


Abstract
Aldehyde oxidase (AOX1) is an enzyme with broad substrate specificity, catalyzing the oxidation of a wide range of endogenous and exogenous aldehydes as well as N-heterocyclic aromatic compounds. In humans, the enzyme’s role in phase I drug metabolism has been established and its importance is now emerging. However, the true physiologic function of AOX1 in mammals is still unknown. Further, numerous single-nucleotide polymorphisms (SNPs) have been identified in human AOX1. SNPs are a major source of interindividual variability in the human population, and SNP-based amino acid exchanges in AOX1 reportedly modulate the catalytic function of the enzyme in either a positive or negative fashion. For the reliable analysis of the effect of amino acid exchanges in human proteins, the existence of reproducible expression systems for the production of active protein in ample amounts for kinetic, spectroscopic, and crystallographic studies is required. In our study we report an optimized expression system for hAOX1 in Escherichia coli using a codon-optimized construct. The codon-optimization resulted in an up to 15-fold increase of protein production and a simplified purification procedure. The optimized expression system was used to study three SNPs that result in amino acid changes C44W, G1269R, and S1271L. In addition, the crystal structure of the S1271L SNP was solved. We demonstrate that the recombinant enzyme can be used for future studies to exploit the role of AOX in drug metabolism, and for the identification and synthesis of new drugs targeting AOX when combined with crystallographic and modeling studies.
Introduction
Aldehyde oxidase (AO, EC1.2.3.1) is a molybdo-flavoenzyme present in the cytosol of many tissues in mammals (Garattini et al., 2008; Garattini et al., 2009). In humans, only one AOX functional gene is present, named AOX1, although several AOX orthologs are found in almost all mammalian organisms (Garattini et al., 2003). Human AOX is a α2 homodimeric enzyme in which each 150-kDa monomer is characterized by three separate domains: the 20-kDa N-terminal domain binds the two distinct [2Fe–2S] clusters, FeSI and FeSII, the 40-kDa central domain binds FAD, and the 80-kDa C-terminal domain binds the molybdenum cofactor (Moco) (Garattini et al., 2003). AOX enzymes generally catalyze oxidation reactions that largely involve nucleophilic attack by electrophilic carbons, including aldehyde groups and carbons of N-heterocycles that are para to or adjacent to the nitrogen atom (Beedham, 1985; Rashidi et al., 1997; Beedham, 1998). However, the true physiologic functions in respect to substrate specificity and the involvement in metabolic pathways of AOX in mammals are still unknown (Garattini et al., 2003). AOX1 in humans has been recognized as an enzyme with an emerging importance in phase I drug metabolism (Garattini and Terao, 2012; Kitamura and Sugihara, 2014). The number of drug molecules found in metabolic studies to be substrates for AOX is increasing and this has become a significant consideration for pharmaceutical companies (Obach et al., 2004; Barr and Jones, 2011). Interest in defining the physiologic functions of AOX enzymes is increasing, with a view toward determining their role in the metabolism of new molecules (drugs, prodrugs) in addition to their application to new biomedical analytical devices (Badalyan, et al., 2013, Badalyan, et al., 2014). Human AOX1 may have an important role in the clearance of a large number of drugs and xenobiotics (Obach et al., 2004; Pryde et al., 2010; Garattini and Terao, 2011), including N-heterocyclic drugs such as methotrexate, 6-mercaptopurine, cinchona alkaloids, and famciclovir substrates, all of which are oxidized by AOX (Clarke et al., 1995). Additionally, AOX1 is involved in the oxidation of intermediary drug metabolites, such as the conversion of cyclic iminium ions arising from the cytochrome P450-catalyzed oxidation of pyrrolidines and piperidines into lactams, or the oxidation of aldehydes derived from alcohol-containing drugs (Beedham, 1997). The multitude of possible physiologic substrates is significantly large and the possible involvement of AOX in several metabolisms was suggested in several of studies (Warner and Finnerty, 1981; Enroth et al., 2000; Kitamura et al., 2006). Additionally, interindividual variability of AOX1 activity has been reported in humans (Kawashima et al., 1999; Al-Salmy, 2001; Beedham et al., 2003; Hartmann et al., 2012). A major source of interindividual variability in the human population is allelic variants of the human AOX1 gene containing single-nucleotide exchanges, resulting in single-nucleotide polymorphisms (SNPs) encoding proteins with increased or decreased catalytic activity (Adachi et al., 2007; Hartmann et al., 2012). Missense single-nucleotide polymorphisms in the coding region of the hAOX1 gene may affect the catalytic function of the enzyme in either a positive or negative fashion. Analyzing the role of AOX in human metabolism, and its substrate specificities, in addition to identifying new inhibitors and the role of single amino acids in catalysis, the production of ample amounts of active enzyme for spectroscopic and crystallographic analyses is a prerequisite. However, it was reported previously that human AOX is poorly expressed using a bacterial expression system in Escherichia coli (Alfaro et al., 2009; Hartmann et al., 2012). Nevertheless, with this recombinant enzyme the crystal structure of the human AOX1 in the substrate-free form and in complex with the substrate phthalazine and the inhibitor thioridazine were solved at 2.6- and 2.7-Å resolution, respectively (Coelho et al., 2015).
To further study the impact of SNPs on the structure, catalytic mechanism, and mode of AOX inhibition, the goal of this study was to optimize the expression of human AOX1 in E. coli. One of the main obstacles for heterologous expression in bacteria might be attributable to the dissimilarity between protein expression machineries and codon usage in bacterial and human cells. Therefore, in this work we report a heterologous expression system in E. coli using a novel codon-optimized gene construct. The codon-optimized expression system led to the production of active enzyme in higher amounts sufficient to allow structural, spectroscopic, and mechanistic studies. Using the optimized expression system, three new SNPs were studied, namely C44W, G1269R, and S1271L. In addition, crystals of the S1271L SNP were obtained and the crystal structure was solved.
Materials and Methods
Cloning, Expression, and Purification.
The cDNA of hAOX1 was amplified using primers designed to permit cloning into the NdeI and SalI sites of the expression vector pTrcHis (Temple et al., 2000). The resulting plasmid, which was designated pSS130, expresses hAOX1 as an N-terminal fusion protein with an His6-tag. Further, a codon-optimized hAOX1 gene was synthesized (GeneArt, Thermo Fisher Scientific, Sunnyvale, CA) that contains a DNA sequence that included codons predominantly used in E. coli but do not result in a change of the amino acid sequence. The synthetic gene was cloned in pTrcHis NdeI-SalI and the resulting plasmid was designated pTHco-hAOX1. For site-directed mutagenesis and construction of the hAOX1 variants C44W, S1271L, and G1269R, the expression vector pTHco-hAOX1 was used as a template, and base-pair exchanges were introduced by polymerase chain reaction mutagenesis.
The hAOX1-wild type (WT) protein and variants C44W, S1271L, and G1269R were expressed and purified as described previously, with minor modifications (Hartmann et al., 2012). For expression in E. coli, the hAOX1 plasmids pTHco-hAOX1-WT, pAF1 (hAOX1 S1271L), pAF2 (hAOX1 C44W), and pAF3 (hAOX1 G1269R) were transformed into E. coli TP1000 (ΔmobAB) cells (Palmer et al., 1996). In general, 26 liters of E. coli cells were grown at 30°C in Luria broth medium supplemented with 150 μg/ml ampicillin, 1 mM molybdate, and 20 μM isopropyl β-d-1-thiogalactopyranoside. Cells were harvested by centrifugation after 24 hours of growth and resuspended in 50 mM sodium phosphate buffer, pH 8.0, containing 300 mM NaCl and frozen at 20°C until purification. After cell lysis, the crude extract containing hAOX1 was first purified using a nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen). The extract was mixed with 0.2 ml of matrix per liter of cell culture, in 10 mM imidazole, 50 mM sodium phosphate, and 300 mM NaCl, pH 8.0, and then the crude extract was stirred gently at 4°C for 20 minutes for protein binding. The crude extract was poured into a column and the resin was washed with 10 column volumes of 10 mM imidazole, 50 mM sodium phosphate, and 300 mM NaCl, pH 8.0, followed by a washing step of 10 column volumes of the same buffer with 20 mM imidazole. His-tagged hAOX was eluted with 250 mM imidazole in 50 mM sodium phosphate, and 300 mM NaCl, pH 8.0.
Human AOX from plasmid pSS130 was further purified by anion exchange chromatography. The column was previously equilibrated with 50 mM Tris × HCl, 1 mM EDTA, pH 8.0. For the purification of the hAOX a Mono Q 5/50 column (GE Healthcare, Piscataway, NJ) was used, on an ÄKTA purifier fast protein liquid chromatography system (GE Healthcare). Human AOX was bound to the matrix and could be eluted using a linear gradient of 1 M NaCl including buffer.
The final purification step (hAOX-WT and SNPs variants) consisted of size-exclusion chromatography using a Superdex 200 10/300 GL Column (GE Healthcare) on an ÄKTA purifier system. After the anion exchange chromatography by Mono-Q column, the protein was concentrated up to 500 μl loaded onto the column, which was equilibrated with 50 mM Tris × HCl, 200 mM NaCl, and 1 mM EDTA, pH 8.0
In Vitro Chemical Sulfuration.
hAOX1-WT and variants were subjected to an in vitro chemical sulfuration after the methods originally reported from Wahl and Rajagopalan (1982), with slight modifications and optimizations for hAOX1. The proteins were incubated under reducing conditions (500 μM dithionite) in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) in addition to sodium sulfide (2 mM) as sulfur source. Methyl viologen (25 μM) was used to confirm the absence of oxygen during the reaction. After a 30-minute incubation at room temperature (RT) the reaction was stopped and the enzyme was exchanged into buffer of 50 mM Tris × HCl, 200 mM NaCl, and 1 mM EDTA, pH 8.0, by using gel filtration columns.
SDS-PAGE.
Proteins were qualitatively separated by SDS-PAGE under denaturing and reducing conditions according to their molecular weight in discontinuous Tris/Glycine-SDS gels, as described after Laemmli (1970). Staining of the proteins after separation on the polyacrylamide gels was performed with Coomassie Blue R. Proteins samples were mixed with SDS-PAGE loading buffer and denatured for 5 minutes at 95°C. Molecular weight of the separated proteins was determined by comparison with a protein standard (molecular weight marker, MWM; Fermentas/Thermo Fisher Scientific). Gels were run at constant of 20 mA for one hour at room temperature. Cause of the hAOX1 monomer mass of 150 kDa 10% gels were used to obtain an effective separation.
Ultraviolet–Visible Spectroscopy Absorption Spectra.
The concentration of hAOX1 was determined at 450 nm on a Shimadzu UV-2401PC spectrophotometer (Somerset, NJ) using the specific molar extinction coefficient of 21,100 M−1 cm−1 at 450 nm. Absorption spectra were recorded in 50 mM Tris × HCl, 200 mM NaCl, and 1 mM EDTA, pH 8.0.
Metal Quantification.
Inductively coupled plasma optical emission spectrometry with an Optima 2100 DV (PerkinElmer Life and Analytical Sciences, Waltham, MA) was used to measure the metal content. Five-hundred microliters of purified hAOX1 (about 10 μM) and an equal volume of 65% nitric acid were mixed to wet ash the protein at 100°C overnight. The samples were diluted with 4 ml of water. The buffer used as reference was 50 mM Tris HCl, 200 mM NaCl, and 1 mM EDTA, pH 8.0. The detection was at wavelengths of 203.845 nm, 202.031 nm, and 204.597 nm for molybdenum and 238.204 nm, 239.562 nm, and 259.939 nm for iron. A standard was used for calibration and quantification of the detected metals (standard solution XVI; MerckMillipore, Darmstadt, Germany). The resulting mass concentrations were calculated and related as percent of protein saturated with molybdenum and iron corresponding to the two [2Fe–2S] clusters.
Steady-State Kinetics.
Steady-state enzyme kinetics were performed with purified hAOX1 in 50 mM Tris buffer, pH 8.0, containing 200 mM NaCl and 1 mM EDTA at 25°C in a final volume of 500 μl. The substrates benzaldehyde and phthalazine were used in a range of 1–400 μM using dichlorophenolindophenol (DCPIP; 100 μM) or ferricyanide (1 mM) as electron acceptors. For the substrate phenanthridine, in addition to ferricyanide and DCPIP, molecular oxygen was used as an electron acceptor and the product phenanthridone was detected at 321 nm. The substrate phenanthridine was used in a range of 1–100 μM. Total enzyme concentration varied between 100–200 nM. Reactions monitored over a range of 30 seconds. Activities were calculated using the extinction coefficient of 16,100 M–1cm−1 at 600 nm for DCPIP, 1080 M–1cm−1 at 420 nm for ferricyanide, and 4,775 M–1cm−1 at 321 nm for phenanthridone. The obtained data from three individual measurements were fitted nonlinear using the equation of Michaelis-Menten (eq. 1) to obtain the kinetic constants KM and turnover numbers (eq. 2). (1)
(1) (2)Fitting of the Michaelis-Menten curves was carried out using the software OriginPro 8.1G (Waltham, MA). The enzyme kinetics assays were performed on a Shimadzu UV-2401PC photometer.
(2)Fitting of the Michaelis-Menten curves was carried out using the software OriginPro 8.1G (Waltham, MA). The enzyme kinetics assays were performed on a Shimadzu UV-2401PC photometer.
Crystallization.
Crystals of the hAOX1-S1271L variant were obtained at 20°C using polyethylene glycol 4000 as precipitant. Crystals appeared a few hours after setting up the crystallization drops and grew to their maximum size (∼0.10 × 0.15 × 0.05 mm3) within 24 hours. The hAOX1-S1271L crystals were very sensitive to manipulation, as in the case of the hAOX1-free crystals (Coelho et al., 2015), and they were immediately flash-cooled in liquid nitrogen using a cryo-solution containing 15% glycerol. Several crystals of the hAOX1-S1271L variant were tested at different synchrotron radiation sources (ESRF, DLS, and SLS), but only a few diffracted to useful resolution. Crystals of the hAOX1-G1269R variant were also obtained under similar crystallization conditions but only grew to a maximum size of ∼0.06 × 0.10 × 0.03 mm3 and did not provide usable diffraction data.
Data Collection and Processing.
The hAOX1-S1271L crystals diffracted beyond 3.37 Å, and a complete dataset was collected at beamline I03 of the Diamond Light Source (Didcot, UK) at 0.976Å wavelength. As for the wild type, the crystals belong to space group P42212, with unit cell dimensions a = b = 147.4 Å and c = 131.7 Å. All data sets were processed using XDS (Max Planck Institute, Heidelberg, Germany; Kabsch, 2010) and Aimless [MRC Laboratory of Molecular Biology (LMB); Cambridge, UK; Evans, 2006]. Data collection statistics are present in Table 1.
Data collection and refinement statistics for crystals of variant hAOX1-S1271L
Structure Solution and Refinement.
The hAOX1-S1271L structure was solved by molecular replacement with Phaser (Phaser Scientific Software, Miami, FL; McCoy et al., 2005), using the structure of the hAOX1-free protein as a search model (PDB ID: 4UHW). Structure refinement was carried out in Refmac (LMB; Murshudov et al., 1997) followed by several rounds of rebuilding in Coot (Emsley et al., 2010), where water molecules were manually inspected. The final model contained residues 4–1336 and 52 water molecules. The following loops had no electron density and were excluded from the model: 168–198, 570–571, 655–660, and 881–882. Final Rwork and Rfree values were 16.20 and 25.31, respectively, and the coordinates were deposited in the Protein Data Bank (PDB) under the accession code (PDB ID: 5EPG).
Results
Effect of Codon Optimization on hAOX1 Expression and Activity.
The heterologous expression of human genes in E. coli can result in a low yield of protein, because of different codon usage by humans and E. coli. Here, we used an approach in which a synthetic hAOX1 gene was constructed using the genetic code with the most frequently used codons in E. coli. The construct resulted in a protein with an N-terminal–His6 tag fusion that facilitates protein purification. The expression from the optimized gene sequence was conducted in E. coli TP1000 (ΔmobAB) cells (Palmer et al., 1996) shown previously to be optimal for the expression of Mo-molybdopterin–containing proteins owing to their inability to synthesize the bis-molybdopterin guanine dinucleotide cofactor present in most E. coli molybdo-enzymes (Temple et al., 2000). The expression and yield after protein purification of the codon-optimized hAOX1 construct was compared with the non–codon optimized gene sequence containing a gene sequence that was isolated from human cDNA. This gene was cloned into the same vector for better comparison. After expression, the enzyme was purified in a two-step purification by Ni-NTA (Thermo Fisher Scientific) and size-exclusion chromatography. A further anion exchange chromatography purification step was only necessary for the native, non–codon optimized hAOX1 construct, owing to more contaminants present in the purified protein. The yield of the proteins expressed from construct pTrcHis-native hAOX1 and pTrcHis-codon–optimized hAOX1 (co-hAOX1) was compared (Table 2). The results show that the codon-optimized construct led to a 10- to15-fold higher protein yield than the construct containing the human codon sequence, with an average of 1.1 ± 0.3 mg protein per liter of E. coli culture. The protein expressed using the native sequence construct was determined to be 85–90% pure, and the protein from the co-hAOX1 construct was purified to 95%, determined after SDS-PAGE. The specific activity of both proteins after purification was comparable, with 141.4 ± 29.8 mU/mg (co-hAOX1) and 113.6 ± 34.1 mU/mg (hAOX1). However, both enzymes as purified showed poor levels of activity owing to low insertion of the equatorial sulfido ligand at the molybdenum atom in the active site because of the expression in E. coli. To increase the portion of the active protein, an in vitro chemical sulfuration step was performed as described previously (Wahl and Rajagopalan, 1982). The results in Table 2 show that the chemical sulfuration resulted in a 10-fold increase of the specific activity for both proteins. Since the molybdenum and iron saturations of both hAOX1 and co-hAOX1 are comparable, with 55–66% molybdenum and 71–76% iron saturation, the specific activities can be directly compared. After size-exclusion chromatography, two major peaks were detected for the native construct–expressed hAOX1 corresponding to calculated molecular masses of 300 and 150 kDa, which represent the dimeric and monomeric forms of hAOX1, respectively (Fig. 1A). In contrast, the codon-optimized hAOX1 construct yielded mainly the dimeric form of the protein (Fig. 1B).
Characterization of purified hAOX1-WT, the codon-optimized co-hAOX1 protein, and hAOX1 variants C44W, S1271L, and G1269R after expression in E. coli TP1000 cells

Characterization of hAOX1-WT and the protein derived from a codon-optimized gene sequence (co-hAOX). (A) Size-exclusion chromatogram (absorbance at 450 nm), UV/Vis spectrum, and a 10% SDS-polyacrylamide gel of purified hAOX-WT. (B) Size-exclusion chromatogram (absorbance at 450 nm) and a 10% SDS-polyacrylamide gel of purified co-hAOX expressed using the codon-optimized gene sequence. The UV-Vis spectra were determined using purified air-oxidized hAOX1 proteins in 50 mM Tris (pH 8.0) at RT. The elution profiles of the size-exclusion chromatography in 50 mM Tris, 200 mM NaCl, and 1 mM EDTA, pH 8.0, on a Superdex 200 column show different peaks of wild-type hAOX1 protein corresponding to multimeric (600 kDa), dimeric (300 kDa), and monomeric (150 kDa) forms. The majority of the lower bands were identified by mass spectrometry as degradation products of hAOX1 (data not shown).
The ultraviolet–visible spectroscopy (UV-Vis) spectra of hAOX1 and co-hAOX1 in their oxidized form displayed the typical features of molybdo-flavoenzymes (Fig. 1, A and B insets). The 280:450-nm absorbance ratio of 5.2 is indicative of the high purity of the enzymes, and the 450:550-nm absorbance ratio, which was calculated to be 3.1, demonstrates full saturation with FAD.
These results show that the codon-optimization procedure enabled a 10-fold higher yield of protein by facilitating protein purification that lead to a more pure protein. The activity of the purified protein was not affected by the alteration of the codon sequence.
Characterization of Single-Nucleotide Polymorphisms in hAOX1.
With the codon-optimized hAOX1 construct the studies of single-nucleotide polymorphisms were facilitated, owing to the higher expression levels and protein yield. We selected for further characterization three hAOX1 SNP variants reported before (Hartmann et al., 2012) and present in the NCBI SNP database (http://www.ncbi.nlm.nih.gov/snp). The C44W, S1271L, and G1269R amino acid exchanges were considered interesting because of their localization: Cys44 serves as a ligand to FeSII, while S1271 and G1269 are located directly adjacent to the highly conserved active-site glutamate (E1270) that is essential for the catalytic activity of AOX (Fig. 2). The S1271L exchange was identified on the basis of the most frequent heterozygous SNP in a screening of 180 healthy volunteers representative of the Italian population (Hartmann et al., 2012).

Location of the amino acid exchanges in the hAOX structure as a result of SNPs identified in the hAOX1 gene. Close-up of the hAOX active site (PDB ID: 4UHW). Highlighted is the localization of the three SNPs discussed in this paper: Cys44 in pink, Gly1269 in green, and Ser1271 in orange. The Moco and the two FeS centers (and the corresponding Cys residues of FeSII: Cys49, Cys52 and Cys74) are also represented, as well as residues Glu1270 and Gln 1203. The figure was prepared using PyMOL v1.7.2 (Schrödinger, Cambridge, MA).
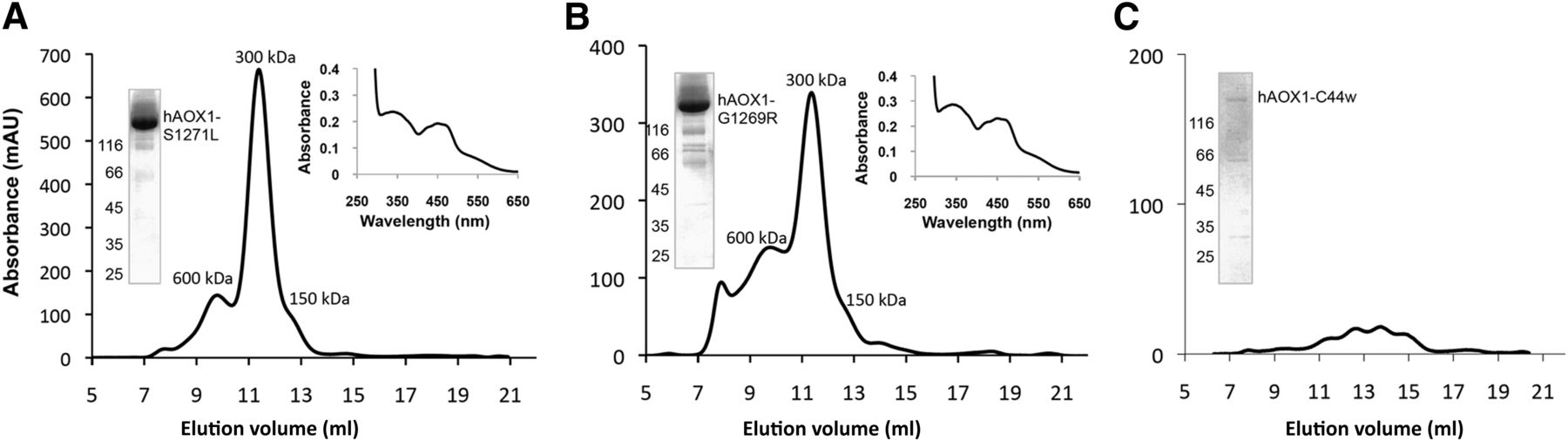
The three hAOX1 variants were expressed and purified under similar conditions as the co-hAOX1-WT protein. Differences in the oligomerization states of the variants and wild-type hAOX1 were analyzed by size-exclusion chromatography. The hAOX1 variants S1271L and G1269R showed similar protein yield after purification (Table 2) and a dimer/monomer ratio comparable to the co-hAOX1-WT protein (Fig. 3, A and B). Both protein variants eluted mainly as dimers from the size-exclusion chromatography, showing, however, some aggregate formation (Fig. 3). In contrast, the C44W variant did not give rise to a stable protein that could be purified (Fig. 3C).

Characterization of hAOX1 SNP. Size-exclusion chromatogram (absorbance at 450 nm), UV/Vis spectrum, and a 10% SDS-polyacrylamide gel of the variants (A) hAOX-S1271L, (B) hAOX-G1269R, and (C) hAOX1-C44W. The UV/Vis spectra were determined using purified air-oxidized hAOX1 proteins in 50 mM Tris (pH 8.0) at RT. The elution profiles of size-exclusion chromatography in 50 mM Tris, 200 mM NaCl, 1 mM EDTA, pH 8.0 on a Superdex 200 column show different peaks of hAOX variants corresponding to the multimeric (600 kDa), dimeric (300 kDa), and monomeric (150 kDa) forms.
The molybdenum saturation in the S1271L and G1269R variants was slightly decreased to 41% and 31%, respectively, whereas the iron saturation varied between 58% (S1271L) and 72% (G1269R) (Table 2). The S1271L and G1269R proteins showed purities comparable to that of the wild-type protein, and the UV/Vis absorption spectra of both purified variants showed the same characteristic features as wild-type hAOX1 (Fig. 3). However, when the specific activities of both protein variants were determined, measurable activities were only obtained for the S1271L variant. Specific activity of 1132 U/mg was detected after chemical sulfuration, which is comparable to the activity obtained for the co-hAOX1 wild-type protein when related to a full complement of Moco (on the basis of the molybdenum concentration). In contrast, the G1269R variant was completely inactive.
Steady-State Kinetics and pH Optimum of hAOX1-WT and the S1271L Variant.
To test the influence of the S1271L SNP variant on the optimum pH and the catalytic activity, the pH-dependent catalytic profiles were determined in comparison with the co-hAOX1 wild-type protein. In general, the pH optimum of the co-hAOX1-WT and the S1271L variant were comparable, with a pH optimum of around 8.0 for the wild-type enzyme with phenanthridine as substrate and molecular oxygen as electron acceptor. The pH optimum was only very slightly shifted to pH 8.3 for the S1271L variant under the assay conditions (Fig. 4). Therefore, all further steady-state kinetics were determined at pH 8.0.

pH optimum of hAOX1-WT and hAOX1-S1271L. The pH optimum was determined for (A) hAOX1-WT and (B) hAOX1-S1271L by measuring activities of hAOX1 with phenanthridine as substrate and molecular oxygen as electron acceptor, in the pH range of 6.5–10 in 50 mM Tris/HCl buffer. Activity measurements were performed at RT.
To determine the kinetic parameters, the co-hAOX1-WT and S1271L variant were assayed with phenanthridine, phthalazine, and benzaldehyde as substrates using molecular oxygen (only with phenanthridine) and potassium ferricyanide or 2,6-dichlorophenol indophenol (DCPIP) as electron acceptors (with phenanthridine, phthalazine, and benzaldehyde). Kinetic experiments were performed by spectrophotometric assay following product formation (phenanthridine:oxygen) or the reduction of the electron acceptor (ferricyanide or DCPIP).
For all three protein variants, the kinetic constants are shown in Table 3 and are related to a 100% molybdenum saturation for better comparability. In general, the results show that all three variants have kinetic comparable constants with all substrates and electron acceptors analyzed. Minor differences in the kinetic constants might have resulted from slight differences in the sulfuration level after chemical sulfuration.
Steady-state kinetics of hAOX1-WT and SNP variant S1271L
Steady-state kinetic parameters were calculated and related to a molybdenum saturation of 100% for hAOX1-WT and hAOX1-S1271L under assay conditions using molecular oxygen (phenanthridine), 1 mM ferricyanide, and 100 μM 2,6 dichlorophenolindophenol as electron acceptors. Data are mean values from three independent measurements (±S.D.).
The fact that hAOX1 and co-hAOX1 showed comparable kinetic constants indicates that the codon-optimization of the gene and the resulting higher expression levels did not influence the correct folding and cofactor insertion of the protein during expression in E. coli cells, showing that the codon-optimized construct is suitable for further studies.
Therefore, the enzymatic activity of the variant S1271L was determined. The kinetic constants with all substrates determined for the S1271L variant generally showed no major differences compared to the wild-type enzyme. However, whereas the kcat values were in general around 60–80% compared to the values determined for the same substrates with the co-hAOX1 WT construct, the KM values were also decreased to the same extent (60–80% of co-hAOX1 WT), resulting overall in the same catalytic efficiencies determined for both protein variants (Table 3). Only with the substrate benzaldehyde a decrease in kcat and KM of about 50% was determined for the variant, resulting in a 2-fold decreased catalytic efficiency. Thus, the amino acid exchange adjacent to the active-site glutamate residue generally does not affect substrate binding negatively or the turnover of bulkier substrates at the active site.
The steady-state kinetics showed that hAOX1, co-hAOX1, and S1271L had the highest kcat values with phenanthridine as substrate when molecular oxygen was used as electron acceptor, whereas with electron acceptors like ferricyanide and DCPIP, the kcat values were decreased. Molecular oxygen as physiologic electron acceptor in general resulted in a higher kcat compared with ferricyanide or DCPIP as electron acceptor. However, in all cases DCPIP drastically decreased the kcat and KM for all three substrates tested. To characterize the influence of DCPIP on the catalytic constants of hAOX, inhibition studies with varying concentrations of DCPIP were performed. Using co-hAOX1 and cinnamaldehyde as substrate (substrate consumption clearly detectable at 295 nm), inhibition studies showed an uncompetitive inhibition pattern for hAOX1-DCPIP (Fig. 5) with a Ki value of 41 ± 8 μM. These data are consistent with previously reported parameters (Barr and Jones, 2011).

Lineweaver-Burk plots of hAOX1 inhibition by DCPIP. Inhibition of hAOX1 using DCPIP concentrations between 0 and 200 μM were measured: 200 μM (diamond), 100 μM (filled square), 50 μM (triangle), 10 μM (empty circle), 1 μM (empty square), and 0 μM (filled circle). Inset: secondary plot of 1/Vmaxapp and inhibitor concentration to determinate Ki value. The product formation was measured with 5–100 μM cinnamaldehyde as substrate using molecular oxygen as electron acceptor.
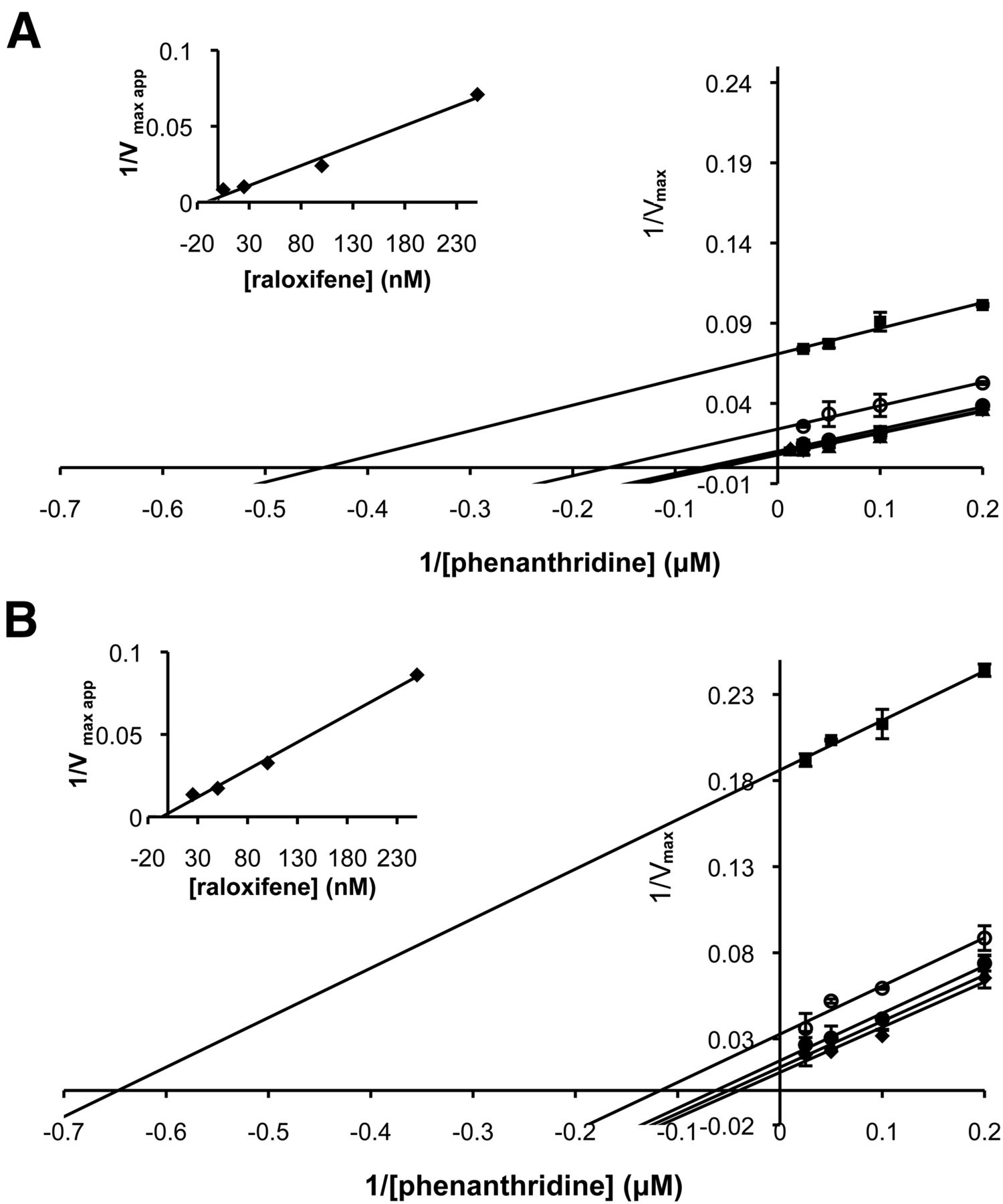
Additionally, we determined whether the inhibition of the potent inhibitor raloxifene was affected by the S1271L amino acid exchange in the active site. As shown in Fig. 6, the inhibitor showed an uncompetitive binding mode with a Ki value of 7.25 ± 3 nM for hAOX1–WT. The uncompetitive inhibition pattern was retained in the hAOX1-S1271L variant, with only a slight decrease of the Ki value to 5.5 ± 2 nM. Since the inhibition mode is uncompetitive, the inhibitor probably binds at a position different from the active site; thus, amino acid variations in the active site do not affect the inhibitor affinity.

Lineweaver-Burk plots of the inhibition of hAOX1-WT and variant hAOX1-S1271L by raloxifene. Lineweaver-Burk plots of hAOX1-WT (A) and hAOX1-S1271L (B) inhibition by raloxifene. Inhibition of human AOX1s using raloxifene concentrations between 0 and 250 nM were measured: 250 nM (square), 100 nM (empty circle), 25 nM (filled circle), 5 nM (triangle), and 0 nM (diamond). Inset: secondary plot inhibitor concentration to determinate Ki value. Phenanthridine as substrate and molecular oxygen as electron acceptor were used. Assays were performed at 25°C in Tris 50 mM, NaCl 200 mM, and 1 mM EDTA buffer, pH 8.0. Data are mean values from three independent measurements (± S.D.).
Crystal Structure of hAOX-S1271L Variant.
To further characterize the S1271L variant, we crystallized the protein and determined its structure, using diffraction data to 3.37 Å resolution. The structure of the hAOX1-S1271L variant is very similar to the structure of the wild-type hAOX1 recently determined at 2.6 Å resolution (Coelho et al., 2015) with a root-mean-square deviation of 0.36 Å for 2386-superimposed Cα, indicating that the structural integrity of the protein has been preserved in the variant. In the wild-type hAOX1 the Ser1271 residue is located in the Moco active site region (Fig. 7, A and B) immediately after the catalytic Glu1270. In the hAOX1-S1271L structure, in spite of the modest resolution, the electron density maps and the analysis of the B-factor for the surrounding residues confirm the presence of the single point mutation S1271L (Fig. 7A). Upon refinement, the position of the catalytic Glu1270 side chain in the variant has slightly moved in comparison with the wild-type, but this alteration should have no biologic relevance since there is no significant change in the kinetic data between the wild type and the S1271L variant. The G1269R variant was also crystallized, but no usable diffraction data could be obtained.

Crystal structure of the hAOX1-S1271L variant. Close up of the hAOX1-S1271L variant active site. (A) Details of Leu1271 (orange) immediately after the catalytic Glu1270 residue are shown with the 2mFo–DFc map contoured at 1σ. (B) Comparison of hAOX1-S1271L (blue) and the hAOX1-WT (Coelho et al., 2015) for the Leu1271 and Ser1271 residue, respectively. Also shown is the localization of the G1269 SNP (green) and the Moco and FeSI cofactors. The figure was created using PyMOL v1.7.2.
Discussion
Previously established expression systems for the heterologous expression of hAOX1 in E. coli resulted in a low yield of protein of about 50 μg per liter of E. coli (Hartmann et al., 2012). Here, we have analyzed the effect of a synthetic, codon-optimized gene for hAOX1 on the expression yield and activity of the purified protein. The results obtained highlight the importance in the selection of strategies for heterologous expression of complex human cofactor–containing proteins in E. coli. In the case of hAOX1, the amount of rare codons results in low expression levels of the gene, which can be overcome by introducing codons more commonly used in E. coli into the respective gene sequence. The expression of the codon-optimized hAOX1 resulted in a protein yield of 1–1.5 mg per liter of E. coli culture, which is about 10- to 15-fold higher than the yield obtained using the native gene sequence in the same expression vector. With this higher level of protein expression and higher protein concentration in the E. coli extracts, the purification of the protein was additionally facilitated and a two-step purification by Ni-NTA chromatography and size exclusion chromatography was sufficient to yield a 95% pure protein, which could be used for kinetic and crystallographic studies.
However, the main problem remained—obtaining a 100% active enzyme after expression of the complex molybdo-flavoenzyme in E. coli. This is related to the low saturation of the sulfurated molybdenum cofactor since the specific system for Moco sulfuration of the human protein is not present in E. coli (Mendel and Leimkuhler, 2015). The sulfuration is the last important step of the biosynthesis of the mono-oxo molybdenum cofactor in the xanthine oxidase family of molybdo-enzymes. To have an active enzyme it is necessary that it become fully sulfurated, with a sulfur ligand bound to the molybdenum atom at the equatorial position. Unfortunately, attempts to coexpress the human Moco sulfurase (Peretz et al., 2007) in E. coli to increase the sulfuration levels have failed, owing to its expression in inclusion bodies (data not shown). To overcome this low enzyme activity of the purified protein, a chemical sulfuration step was included during the purification procedure (Massey and Edmondson, 1970), (Wahl and Rajagopalan, 1982). The chemical sulfuration proved to be successful and resulted in a 10-fold increase in enzyme activity.
The direct comparison of the purified protein from the codon-optimized construct and the native hAOX1 gene showed that the folding of the protein, the insertion of the cofactors (Moco, FAD, and the 2× [2Fe–2S] clusters), in addition to the level of Moco sulfuration and the overall activities, were not affected, since both proteins showed similar characteristic spectroscopic features and comparable kinetic constants. Also, the chemical sulfuration of both proteins yielded the same 10-fold increase of enzyme activity. The results show, in general, that codon optimization is feasible for this complex human protein.
The higher protein yield of the codon-optimized construct may benefit future studies of the protein and further a better understanding of its mode of action. We investigated three SNPs occurring in the AOX gene of human individuals. We selected three SNPs that resulted as amino acid exchanges in a ligand of the FeSII (C44W) and two amino acid residues near the substrate binding site (S1271L and G1269R). Whereas the C44W variant was shown to be unstable/not expressed, the S1271L and G1269R variants were successfully expressed and purified. Both proteins showed similar cofactors saturation; however, only the S1271L variant was determined to be active and could be further characterized. The G1269R variant, in contrast, was shown to be completely inactive. These results suggest that homozygous human individuals presenting the SNPs resulting from amino acid exchanges C44W and G1269R do not contain an active form of the hAOX1 enzyme.
The inability to synthesize a stable C44W protein variant can be explained by the fact that the replacement of a cysteine by a tryptophan in the position 44 results in a loss of the FeSII insertion into the protein. The assembly of the iron-sulfur clusters is one of the first and crucial steps of the protein maturation. The lack of FeSII probably led to the production of an instable and structurally disordered enzyme, in which the other cofactors could not be inserted. As a consequence, the protein was rapidly degraded in the cell. These results reveal the importance of this SNP in individuals, who consequently do not have an active AOX1 protein. In these individuals the lack of AOX1 will negatively affect drug metabolism and other detoxification events mainly catalyzed by AOX1. The same effect should be considered for the SNP resulting in the amino acid exchange G1269R. Here, a protein was obtained in which all cofactors were inserted; however, the protein was shown to be inactive. Since the glycine is located next to the catalytically essential glutamate, it might therefore negatively affect the first step of catalysis, which is the proton abstraction of the Mo-OH group by the deprotonated glutamate (E1270), which acts as an active-site base at the pH optimum of 8.0 of the enzyme (Coelho et al., 2012; Mahro et al., 2013). The generated Mo-O– group then performs a nucleophilic attack on the carbon atom of the substrate in the second step of the reaction mechanism. Concomitantly, there is a hydride transfer to the sulfido ligand of the molybdenum atom, and an intermediate species is formed. Further, the product is released from the reduced molybdenum site and a water molecule replenishes the vacant coordination position. The reaction cycle is completed once molybdenum is reoxidized and the two reducing equivalents are transferred to molecular oxygen via the two [2Fe2S] and the FAD cofactor (Huber et al., 1996; Hille et al., 2014). In the G1269R variant the positive charge probably interferes with the negative charge of the active-site glutamate (E1270), thus inhibiting the first step of the reaction mechanism. In addition, the bulkier arginine in comparison with the glycine probably results in a steric hindrance of the glutamate side chain, which probably has to change its position, interfering with its catalytic role as a general base.
The kinetic constants were only obtained for variant S1271L, which is a frequent SNP, but was so far only identified to be heterozygous in the individuals tested (Hartmann et al., 2012). However, the SNP variant showed the same characteristics as the wild-type protein with the substrates phenanthridine, phthalazine, and benzaldehyde. We were also able to obtain the crystal structure of this variant and compare it with the structure of the substrate-free enzyme. No structural deviations were observed, which is in accordance with the kinetic data obtained, similar for both hAOX and hAOX-S1271L. Crystals of the G1269R variant were also obtained, using similar crystallization conditions as for S1271L, but in this case the X-ray diffraction was very poor and insufficient for the structure solution.
The steady-state kinetics experiments and the inhibition studies showed that DCPIP acts as an uncompetitive inhibitor of the human AOX1. An inhibition of bovine xanthine oxidase by DCPIP has been reported before (Barr and Jones, 2011). DCPIP is frequently used as electron acceptor in enzyme-catalyzed oxidation reactions. For bovine xanthine oxidase, the rate of electron transfer from conventional substrates to DCPIP approximates that for electron transfer to the physiologic acceptor, molecular oxygen (Avis et al., 1956). However, the electron acceptors receive their electrons at fundamentally different cofactors: the electron transfer to oxygen takes place at the FAD site (Komai et al., 1969), whereas DCPIP or other artificial acceptors (e.g., phenazine methosulfate or ferricyanide) directly receive the electrons from the reduced molybdenum atom (Massey et al., 1970). For xanthine oxidase, the mechanism of inhibition of DCPIP was shown to be noncompetitive (Gurtoo and Johns, 1971), showing that DCPIP binds to the reduced and oxidized enzyme with the same rate. In hAOX1, the mode of inhibition was shown to be uncompetitive, and DCPIP probably binds only to the reduced enzyme. Here, the differences between hAOX1 and bovine XO can be explained by the different architectures of the active site and resulting charge differences, also revealed by the different substrate specificity of both enzymes. In general, AOX enzymes have a broader substrate specificity, acting on aldehydes, purines, and heterocyclic compounds as substrates, whereas the selectivity of XO enzymes is restricted to manly purines as specific substrates.
In total, our results show that the codon-optimized construct for the expression of hAOX1 in E. coli gave rise to an active enzyme that could be used for site-directed mutagenesis studies and kinetic and X-ray crystallography studies in a reproducible manner.
Our data provide important evidence for the role of amino acids in the active site of AOX1 and show that individuals with SNPs exist in whom the base-pair exchange of the SNP leads to loss of enzyme activity. We demonstrate that the recombinant enzyme can be used for future studies to exploit the role of AOX1 in drug metabolism, and for the identification and synthesis of new drugs targeting AOX in combination with crystallographic and modeling studies.
Acknowledgments
The authors thank the staff of the Diamond Light Source (Didcot, United Kingdom) for assistance during data collection.
Authorship Contributions
Participated in research design: Foti, Hartmann, Leimkühler.
Conducted experiments: Foti, Hartmann, Coelho.
Performed data analysis: Foti, Coelho, Santos-Silva, Romão, Leimkühler.
Wrote or contributed to the writing of the manuscript: Foti, Coelho, Santos-Silva, Romão, Leimkühler.
Footnotes
- Received November 16, 2015.
- Accepted February 1, 2016.
This work was financially supported by the Deutsche Forschungsgemeinschaft [Grant LE1171/8-1], the Fundação para a Ciência e Tecnologia [Grants UID/Multi/04378/2013, EXCL/QEQ-COM/0394/2012, PTDC/BIA-PRO/118377/2010, SFRH/BPD/84581/2012], and the Deutscher Akademischer Austauschdienst [PPP 441.00].
Abbreviations
- AOX
- aldehyde oxidase
- DCPIP
- dichlorophenolindophenol
- Moco
- molybdenum cofactor
- RT
- room temperature
- SNP
- single-nucleotide polymorphisms
- UV-Vis
- ultraviolet–visible spectroscopy
- WT
- wild type
- XO
- xanthine oxidase
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}