


Abstract
Nonspecific binding or sequestration results in differences between free and total drug concentrations, both in vitro and in vivo. Membrane partitioning and not protein binding is the primary mechanism of drug sequestration. Therefore, physicochemical properties, e.g., LogP can be used to predict drug sequestration in membrane and cell-based assays. The concentration of drug in a membrane is determined by the both the rate in and out of the membrane. In contrast, membrane permeability is a function of the rate in only. This commentary discusses the origins of membrane partitioning and permeability and their impact on cellular disposition.
Introduction
The need to determine free drug concentrations in drug discovery and development is increasingly recognized (Obach, 1997, 1999; Lam et al., 2006; Fan et al., 2010; Giacomini et al., 2010). The critical impact of plasma protein binding on drug disposition is clearly understood. The purpose of this commentary is to clarify the origin and impact of nonspecific binding. Nonspecific binding or sequestration results in differences between free and total drug concentrations, both in vitro and in vivo. In an in vitro system, several types of sequestration are possible. Drug can bind to specific proteins, partition into membranes, partition into lysosomes, bind nonspecifically to proteins, or even the apparatus (see Fig. 1). In all cases, the impact is to essentially increase the apparent volume of distribution of the incubation. That is, the amount of drug in the incubation divided by the free concentration is larger than the physical volume of the incubation. A decrease in the free concentration due to nonspecific sequestration results in higher Km, Ki, EC50, and IC50 estimates, and underpredicted rates and clearances.

A, protein binding versus membrane partitioning in a microsome. B, membrane partitioning versus membrane permeability in a cell. Internal membrane partitioning is also depicted. Hatched polar head groups represent phosphatidylcholine, and gray polar head groups represent phosphatidylserine. CYP, cytochrome P450.
The free fraction of drug can be determined experimentally. Equilibrium dialysis can be used for microsomal or protein incubations. For cell preparations in serum-free media, simple centrifugation can be used to determine free fraction. When serum is present, the partition constant for serum also determines free fraction. Experimentally, all types of sequestration (protein binding, membrane partitioning, binding to apparatus, etc.) will have the same impact on the volume of distribution of the incubation.
Although the impact of nonspecific binding on free drug concentration is the same for different kinds of sequestration, the mechanism of sequestration is important. Nonspecific binding to proteins or apparatus can be erratic (Minder et al., 1970; Palmgrén et al., 2006), whereas partitioning into membranes is governed by physicochemical interactions (Francesco and Bickel, 1977; Römer and Bickel, 1979; Austin et al., 2002; Hallifax and Houston, 2006; Abraham and Austin, 2012). In fact, partitioning into membranes is an integral part of membrane permeability.
Microsomal Protein Binding versus Membrane Partitioning.
Microsomes, the term used for the smooth endoplasmic reticulum (ER) fraction, have been used as a drug metabolism enzyme source for over 50 years (Axelrod, 1955a,b; Brodie et al., 1955; De Duve and Beaufay, 1981; Murphy, 2008). Initially, liver microsomes were invaluable for determining metabolic pathways and characterizing the kinetics of the cytochromes P450. Today, microsomes are still used as an essential reagent when predicting clearance and drug interactions. One of the key variables in a microsomal incubation is the microsomal content, usually measured in terms of total protein concentration (milligram of microsomal protein per milliliter). It has been recognized that observed kinetic constants can be greatly impacted by this variable (Tran et al., 2002; Margolis and Obach, 2003; Riley et al., 2005). The actual kinetic constants are not dependent on microsomal content, but instead sequestration changes the free concentration of drug. For example, the term [I]/Ki is used to predict the propensity for inhibition of saturable processes. In a microsomal cytochrome P450 inhibition assay, the free inhibitor concentration must be used to prevent underestimation of Ki when the inhibitor is sequestered by the microsomes.
Clearances are also underpredicted when the drug is sequestered by microsomes. This can be observed when the amount of microsomal protein is increased to increase metabolism in a substrate-depletion assay. Highly partitioned molecules will show a minimal increase in the rate of metabolism because the additional microsomes will further decrease the free fraction of drug.
Although we discuss microsomal content in terms of milligrams of protein, microsomes consist of both proteins and phospholipid membranes of the ER. Phospholipid content of rat liver microsomes has been reported to be 0.5 mg phospholipid/mg protein (Ernster et al., 1962). Although frequently discussed as microsomal protein binding, as discussed below, nonspecific interactions with microsomes are almost exclusively due to partitioning of drugs into membranes.
Membrane Partitioning Is More Common than Nonspecific Protein Binding.
Nonspecific interactions with membranes, proteins, lysosomes, or the experimental apparatus will decrease the free-drug fraction and may result in inaccurate kinetic parameter estimates. Evidence suggests that, by far, the most common cause of drug sequestration is partitioning into phospholipid membranes. Early work by Bickel showed that basic lipophilic drugs such as imipramine, chlorpromazine, and other drugs predominantly sequester into phospholipid membranes, specifically via hydrophobic interactions with membrane lipids such as phosphatidylcholine (Bickel and Steele, 1974; Francesco and Bickel, 1977; Römer and Bickel, 1979). Margolis and Obach (2003) compared binding of a number of drugs to microsomes and phospholipid vesicles and determined that sequestration was associated with the phospholipid component. In addition, the extent of drug partitioning into microsomes is highly correlated with partitioning into hepatocytes (Riley et al., 2005; Kilford et al., 2008). Although active transport can alter hepatocyte uptake (Giacomini et al., 2010), similar results were obtained with nonviable hepatocytes. In addition, species differences are minimal for microsomal partitioning (Obach, 1997).
When considering partitioning, the composition of the lipid bilayer is important. Phospholipid composition is uniform in both leaflets of the ER membrane and, therefore, in microsomes (Alberts et al., 2002). Plasma membrane leaflets are asymmetric in their phospholipid composition, with higher phosphatidylserine and phosphatidylethanolamine in the inner leaflet and greater phosphatidylcholine in the outer leaflet (Janmey and Kinnunen, 2006). It has been shown that phosphatidylserine membranes have higher affinity for basic compounds and lower affinity for acidic membranes, presumably due to the additional negative charge (Osterberg et al., 2001). However, microsomes may adequately represent overall membrane partitioning, because the overall phospholipid composition of microsomes and plasma membrane is similar.
Physicochemical Properties Predict Membrane Partitioning and Permeability.
Hydrophobicity and charge are highly correlated with microsomal partitioning (Abraham and Austin, 2012). Hydrophilic compounds show minimal partitioning and, as expected, poor membrane permeability. Organic anions have minimal affinity for the membrane, whereas hydrophobic amines partition extensively. This is presumably because the amine can interact with the negatively charged phospholipids (Smith et al., 2012). Figure 2A depicts the relationships between the partition coefficient (KL) in microsomes, LogP, and charge. For each class of molecules (acids, bases, and neutral molecules), partitioning generally increases with increasing LogP. However, the slopes for each class appear to be different (Fig. 2A). To obtain a better fit for the combined set, LogD (Fig. 2B) is used for acidic compounds (Austin et al., 2002). A quadratic equation was also used to describe the combined LogP(D) dataset, and it provided a lower overall error than a single linear fit (Hallifax and Houston, 2006).
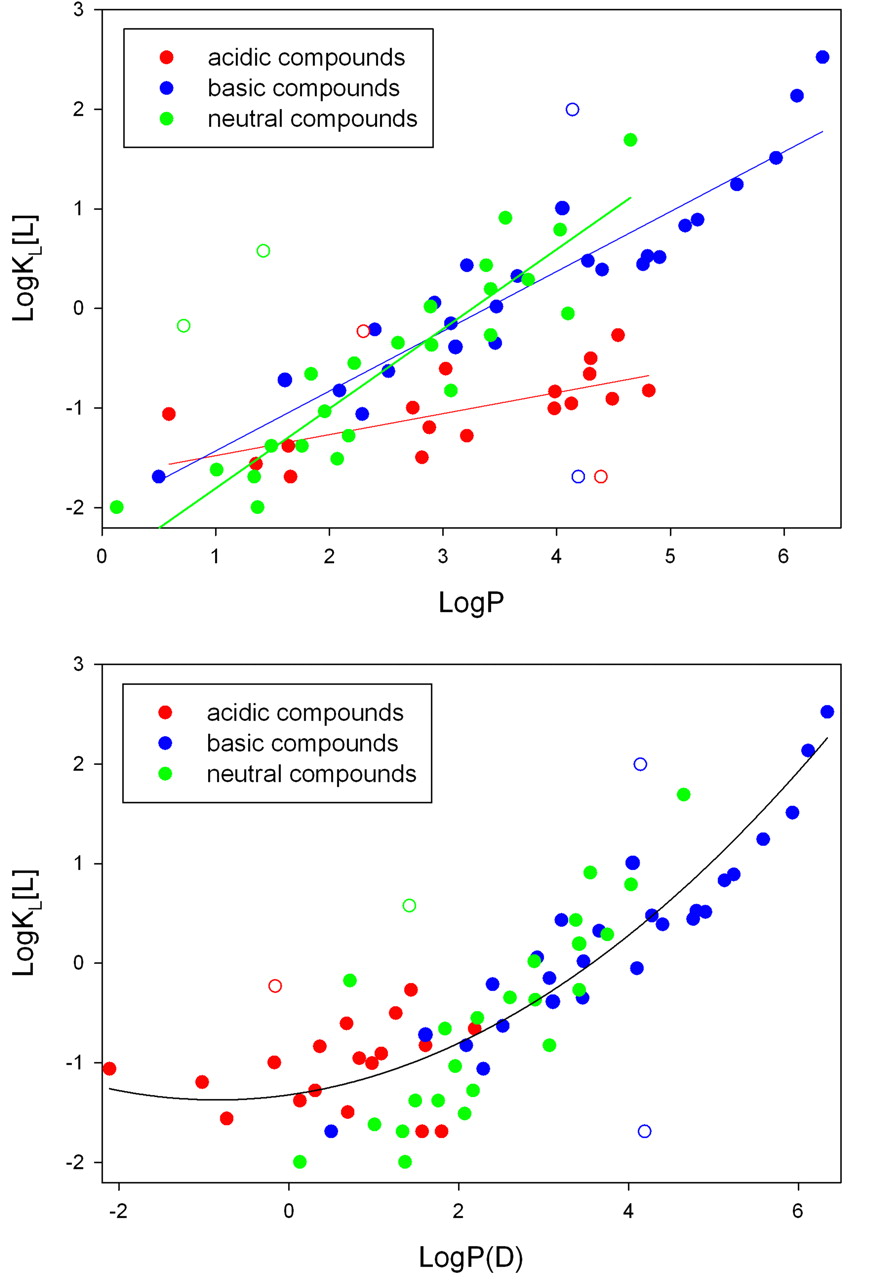
A, relationship between LogP and KL[L] (membrane partitioning). Data are drawn on the basis of the findings in Obach, 1999, Austin et al., 2002, Hallifax and Houston, 2006, and Gao et al., 2008. When values for a compound are reported in multiple references, averages were used. Outliers identified by Cook's distances >4/n are depicted by open circles. B, a single quadratic equation fit to data from Fig. 2A with LogD values instead of LogP values for acids.
The octanol/water partition coefficient was developed to provide an experimental measure of general hydrophobicity for use in linear free-energy relationships (LFERs). For all LFERs, there are parameters and susceptibility constants (for example, σ and ▵ for the Hammett equation). LogP(D) values are the hydrophobicity parameters, and the slopes in Fig. 2A are the susceptibility constants, i.e., the impact (slope) of logP on the partition constant. LogP(D) was never expected to provide a single correlation across different chemical series (Hansch, 1969), and the different slopes in Fig. 2A might be expected. Although the quadratic equation in Fig. 2B appears to describe all the data with a single equation, the residuals for the neutral compounds are nonrandom. The relationship between hydrophobicity and membrane partitioning for one or more classes of compounds may well exhibit curvature. However, paucity of currently available data precludes selection of nonlinear models. As additional data become available, the exact nature of the relationship between LogP(D) and membrane partitioning should become more clear.
Membrane Permeability: It Is All about the Rate in.
The membrane partition constant for the free drug (Kp,u) is defined by the clearance into the membrane, CLi, divided by the clearance out of the membrane, CLo (see Fig. 3). Upon exposure to a drug, the membrane concentration will increase to a level determined by the membrane partition constant. Thus, both CLi and CLo determine the equilibrium membrane concentration. This is not the case for membrane permeability.

Modeling membrane permeability. Ordinary differential equations describe diffusional clearance CLi and CLo (eqs. 1–4) for a three-compartment model and CLd for a two-compartment model (eqs. 5 and 6). Comparison of eqs. 5 and 6 with eq. 4 shows that CLd = CLi/2. Equation 7 can also be derived by comparing explicit integrated solutions of differential equations and taking the limit as Vm → 0. Cp, Cm, and Ccell are unbound drug concentrations outside the cell (e.g., plasma), inside the membrane, and inside the cell, respectively. Vp, Vm, and Vcell are apparent volumes of distribution of the drug in plasma, membrane, and cell compartments. The membrane partition coefficient of the unbound drug Kp,u is defined as CLi/CLo.
Membrane partitioning is an equilibrium distribution process, whereas membrane permeability is a rate. Membrane permeability is required to achieve intracellular drug concentrations, the effect-site concentration for most drug targets. Before reaching steady-state, the rate out of the membrane will increase as the membrane concentration increases, until the rate out equals the rate in. At steady state, passive clearance, CLd (the permeability × surface area) will equal CLi/2 (see Fig. 3 and accompanying derivation). CLi is divided by 2 because the drug going in can either progress forward or diffuse back. Thus, whereas both CLi and CLo determine the concentration of drug in the membrane, passive drug permeability is determined by only CLi.
Therefore, compounds that do not partition into membranes have poor permeability. Compounds that partition extensively can be highly permeable across a single membrane but exhibit lag times in permeability experiments across cells. Observed lag times are presumably due to the saturation of internal membranes during passage across the cell. Models that do not explicitly include membrane partitioning will predict no lag time for cellular permeability (Korzekwa et al., 2012).
Transport and Metabolism Alter Intracellular Concentrations.
Because membrane concentrations can be several thousand-fold higher than free concentrations, membrane partitioning becomes an inseparable component of cellular disposition (Rowland and Tozer, 2010; Korzekwa et al., 2012). Transporters move compounds in or out of cells, and they obviously affect cellular disposition. Metabolizing enzymes can also decrease cellular concentrations when the rate of metabolism is fast relative to the rate of equilibration. Some apical efflux transporters [e.g., P-glycoprotein (Pgp)] presumably transport drug directly from the membrane, whereas transporters for hydrophilic molecules presumably transport free drug (Gottesman et al., 1995, 1996; Hennessy and Spiers, 2007). Although not generally accepted, it has been suggested that the cytochromes P450 extract drug molecules directly from the membrane (Cojocaru et al., 2007). For the cytochromes P450, similar kinetics would be observed irrespective of access to the active site from the membrane or from the cytosol. The decrease in intracellular concentration (free and total) would depend on the rate of metabolism relative to the rate of equilibration. This is also true for efflux transporters that transport drug directly from the cytosol. However, if transport occurs from the plasma membrane, dramatic differences in intracellular concentrations can be observed depending on 1) the localization of the transporter (apical or basolateral membrane) and 2) the membrane exposed to the drug. Thus, apical drug exposure to a membrane with an apical efflux transporter (e.g., Pgp at the blood-brain barrier) will result in dramatic decreases in intracellular concentrations. In contrast, basolateral drug exposure would result in minimal changes in intracellular concentrations (e.g., Pgp in the liver) (Agarwal et al., 2011; Korzekwa et al., 2012).
Looking Inside the Black Box.
Using in vitro data to predict in vivo responses requires knowledge of unbound concentrations at the target site both in vitro and in vivo. For compounds with poor permeability, high transporter activity, or high metabolic activity, the unbound intracellular concentration may be different from the unbound extracellular concentration. Although the details are outside the scope of this commentary, membrane permeability and partitioning are important determinants of free extracellular, free intracellular, and total cellular concentrations. Therefore, although a black-box approach (i.e., ignoring the mechanism of drug sequestration) can adequately describe free fraction, a mechanistic approach is necessary to describe cellular disposition (e.g., free intracellular concentration).
In summary, membrane partitioning and not nonspecific protein binding is the primary mechanism of drug sequestration. Therefore, physicochemical properties (e.g., LogP) can be used to predict drug sequestration in membrane and cell-based assays. The concentration of drug in a membrane is determined by both the rate in and out of the membrane. In contrast, membrane permeability is a function of the rate in only. Understanding these relationships can result in better in vitro protocols, improved models for prediction, and ultimately more rapid drug design and optimization.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Nagar and Korzekwa.
Acknowledgments
We thank Drs. Donald Tweedie and Upendra Argikar for their careful review of this manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
ABBREVIATIONS:
- ER
- endoplasmic reticulum
- LFER
- linear free-energy relationship
- CLi
- clearance into the membrane
- CLo
- clearance out of the membrane
- Pgp
- P-glycoprotein.
- Received May 2, 2012.
- Accepted June 18, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}