabolism of dasabuvir were evaluated in four healthy men. After administration of a single 400-mg oral dose of [14C]dasabuvir, the mean total recovery of the administered radioactive dose was 96.6%, with recovery in individual subjects ranging from 90.8% to 103%. Nearly all of the administered radioactive dose (94.4%) was recovered in feces, with a limited amount of radioactivity (2.2%) recovered in urine through the last collection interval, indicating that dasabuvir and metabolites were predominantly eliminated in humans through feces and minimally through renal clearance.
LC-MS characterization of dasabuvir metabolites in urine and feces revealed that a total of 11 metabolites were present in human excreta. Biotransformation of dasabuvir in humans primarily involves P450-mediated hydroxylation at the tert-butyl group to form the active metabolite M1 (approximately similar activity against genotype 1 HCV infection as dasabuvir), which is also produced in preclinical toxicology rodent species with adequate safety coverage (Abbvie internal data). M1 is further oxidized to M4 (tert-butyl aldehyde) and M5 (tert-butyl acid). Subsequent glucuronidation of M1 and M5 produces metabolite M3 (tert-butyl ether glucuronide) and M6 (tert-butyl acid glucuronide), respectively; M2 is derived from the sulfation of M1. Reduction of M5, M1, and M2 at the pyrimidine-2,4(1H,3H)-dione moiety leads to formation of M8, M9, and M10, respectively. Metabolites M8, M9, and M10 were only observed in human feces. Trace levels of M7 (a reduction product of M6) and M11 (O-desmethyl metabolite of M1) were also detected in human urine.
Of the total radioactivity excreted in human feces and urine, the unchanged parent drug constituted 26.2% of the total dose and dasabuvir metabolites accounted for approximately 70% of the dose, suggesting that the absorption of [14C]dasabuvir in humans was > 70% of the dose. M1 was the most abundant radiochemical component in human feces, accounting for 31.5% of the total dose, followed by M2 (15.2%) and M5 (11.1%). The total amount of M1-related conjugates in human urine and feces accounted for 23.9% of the dose. Secondary oxidative metabolites of M1 and subsequent conjugates comprised 13.8% of the dose, indicating that M1 is mainly cleared through fecal elimination as unmodified M1 or conjugates of M1; subsequent oxidation of M1 plays a secondary role.
Unchanged dasabuvir was the major component of total drug-related radioactivity in plasma. Seven metabolites were identified in plasma, including the tert-butyl hydroxylate M1 and minor metabolites M2, M3, M4, M5, M6, and M11. The unchanged parent drug was the principal component in plasma, with an AUC0–∞ of 6290 ng⋅h/ml. The AUC0–∞ of the most abundant plasma metabolite M1 was 2280 ng⋅h/ml. Metabolite profiling indicated that dasabuvir represented approximately 58% of the total radioactivity and M1 accounted for about 21% that of total drug-related materials. Other metabolites, including M2, M3, M4, M5, M6, and M11, were relatively minor, ranging from trace levels up to 6.6% of the total drug in plasma.
Metabolism involving tert-butyl oxidation to form M1 is one of the primary clearance pathways for elimination of dasabuvir in humans. P450 enzyme characterization of this pathway is essential to understanding clinical drug–drug interactions. The in vitro P450 reaction phenotyping results indicated the importance of CYP2C8, followed by CYP3A4, in dasabuvir metabolism, with quercetin inhibiting approximately 60% of dasabuvir hepatic metabolism in vitro. These results are consistent with clinical findings (Menon et al., 2014) showing that CYP2C8 inhibition increased dasabuvir exposure (2-fold Cmax, 11-fold AUC0–72 h increase) after coadministration with gemfibrozil, whereas the M1 metabolite Cmax decreased by 20-fold and the AUC0–72 h decreased by 4-fold after administration of dasabuvir alone. These results indicate that CYP2C8 plays an important role in dasabuvir metabolism in humans. M1 is mainly cleared through direct biliary/fecal elimination and through further oxidation to M5 (tert-butyl acid) or glucuronidation to M2 (tert-butylhydroxyl glucuronide). A recombinant P450 phenotyping study indicated that M1 is predominantly metabolized by CYP3A4, with no metabolism by other P450 enzymes observed. M5 is an inactive metabolite, present at low levels in circulation. Detailed characterization of in vitro CYP450 and transporter assays and physiologically based pharmacokinetic models to provide mechanistic understanding of potential drug–drug interactions involving dasabuvir, M1 metabolite, and other DAA components were reported elsewhere (M. Shebley et al., manuscript in preparation).
In summary, the overall disposition and metabolism of dasabuvir has been determined in healthy human volunteers. Dasabuvir is well absorbed and extensively metabolized through tert-butyl hydroxylation to metabolite M1, followed by glucuronidation or sulfation of M1 or subsequent secondary oxidation pathways. Dasabuvir is eliminated by the biliary-fecal route. Renal excretion of dasabuvir and metabolites is deemed to be negligible. All of the metabolites identified in this study were also present in the preclinical safety species.
Acknowledgments
The authors thank Rich Voorman for contributions to study planning and valuable discussion, Dachun Liu for preparation of M1, and John Pratt for providing M5.
Authorship Contributions
Participated in research design: Shen, Menon, Kavetskaia, Fischer.
Conducted experiments: Serby, Zhang, Wan.
Contributed new reagents or analytic tools: Serby, Reed.
Performed data analysis: Shen, Serby, Menon, Marsh, Wan.
Wrote or contributed to the writing of the manuscript: Shen, Serby, Reed, Lee, Menon, Zhang, Marsh, Wan, Kavetskaia, Fischer.
Footnotes
- Received October 1, 2015.
- Accepted May 12, 2016.
↵1 Current affiliation: Global Clinical Pharmacology, Pfizer, Groton, Connecticut.
This research was supported by AbbVie. AbbVie participated in the interpretation of data, writing, review, and approving the publication.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ABT-333
- N-(6-(3-(tert-butyl)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (dasabuvir)
- amu
- atomic mass unit
- AUC
- area under the curve
- AUC0–∞
- area under the concentration time curve from time zero to infinity
- CID
- collisional induced dissociation
- AUC0–t
- area under the concentration time curve from time zero to the last measurable time point
- DAA
- direct-acting antiviral agent
- DMSO
- dimethylsulfoxide
- FMO
- flavin-containing monooxygenase
- HCV
- hepatitis C virus
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- LSC
- liquid scintillation counting
- M1
- N-(6-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(1-hydroxy-2-methylpropan-2-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
- M5
- 2-(5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxy-3-(6-(methylsulfonamido)naphthalen-2-yl)phenyl)-2-methylpropanoic acid
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- m/z
- mass-to-charge ratio
- NS
- nonstructural protein
- P450
- cytochrome P450
- SPE
- solid-phase extraction
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}