


Abstract
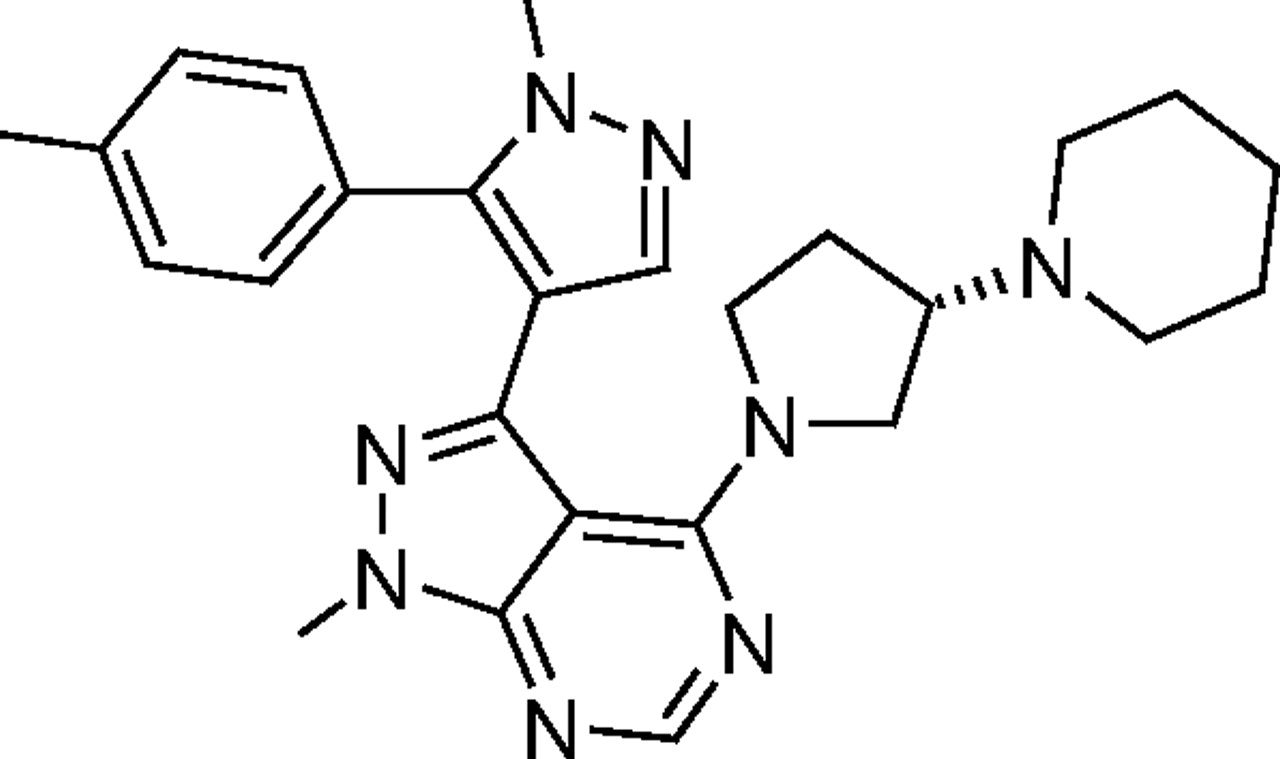
CYP3cide (PF-4981517; 1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine) is a potent, efficient, and specific time-dependent inactivator of human CYP3A4. When investigating its inhibitory properties, an extreme metabolic inactivation efficiency (kinact/KI) of 3300 to 3800 ml · min−1 · μmol−1 was observed using human liver microsomes from donors of nonfunctioning CYP3A5 (CYP3A5 *3/*3). This observed efficiency equated to an apparent KI between 420 and 480 nM with a maximal inactivation rate (kinact) equal to 1.6 min−1. Similar results were achieved with testosterone, another CYP3A substrate, and other sources of the CYP3A4 enzyme. To further illustrate the abilities of CYP3cide, its partition ratio of inactivation was determined with recombinant CYP3A4. These studies produced a partition ratio approaching unity, thus underscoring the inactivation capacity of CYP3cide. When CYP3cide was tested at a concentration and preincubation time to completely inhibit CYP3A4 in a library of genotyped polymorphic CYP3A5 microsomes, the correlation of the remaining midazolam 1′-hydroxylase activity to CYP3A5 abundance was significant (R2 value equal to 0.51, p value of <0.0001). The work presented here supports these findings by fully characterizing the inhibitory properties and exploring CYP3cide's mechanism of action. To aid the researcher, multiple commercially available sources of CYP3cide were established, and a protocol was developed to quantitatively determine CYP3A4 contribution to the metabolism of an investigational compound. Through the establishment of this protocol and the evidence provided here, we believe that CYP3cide is a very useful tool for understanding the relative roles of CYP3A4 versus CYP3A5 and the impact of CYP3A5 genetic polymorphism on a compound's pharmacokinetics.
Introduction
Pharmacokinetic modeling based on in vitro data has led to improvements in the prediction of the metabolic fate of drugs eliminated primarily via oxidative metabolism (Rostami-Hodjegan and Tucker, 2007). A key requisite to the success of such approaches is the ability to determine the quantitative contribution of individual cytochrome P450 (P450) isoforms to the metabolism of a drug. Such P450 reaction phenotyping experiments have now become standard practice in the evaluation of any new chemical entity believed to be metabolized by the P450 enzymes (Harper and Brassil, 2008; Emoto et al., 2010). Of the P450 enzymes, the CYP3A family is the most abundant and is responsible for the metabolism of a wide range of xenobiotics (Shimada et al., 1994; Wilkinson, 1996; Paine et al., 1997; Thummel and Wilkinson, 1998; Bu, 2006; Niwa et al., 2008). Four members of the family have been reported in humans (CYP3A4, CYP3A5, CYP3A7, and CYP3A43) (Daly, 2006). CYP3A4 is generally thought to be the predominant form expressed in the liver and intestine (Paine et al., 1997; von Richter et al., 2004; Kawakami et al., 2011) and is the most important enzyme in drug metabolism. CYP3A5 is a polymorphically expressed enzyme with the frequency of the wild-type, CYP3A5*1, being only 5 to 15% in the white population, 25 to 40% in various Asian ethnic groups, and approximately 40 to 60% in Africans and African Americans (Kuehl et al., 2001; Daly, 2006). Thus, a high proportion of white people do not express functional CYP3A5. CYP3A7 is predominantly fetal, but expression in adult human liver has also been measured (Sim et al., 2005), and CYP3A43 is expressed at very low levels (Daly, 2006).
The clinical impact of the CYP3A5 polymorphism is ambiguous, with some CYP3A substrates reportedly affected by the CYP3A5 genotype such as tacrolimus (Barry and Levine, 2010), verapamil (Jin et al., 2007), and cyclosporine (Zhu et al., 2010) and others not affected such as midazolam (Fromm et al., 2007), alfentanil (Kharasch et al., 2007), and nifedipine (Fukuda et al., 2004). The CYP3A5 polymorphism is also a likely contributor to the large interindividual variability in pharmacokinetics observed for some CYP3A substrates. CYP3A5 demonstrates 84% similarity to CYP3A4 in terms of amino acid sequence, and there is a considerable overlap in substrate specificity for the two enzymes (Wrighton and Stevens, 1992; Liu et al., 2007). This makes it difficult to distinguish the enzymatic contribution of CYP3A4 from that of CYP3A5 in vitro and therefore challenging to predict the clinical impact of the CYP3A5 genotype on the pharmacokinetics of a CYP3A substrate. Because many drugs are metabolized by CYP3A (Rendic, 2002; Williams et al., 2004), it would be extremely beneficial to have the appropriate in vitro tools to understand the relative roles of CYP3A4 and CYP3A5 in the metabolism of various drugs and to quantitatively predict the impact of genetic polymorphism, if any, on their pharmacokinetics.
The objective of this study was to seek a compound that would possess adequate selectivity for inhibition of CYP3A4 relative to CYP3A5 (or vice versa) that could be used as an in vitro tool to distinguish the relative contributions of these two closely related enzymes to the metabolism of drugs. We began with a high-throughput screen of our compound libraries using recombinant (r) human CYP3A4 and CYP3A5 in a midazolam 1′-hydroxylase activity assay. Initial leads showing promise of selectivity were further examined, and close-in analogs of these were also tested. This yielded 1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine(PF-04981517; CYP3cide; Fig. 1), a CYP3A4 selective compound that showed the greatest promise of all compounds tested. Herein described is a detailed characterization of the inhibitory properties of this compound, exploration of its mechanism of action, and demonstration of its practical use as a reaction phenotyping tool that can delineate the relative contributions of CYP3A4 versus CYP3A5 to the metabolism of drugs in human liver microsomes.
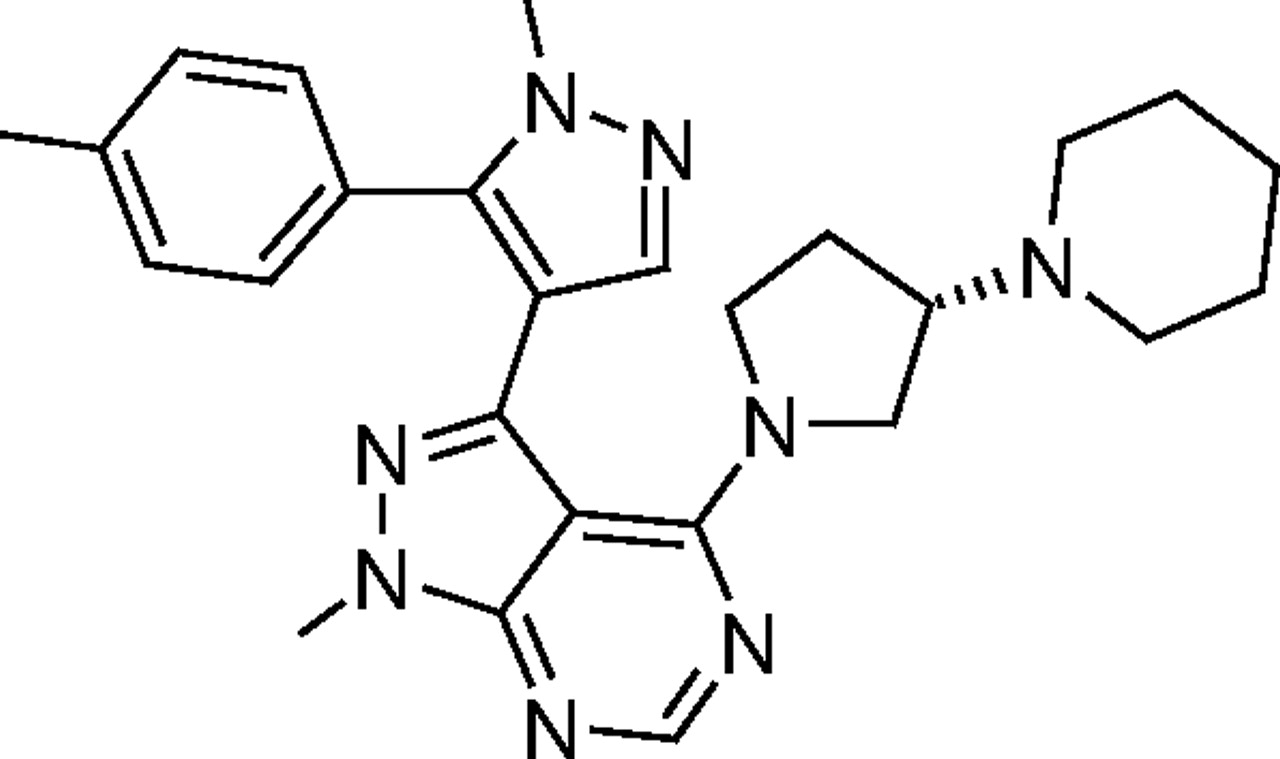
Structure of CYP3cide (1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine).
Materials and Methods
Materials.
P450 substrates, metabolites, internal standards, and incubation reagents were the same as those described previously (Walsky and Obach, 2004). CYP3cide was synthesized at Pfizer, Inc. (Groton, CT). It is now commercially available from Sigma-Aldrich (St. Louis, MO), Tocris Bioscience (Bristol, UK), or Toronto Research Chemicals Inc. (North York, ON, Canada). Pooled human liver microsomes (HLM) from a mixed pool of 50 donors and a single genotyped donor [CYP3A5 *1/*1: BD Biosciences (BD) HH47, HH86, HH103, HH104, HH107, BD HH785, BD HH860, and BD HH867; CYP3A5 *1/*3: HH1, HH2, HH8, HH9, HH48, HH80, HH89, HH90, HH91, HH92, HH100, HH108, and HH117; and CYP3A5 *3/*3: HH25, HH74, HH75, HH97, HH98, HH101, HH116, HH118, and BD HH189] were obtained from BD Biosciences (San Jose, CA). The BD designation in front of some microsomal donor samples indicates the sample has been genotyped and immunoquantified for CYP3A4 and CYP3A5 by BD Biosciences; otherwise, the donor characterizations were conducted by Pfizer, Inc. rCYP3A4 with human P450 oxidoreductase (POR) and b5, CYP3A5 with human POR and b5, CYP3A7 with human b5, and rabbit POR were also obtained from BD Biosciences. rCYP3A4, rCYP3A5, and rCYP3A7 enzymes were heterologously expressed from human cDNA using a baculovirus expression system. The rCYP3A4 protein used in the spectral binding studies was produced by Invitrogen (Carlsbad, CA) from overexpressed plasmid in Escherichia coli. Other reagents were obtained from common commercial suppliers.
Reversible Inhibition and Selectivity of CYP3cide.
The general incubation methodologies were as reported previously (Walsky and Obach, 2004). The enzyme source of these reactions was either pooled HLM, rCYP3A4, rCYP3A5, or rCYP3A7 with POR and cytochrome b5. For the recombinant enzymes, the P450/oxdioreductase/b5 ratio was calculated to be 1:5:7, 1:13:7, and 1:3:2, respectively. The P450 and POR concentrations were experimentally derived by the vendor and noted from the product specifications sheet. The cytochrome b5 concentration was calculated using an assumed specific activity of 3200 nmol of reduced cytochrome c min−1 per nmol of reductase and applying it to the reduced cytochrome c rate reported by the vendor (Yasukochi and Masters, 1976). The ratio of all three enzymes was stated using the reported P450 concentration as the denominator. P450 isoform-specific substrates testing was conducted at an experimentally derived KM value to establish IC50 values; thus, the observed results were never more than 2-fold from the Ki (Cheng and Prusoff, 1973). All IC50 determinations were repeated in duplicate. Incubation mixtures typically contained CYP3cide at concentrations of 0, 0.32, 1, 3.2, 10, 32, and 100 μM. Stock solutions of CYP3cide were prepared in 50:50 acetonitrile/water. Kinetic constants were estimated using the SigmaPlot version 12 curve-fitting software (Systat Software, Inc., San Jose, CA).
Time-Dependent Inhibition Kinetics and Selectivity of CYP3cide.
Single Point Inactivation (IC25-Shift) Experiments.
To assess whether CYP3cide exhibited time-dependent inhibition of major P450 enzymes, an IC25-shift experiment was conducted using the method of Obach et al. (2007). In this method, pooled HLM (0.1–1.0 mg/ml, depending on isozyme assessed; Table 1) were preincubated with CYP3cide at the preincubation concentrations indicated in Table 1 in the presence and absence of NADPH (1.3 mM). Preincubations (n = 2) were performed for 30 min at 37°C. After preincubation, a 10-fold dilution of the preincubation mixture (0.02 ml) was added to a mixture (0.18 ml) containing a probe P450 isozyme substrate (concentration ∼KM), incubated at 37°C, and reactions were terminated and then analyzed as described previously (Walsky and Obach, 2004; Obach et al., 2007). Results were calculated as percentage of control activity remaining and were reported as an observed decrease in enzyme activity.
Selectivity of reversible and time-dependent inhibition of human P450 enzymes by CYP3cide
Inactivation Kinetic Experiments to Determine KI and kinact.
Inactivation kinetic experiments were conducted according to methods described previously (Obach et al., 2007; Rowland Yeo et al., 2011). In the inactivation preincubation, various concentrations of CYP3cide were preincubated at 37°C with pooled HLM, genotyped individual HLM, rCYP3A4, or rCYP3A5 (0.1 mg/ml), MgCl2 (3.3 mM), and NADPH (1.3 mM), in phosphate buffer (100 mM, pH 7.4). At six time points, aliquots of the inactivation preincubation mixture (0.01 ml) were removed and added to a second mixture (0.19 ml) containing either midazolam or testosterone at concentrations approximately 10-fold the KM, MgCl2 (3.3 mM) and NADPH (1.3 mM), in phosphate buffer (100 mM, pH 7.4), at 37°C. Terminated incubation mixtures were filtered and analyzed by liquid chromatography (LC)-tandem mass spectrometry (MS/MS) for metabolites as described previously (Walsky and Obach, 2004). To determine kobs,app values, the decrease in natural logarithm of the activity over time was plotted for each inactivator concentration, and kobs,app values were described as the negative slopes of the lines. Inactivation kinetic parameters were determined using nonlinear regression of the data to the expression in eq. 1:
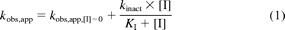 where [I] refers to the concentrations of CYP3cide in the inactivation preincubations, kobs,app are the negative values of the slopes of the natural logarithm of the percentage activity remaining versus inactivation incubation time at various [I], kobs,app,[I] = 0 is the apparent inactivation rate constant measured in the absence of inactivator, kinact is the limit maximal inactivation rate constant as [I]→∞, and KI is the inactivator concentration yielding kobs,app at the sum of kobs,app,[I] = 0 and 0.5 × kinact.
where [I] refers to the concentrations of CYP3cide in the inactivation preincubations, kobs,app are the negative values of the slopes of the natural logarithm of the percentage activity remaining versus inactivation incubation time at various [I], kobs,app,[I] = 0 is the apparent inactivation rate constant measured in the absence of inactivator, kinact is the limit maximal inactivation rate constant as [I]→∞, and KI is the inactivator concentration yielding kobs,app at the sum of kobs,app,[I] = 0 and 0.5 × kinact.
Partition Ratio Experiment.
Partition ratio experiments were conducted in a manner similar to those described previously (Silverman, 1995) using the midazolam 1′-hydroxylase KI/kinact methodology described under Inactivation Kinetics Experiments to Determine KI and Kinact and altered with a 30-min preincubation. The preincubation mixture was prepared using rCYP3A4 (0.1 mg/ml, 0.01 μM) and CYP3cide preincubation concentrations of 0, 0.000895, 0.002, 0.0045, 0.022, 0.05, 0.11, 0.25, 0.56, and 1.3 μM (n = 4). Results were calculated as a percentage of control activity remaining based on vehicle control at each CYP3cide concentration.
Biotransformation of CYP3cide.
The incubation mixture consisted of NADPH (final concentration, 1 mM) and CYP3cide (final concentration, 10 μM) in phosphate buffer (100 mM, pH 7.4). To trap potential reactive metabolites, a final concentration of 200 μM potassium cyanide (KCN) or 5 mM GSH was added to the incubation mixture. The reaction was initiated by addition of rCYP3A4 or rCYP3A5 (final concentration, 50 pmol/ml) without preincubation. The incubation mixture (1 ml) was then incubated at 37°C for 1 h. The reaction was terminated by the addition of acetonitrile (5 ml) followed by vortexing for 1 min and then was centrifuged. The supernatants were transferred into conical glass tubes for evaporation to dryness under nitrogen at 37°C. The residues were reconstituted in 0.2 ml of 30:70 (v/v) acetonitrile/water (0.1% formic acid), and aliquots (0.02 ml) were injected for LC/MS analysis.
Metabolite profiling and structure elucidation were performed using high-performance liquid chromatography (HPLC) coupled in-line with mass spectrometry detection with electrospray ionization (ESI) source in positive ion mode. The instrumental components for the methods are as follows: surveyor HPLC system and LTQ Orbitrap (Thermo Fisher Scientific, Waltham, MA), Phenomenex C18 column (100 × 2 mm; Phenomenex, Torrance, CA). The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The flow rate was maintained at 0.2 ml/min. Parent drug and its metabolites were eluted using a linear gradient in mobile-phase composition summarized as follows: equilibration for 5 min at 1% of solvent B, linear gradient to 30% of solvent B from 5 to 25 min, linear gradient to 50% of solvent B from 25 to 35 min, linear gradient to 90% of solvent B from 35 to 40 min, hold at 90% of solvent B from 40 to 43 min, linear gradient to 1% of solvent B from 43 to 45 min, and hold at 1% of solvent B from 45 to 50 min. The major operating parameters for the Orbitrap ESI-MS methods are as follows: spray voltage, 3.5 kV; capillary voltage, 33 V; tube lens, 120 V; capillary temperature, 325°C; sheath gas flow rate, 40 (arbitrary); auxiliary gas flow rate, 0 (arbitrary); resolution, 30,000; ion trap and Orbitrap maximal injection time, 500 ms; scan range, m/z 100 to 1000 atomic mass units (amu). Ion-trap LC/MSn (n = 2–4) experiments were performed to generate multistage mass spectra for selected molecular ions representing possible metabolites. At a constant pressure of 40 psi, helium was used as the damping and collision gas for all MSn experiments. Precursor isolation window, activation amplitude, activation Q, and activation time were set at 2 amu, 35%, 0.25 ms, and 30 ms, respectively.
To gather more definitive structural information for the α-cyanoamine metabolites, these were synthesized using a photochemical approach. CYP3cide (2.4 μmol) was incubated with methylene blue (0.15 μM), sodium pyruvate (2.4 μM), and KCN (4.6 μM) in methanol (0.12 ml) in front of a fluorescent lamp. The material was incubated for 4 days, during which additional aliquots of methylene blue were added as the color of the solution faded. The solution was evaporated under N2 and redissolved in 0.2 ml of CH3CN, followed by injection on preparative HPLC. Separation was effected on an Agilent Zorbax RX C8 column (9.4 × 250 mm; 5 μ) (Agilent Technologies, Santa Clara, CA) equilibrated in 0.1% formic acid containing 20% of CH3CN at a flow rate of 4 ml/min. The mobile-phase composition was increased to 50% of CH3CN at 50 min. Fractions (1 min) were collected into 0.1 ml of 10% NH4OH, and those containing the α-cyanoamine diastereomers were evaporated in vacuo for nuclear magnetic resonance (NMR) analysis.
NMR spectra of α-cyanoamine metabolites were recorded on a Bruker Avance (Milan, Italy) 600-MHz system controlled by TopSpin version 2.1, equipped with a 1.7-mm CPTCI CryoProbe. One-dimensional spectra were recorded using a sweep width of 12,000 Hz and a total recycle time of 7.2 s. The resulting time-averaged free induction decays were transformed using an exponential line broadening of 1.0 Hz to enhance signal to noise. The sample was dissolved in 0.05 ml of dimethyl sulfoxide (DMSO)-d6, 100% (Cambridge Isotope Laboratories, Inc., Andover, MA), and was placed in a 1.7-mm diameter tube. All spectra were referenced using residual DMSO-d6 (1H δ = 2.5 ppm and 13C δ = 39.5 relative to tetramethylsilane δ = 0.00).
Dialysis of CYP3cide-Treated rCYP3A4.
To confirm the irreversible character of CYP3cide inactivation, a dialysis of CYP3cide-treated rCYP3A4 was performed. A 5-min preincubation of CYP3cide (0.56 μM) with rCYP3A4 (0.1 mg/ml) and MgCl2 (3.3 mM), in phosphate buffer (100 mM, pH 7.4) in the presence and absence of NADPH (1.3 mM), was performed at 37°C, and the samples were placed on ice. Aliquots (0.5 ml, n = 2) of each preincubation mixture were transferred into Slide-A-Lyzer dialysis cassettes with a molecular weight cut-off of 10,000 (Thermo Fisher Scientific). The cassettes were dialyzed (2 × 2000 ml) overnight in phosphate buffer (100 mM, pH 7.4) at 4°C with gentle stirring. After dialysis, aliquots of dialysate (0.01 ml, n = 3) were transferred to a mixture (0.19 ml) containing midazolam (6.2 μM), MgCl2 (3.3 mM), and NADPH (1.3 mM), in phosphate buffer (100 mM, pH 7.4) and were incubated at 37°C for 6 min following the midazolam 1′-hydroxylase analysis methodology described under Inactivation Kinetics Experiments to Determine KI and Kinact and altered. Results were calculated as a percentage of control activity remaining based on vehicle controls.
CYP3cide Spectral Studies.
To understand the mechanism of inhibition, and to help assess the usefulness of the inhibitor against many CYP3A substrates, a spectral binding and a CO-reduced difference spectrum experiment was conducted. CYP3cide binding affinity for CYP3A4 was determined using purified CYP3A4. Enzymes were diluted into phosphate buffer (100 mM, pH 7.4) to a concentration of 0.5 μM and were evenly divided into 1.5-ml, 10-mm semimicro black-walled cuvettes (final volume, 0.65 ml), and a baseline measurement was acquired. A solution of CYP3cide was prepared in DMSO/acetonitrile 10:90% at various concentrations, and 1-μl aliquots were titrated into the sample cuvette (0.02–4.5 μM), with 1 μl of vehicle (DMSO/acetonitrile 10:90%) added similarly to the reference cuvette [DMSO final concentration, 0.2% (v/v)]. After mixing to a homogeneous solution, difference spectra were recorded after each aliquot between 350 and 500 nm at room temperature on an Olis Aminco DW-2000 spectrophotometer, and data were analyzed using Olis GlobalWorks version 4.3.0.116 software (Olis, Inc., Bogart, GA).
In the CO-reduced difference spectrum study, 300 pmol/ml of rCYP3A4 from BD with both oxidoreductase and cytochrome b5 was preincubated with 5 μM CYP3cide for 5 min at 37°C and then was further incubated with and without 1 mM NADPH for 15 min. Once the incubation had completed, the samples were reduced using a few grains of sodium dithionite. Aliquots of 0.65 ml of sample were placed into matching cuvettes. A baseline measurement was determined by scanning from 400 to 500 nm on a Olis Aminco DW-2000 spectrophotometer (Olis, Inc.), the sample cuvette then was bubbled with carbon monoxide for 20 s to saturate the sample. Both the reference and sample cuvettes were scanned again, and the data were analyzed via Olis GlobalWorks version 4.3.0.116 software (Olis, Inc.). The rCYP3A4 content was determined using the methods of Omura and Sato (1964a,b).
To further investigate and determine whether heme adducts were being formed, an HPLC/ESI-MS analysis of heme was conducted. Before the heme was reduced with sodium dithionite from the CO-reduced difference spectrum experiment described in this section above, an aliquot of 0.02 ml of the control and NADPH-treated incubation mixtures were obtained for testing. The incubant was introduced to a Phenomenex C18 (100 × 2 mm) LC column coupled to an integrated HPLC pumping system (Thermo Fisher Scientific). These compounds were then eluted and detected by ion trap mass spectrometer LTQ (Thermo Fisher Scientific). For positive-mode MS detection, the mobile phase consisted of solvent A and solvent B. Parent drug and its metabolites were eluted using a linear gradient in mobile-phase composition summarized in Supplemental Table 1. After passing through the diode-array detector, the HPLC effluent was introduced into the mass spectrometer. The major operating parameters for the ion-trap ESI-MS methods are viewable in Supplemental Table 2. Xcalibur software (version 2.1.0; Thermo Fisher Scientific) was used to control both HPLC and MS systems to acquire and process all spectral data. LC/MS spectra were acquired over a mass range of m/z 120 to 1500.
To test CYP3cide for metabolite-intermediate complex formation, purified CYP3A4 (2 nmol) was reconstituted with POR (2 nmol) in 0.11 ml of dilauroylphosphatidylcholine (1 mg/ml). The reconstituted mixture was added to 0.89 ml of phosphate buffer (50 mM, pH 7.4) containing 3.3 mM MgCl2 and 10 μM CYP3cide. The mixture was split between two cuvettes and placed in a Cary 4000 spectrophotometer (Agilent Technologies). NADPH was added to the reaction cuvette and was allowed to stand for 60 min. The spectrum between 400 and 500 nm was recorded.
Optimal Experimental Conditions for Use of CYP3cide as an In Vitro Tool to Distinguish CYP3A4 and CYP3A5 Metabolism in HLM.
Effect of CYP3cide in Genotyped HLMs.
To determine the differential effect of CYP3cide, a pair of genotyped CYP3A5 *1/*1 (HH47) and CYP3A5 *3/*3 (HH189) HLM tested at 0.1 mg/ml was treated with a range of concentrations of CYP3cide. The vendor reported that the genotyped pair of donors had similar total P450 abundances and also reported that HH47 contained 62.5 pmol CYP3A4/mg protein and 9.3 pmol CYP3A5/mg protein, where HH189 contained 120 pmol CYP3A4/mg microsomal protein determined via Western analysis. Each donor microsomal preparation was treated with CYP3cide using the midazolam 1′-hydroxylase KI/kinact methodology mentioned previously under Inactivation Kinetics Experiements to Determine KI and Kinact and modified with a single 10-min preincubation at eight CYP3cide concentrations (0, 0.0089, 0.02, 0.045, 0.1, 0.25, 0.56, and 1.3 μM, n = 3) in the presence and absence of NADPH (1.3 mM). After the primary reaction was concluded, an aliquot was taken and diluted 20-fold into a secondary reaction to measure loss of CYP3A enzyme activity. This secondary assay consisted of a 6-min reaction in the presence of 23 μM midazolam monitoring for 1′-hydroxymidazolam formation. Results were calculated as a percentage of control activity remaining based on vehicle controls and also as pmol of 1′-hydroxymidazolam formation per minute per pmol of total CYP3A.
Effect of CYP3cide on CYP3A4 Activity in 30 Genotyped HLM.
A set of thirty genotyped CYP3A5 polymorphs were examined. CYP3A5 *1/*1 (BD HH47, HH86, HH103, HH104, HH107 BD HH785, BD HH860, and BD HH867), *1/*3 (HH1, HH2, HH8, HH9, HH48, HH80, HH89, HH90, HH91, HH92, HH100, HH108, and HH117), and *3/*3 (HH25, HH74, HH75, HH97, HH98, HH101, HH116, HH118, and BD HH189) HLM (0.1 mg/ml) were each treated with CYP3cide (0.5 μM, n = 3) following the midazolam 1′-hydroxylase KI/kinact methodology described previously in the inactivation kinetic experiments section using a single 5-min preincubation. Similar to the reaction used to compare the genotyped microsomes, the primary reaction was diluted, and in the secondary reaction, the formation of 1′-hydroxymidazolam was monitored. Results were calculated as a percentage of control activity remaining based on vehicle controls and also as pmol of 1′-hydroxymidazolam formed per minute per mg of microsomal protein.
Effect of Protein Concentration on CYP3cide Inactivation.
The effect of increasing microsomal protein concentration on CYP3cide inactivation was determined using the midazolam 1′-hydroxylase KI/kinact methodology described above in the effect of CYP3cide on CYP3A4 activity in 30 genotyped HLM section with a single 5-min preincubation at six CYP3cide concentrations (0, 0.5, 1.1, 2.5, 5.6, and 13 μM, n = 3) at five CYP3A5 *3/*3 (HH101) HLM concentrations (0.1, 0.5, 2, 5, and 10 mg/ml) in the presence and absence of NADPH (1.3 mM). Results were calculated as a percentage of control activity remaining based on vehicle controls for each protein concentration.
Effect of CYP3cide on the Metabolism of Tacrolimus and Otenabant, Compared with Midazolam.
To evaluate a practical method for using CYP3cide for chemical inhibition phenotyping, three CYP3A-specific substrates, midazolam, otenabant, and tacrolimus, were used in a two-step time-dependent inactivation kinetic assessment. Because of the ability of CYP3cide to inactivate CYP3A4 to the IC85 in a short time period, an additional undiluted method was also investigated for compounds that have low clearance but require an assessment of CYP3A5 contributing to their metabolism.
The time-dependent experiment consisted of three legs: one with CYP3cide, a vehicle control to establish an activity benchmark, and finally one using ketoconazole as a competitive pan-CYP3A inhibitor. For the CYP3cide and vehicle legs, the first step of the assessment consisted of the inactivation reaction, where 0.085 ml of 2.4 mg/ml HLM from a single donor genotyped for CYP3A5 (i.e., CYP3A5 *1/*1, *1/*3, *3/*3) was added to 0.005 ml of 1 μM CYP3cide or vehicle in MeOH/water 10:90% (v/v), for a 500 nM final concentration into one reaction vessel and one vehicle control vessel. After 5 min of warming to 37°C, the inactivation reaction was initiated with 0.01 ml of 10 mM NADPH regeneration system (final concentration, 5 mM glucose 6-phosphate, 1 mM NADP+, and 1 U/ml glucose-6-phosphate dehydrogenase in 100 mM potassium phosphate buffer, in the presence of 1 mM MgCl2) and was delivered to the CYP3cide and the vehicle control legs. The final volume of the inactivation reaction was 100 μl. After 5 min of incubation, a 0.02-ml aliquot from the inactivation or vehicle control reaction was taken and added to 0.34 ml of the CYP3A probe substrate at a concentration of 1 μM, in addition to 0.04 ml of 1 mM NADPH regeneration system.
To establish a baseline CYP3A contribution for a test compound, ketoconazole was used as a competitive pan-CYP3A inhibitor and was compared with the CYP3A4 specific inhibitor results. A similar method was also used to test the ability of CYP3cide with low clearance test compounds in an undiluted inactivation reaction. In the ketoconazole study, 0.004 ml of 100 μM ketoconazole was introduced to 0.35 ml of 0.12 mg/ml CYP3A5-genotyped single-donor HLM and 0.008 ml of 50 μM substrate (midazolam, otenabant, and tacrolimus), and after a 5-min preincubation, 0.040 ml of NADPH regeneration system was added to initiate the reaction. The final concentration of the microsomal protein was 0.1 mg/ml.
In all assays described in this section, to ensure linearity and calculate clearance, time points were taken at 0, 5, 10, 20, 30, and 45 min. At each time point, 0.04-ml aliquots of the final reaction were taken and quenched with 0.12 ml of acetonitrile with 0.1 μg/ml internal standard. All samples were centrifuged at 3000 rpm for 10 min. Then 0.04 ml of supernatant was transferred into 0.06 ml of deionized water and was analyzed by LC/MS.
When testing CYP3cide without a dilution step, the reaction was performed similarly to those conducted with ketoconazole with the exception of a preincubation of 5 min in the presence of NADPH regeneration system before the addition of midazolam probe substrate. In brief, 0.004 ml of 25 μM CYP3cide (final concentration, 250 nM) was added to 0.35 ml of 0.12 mg/ml pooled liver microsomes with 0.04 ml of 10 mM NADPH regeneration and was preincubated for 5 min at 37°C. The reactions were initiated with 0.008 ml of midazolam. Time points, quenching, and centrifugation were the same as in the time-dependent inactivation protocol mentioned in the inactivation kinetic experiments section under Materials and Methods.
To monitor parent depletion, midazolam, otenabant, and tacrolimus were introduced to a Synergi Polar-RP (2 × 30 mm 4 μ; Phenomenex) HPLC column with a CTC PAL autosampler (LEAP Technologies, Carrboro, NC) and an integrated HPLC pumping system (Shimadzu Scientific Instruments, Columbia, MD). These compounds were then eluted and detected by an API 4000-triple quadrupole mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA) fitted with a TurboIonSpray interface. Mobile phase A was 0.1% formic acid and 2 mM ammonium acetate in water, mobile phase B was methanol with 0.1% formic acid and 2 mM ammonium acetate, and the flow rate was 0.7 ml/min. The starting condition for the HPLC gradient was 98:2 (A:B). This was held for 0.3 min. From 0.3 to 0.7 min, the mobile-phase composition changed linearly to 2:98 (A:B). This condition was held until 1.5 min. The gradient was returned in a linear fashion to 95:5 (A:B) from 1.5 to 1.6 min and re-equilibrated until 2 min. The injection volume was 0.01 ml. Table 1 lists the ionization mode, m/z transitions, and retention times for all the compounds used in this analysis. The peak area ratio of the analyte to the internal standard (Pfizer internal compound CP-628374) was determined for each injection and was used to measure the substrate remaining in the intrinsic clearance.
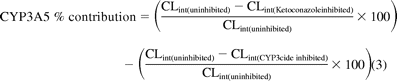
In vitro intrinsic clearance and the percentage contribution attributed to CYP3A5 was calculated using the following eqs. 2 and 3:
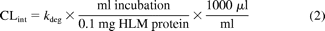
Results
Reversible Inhibition and Selectivity of CYP3cide.
CYP3cide was tested as a reversible inhibitor of several human cytochrome P450 activities in pooled HLM (Table 1). Among the seven enzymes tested, CYP3cide demonstrated only appreciable inhibition of CYP3A activities, with IC50 values ranging between 0.16 and 1.4 μM. Other enzyme activities tested had IC50 values in excess of 43 μM. Examination of rCYP3A4, rCYP3A5, and rCYP3A7 showed that CYP3cide was selective for CYP3A4. IC50 values for midazolam 1′-hydroxylase activity were 0.03 (±0.02), 17 (±2), and 70 (±30) μM for CYP3A4, CYP3A5, and CYP3A7, respectively. For testosterone 6β-hydroxylase activity,the difference between CYP3A4 and CYP3A5 was similar. (Testosterone hydroxylase activity was not tested for CYP3A7.)
Time-Dependent Inhibition Kinetics and Selectivity of CYP3cide.
CYP3cide was examined as a potential time-dependent inhibitor by incubating the compound with HLM and NADPH for 30 min before diluting the mixture 10-fold into an incubation containing marker substrates for human P450 enzymes. The precision of replicate analyses was 7.4% or less. At a concentration as high as 100 μM, no relevant (>20% decrease in activity) time-dependent inhibition was observed for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 activities (Table 1). However, for CYP3A-catalyzed midazolam and testosterone hydroxylase activities, 47 and 53% decreases were observed with preincubation of 1 and 5 μM CYP3cide, respectively. Profound time-dependent inhibition of rCYP3A4 (87%) was observed with as little as 0.05 μM CYP3cide, whereas much greater concentrations were required to generate time-dependent inhibition of rCYP3A5 (2 μM), and time-dependent inhibition was not observed for rCYP3A7.
Inactivation kinetic parameters were measured for CYP3cide in HLM preparations from an individual donor (BD HH189) that had been genotyped as CYP3A5*3 homozygous (i.e., lacking expression of a functional copy of CYP3A5) (Fig. 2A), rCYP3A4 (Fig. 2B), and CYP3A5 (Fig. 2C). For rCYP3A4, KI and kinact values ranged between 0.12 and 0.17 μM and 0.99 and 1.4 min−1, respectively, for the midazolam and testosterone hydroxylase activities. These values were considerably different for rCYP3A5, with a higher KI and lower kinact (Table 2). Comparing kinact/KI as a parameter of inactivator efficiency, inactivation of CYP3A4 by CYP3cide is 200- to 400-fold more efficient than what is observed with CYP3A5. Inactivation kinetic parameters measured in the CYP3A5*3 homozygous liver microsome lots yielded parameters similar to those measured for rCYP3A4 (Table 2). Using rCYP3A4, a determination of the partition ratio was made and was unity, indicating an extremely efficient inactivation (Fig. 2D, inset).

Inactivation of midazolam 1′-hydroxylase activity in a genotyped CYP3A5 *3/*3 HLM donor (HH189) (A), rCYP3A4 from BD (B), CYP3A5 from BD (C) by CYP3cide and the concentration-focused determination of CYP3cide partition ratio using rCYP3A4 from BD (D). The inset depicts the entire concentration range used in the partition ratio experiment. All determinations were a mean of replicates, except for the determination of the partition ratio (n = 4).
Inactivation kinetic parameters for CYP3cide on CYP3A activities in HLM and rCYP3A enzymes
Mechanistic Exploration of CYP3A4 Inactivation by CYP3cide.
To determine whether the time-dependent inhibition was caused by an irreversible inactivation, rCYP3A4 that had been treated with CYP3cide and NADPH were subjected to overnight dialysis (versus 2 × 2000 volumes of buffer) and were tested for residual midazolam 1′-hydroxylase activity, compared with a control in the absence of NADPH. The results showed that after dialysis the treated enzyme had only 16% of the activity of the control, indicating that the time-dependent inhibition is an irreversible inactivation. The spectral binding constant (Ks) of CYP3cide for CYP3A4 was not possible to calculate from the observed difference spectra in which no significant spin shift was observed (Schenkman et al., 1981). Examination of the CO binding difference spectrum in inactivated enzyme versus control showed a substantial decrease in the 450-nm CO binding spectrum (Fig. 3). Incubation of CYP3cide with pure CYP3A4 and NADPH did not yield an absorbance maximum at 455 to 460 nm, which can indicate the generation of a quasi-irreversible metabolite-intermediate complex (Buening and Franklin, 1976; Franklin, 1977; Delaforge et al., 1984), but the only change was an increase in the absorbance at ∼430 nm (Franklin, 1971). Finally, an analysis of inactivated enzyme by HPLC-MS showed that the porphyrin was unaltered after treatment, indicating that CYP3cide does not form a covalent adduct to the heme (Supplemental Data S1). Taken together, these data suggest that CYP3cide is a mechanism-based inactivator; however, the precise mechanism of this inactivation remains undetermined.

CO binding difference spectrum comparison of rCYP3A4 from BD in the presence and absence of CYP3cide and NADPH with a 15-min preincubation. The control is represented by a solid line, and the CYP3cide treated is represented as a dashed line.
Incubations of CYP3cide with rCY3A4 and rCYP3A5 showed that the latter catalyzed extensive metabolism, whereas much lower levels of metabolites were observed for the former. CYP3cide was primarily metabolized via hydroxylation of methylpyrazole (M1) and oxidation of 3-(N-piperidinyl)pyrrolidine portion of the molecule to yield the iminium ion (M5) (Fig. 4A). The lack of metabolites generated by CYP3A4 is consistent with the very low partition ratio for inactivation. Because of CYP3A4 producing very low levels of metabolites, CYP3A5 was used as a surrogate for CYP3A4 in metabolite trapping experiments. In these experiments, KCN or GSH was added to the enzyme incubation to trap potential reactive metabolites. Multiple cyano adducts were observed including diasteromeric α-cyano adducts of M5 (Fig. 4A), although no GSH adducts were detectable. The trapping experiments suggest that the reactive metabolite is a hard electrophile, presumably an iminium ion. A summary of the metabolic scheme can be observed in Fig. 4B. To determine whether the iminium ion was formed on the pyrrolidine or the piperidine, CYP3cide was subjected to photochemically catalyzed oxidation in the presence of cyanide. Two α-cyanoamines were formed that matched the metabolites M10 and M11 by HPLC-MS. Analysis by one-dimensional and two-dimensional NMR was consistent with these modifications being on the piperidine moiety, and the two were diastereomers (Supplemental Data S2).

a, LC/MS chromatogram of CYP3cide incubations with rCYP3A5 with and without KCN treatment and rCYP3A4 with KCN treatment. b, proposed metabolic scheme for rCYP3A5-mediated metabolism of CYP3cide.
Optimal Experimental Conditions for Use of CYP3cide as an In Vitro Tool to Distinguish CYP3A4 and 3A5 Metabolism in HLM.
The effect of CYP3cide on midazolam 1′-hydroxylation activity was assessed in liver microsomes from donors genotyped as CYP3A5*1/*1 homozygous and *3/*3 homozygous. The relationship between CYP3cide concentration in NADPH-driven reactions of HLM and the remaining midazolam hydroxylase activity, both as a percentage of control and CYP3A activity (pmol · min−1 · pmol−1) after treatment is depicted in Fig. 5 and its inset, respectively. In the microsomes from the *3/*3 homozygous donor, >85% of activity was lost when the microsomes were incubated for 10 min with 0.56 μM CYP3cide before dilution into the midazolam activity assay. However, for the *1/*1 sample, only approximately 23% of activity was lost at this concentration, showing the catalysis of midazolam hydroxylation occurring via the unaffected CYP3A5. In practice, the observed magnitude of inhibition can be compared with that from an incubation with ketoconazole, which inhibits both CYP3A4 and CYP3A5. Thus, it can be estimated that the ratio of contributions of CYP3A4 to CYP3A5 to midazolam 1′-hydroxylase in this particular donor was 1:4. The control activities for both genotyped donor microsomes, when adjusted for CYP3A abundance, displayed similar activities on the pmol · min−1 · pmol−1 level. To extend these observations and provide a correlation of the percentage of remaining activity (pmol · min−1 · mg−1) compared with the CYP3A5 abundance, liver microsomes from 30 individual donors were treated with CYP3cide at 0.56 μM, and midazolam 1′-hydroxylase activity was measured and compared with control (no NADPH in the CYP3cide preincubation). The range of relative contributions of CYP3A4 versus CYP3A5 is shown in Fig. 6A, along with genotype. Among expressers of CYP3A5 (i.e., *1/*1 and *1/*3), the range of contributions of CYP3A5 was 36 to 89%. CYP3cide decreased the midazolam hydroxylase activity in liver microsomes from homozygous *3 donors by 79 to 89%. Overall, there is overlap among *1/*1 and *1/*3 liver microsome donors, likely caused by varying expression of CYP3A5 (i.e., some CYP3A5*1/*3 heterozygous donors have very high CYP3A5 expression, compared with *1/*1 homozygous donors). Comparing the activity remaining of donors after treatment with CYP3cide with CYP3A5 abundance, a significant correlation was observed (p < 0.0001) with R2 = 0.51 (Fig. 6B).

The relationship between CYP3cide concentration and midazolam 1′-hydroxylase in HLM from donors genotyped as CYP3A5 *1/*1 (HH47) ▴ and *3/*3 (HH189) ■. Both donors were acquired from BD and have similar total P450 abundance. Inset depicts the same data as absolute activity levels. The solid lines represent the activity of CYP3A5 *1/*1 (HH47), and the dashed lines represent the *3/*3 (HH189) with and without preincubation with a range of CYP3cide concentrations.

A, impact of CYP3cide on midazolam 1′-hydroxylase activity across a range of liver microsomes from genotyped donors. Each donor microsomal preparation was tested in the primary reaction at a concentration of 0.1 mg/ml and was preincubated with 0.56 μM CYP3cide for 5 min, before an aliquot was taken and diluted 20-fold to measure the remaining midazolam 1′-hydroxylase activity. Each value represents the mean of triplicate determination. B, same activity values plotted as pmol · min−1 · mg−1 in the presence of CYP3cide versus the CYP3A5 abundance (pmol/mg) in each donor microsome lot evaluated; Pearson r value = 0.72, and R2 and p value (two-tailed) are stated in the figure.
CYP3cide is a lipophilic compound with a pKa that will render it cationic at physiological pH. Thus, it is possible that if incubated with high concentrations of microsomes, it could nonspecifically partition into microsomal phospholipid, and its apparent potency would be reduced. To test this, the inactivation was measured at varying total microsomal protein concentrations (Fig. 7). At low CYP3cide concentrations (<2.5 μM), there is diminished inactivation activity as the inactivation incubation microsomal protein concentration rises above 5 mg/ml. As CYP3cide concentrations approach 13 μM, the degree of inactivation is not affected by protein concentration.

Relationship between inhibition of midazolam 1′-hydroxylase activity and CYP3cide concentration in genotyped CYP3A5 *3/*3 HLM (HH101) at varying microsomal protein concentrations ranging from 0.1 to 10 mg/ml. Each incubation was conducted in triplicate.
From these data, the optimal condition for using CYP3cide to inactivate CYP3A4 and not affect CYP3A5 in HLM is to use inactivator concentrations up to 0.5 μM for an incubation time of 5 min while keeping microsomal protein below 1 mg/ml. This incubation mixture can then be diluted up to 100-fold into an activity assay. For test compounds that are low clearance and have a CYP3A component, an alternative method is suggested, in which the reaction is run up to 10 mg/ml microsomal protein without a dilution step (Fig. 7). The CYP3cide concentration will need to be adjusted to 5.5 μM because of microsomal binding, in which the free concentration equates to 0.5 μM free [experimental fu,mic at 0.8 mg/ml = 0.56, extrapolated using a method from Austin et al. (2002)].
Effect of CYP3cide on the Metabolism of Tacrolimus and Otenabant, Compared with Midazolam.
To test the usefulness of CYP3cide to delineate the contribution of CYP3A4 versus CYP3A5 metabolism, three drugs known to have a contribution of CYP3A5 to their metabolism were tested in the presence of CYP3cide and liver microsomes from donors possessing genotypes of *1/*1, *1/*3, and *3/*3. The effect of CYP3cide (preincubation for 5 min at 0.25 μM; CYP3A4 inhibition only) was compared with that for ketoconazole (coincubation at 1 μM for inhibition of both CYP3A4 and CYP3A5) in the same experiment to estimate the residual contribution of CYP3A5. For the *3/*3 homozygous liver microsomes, the effects of ketoconazole and CYP3cide were similar, as is expected because only CYP3A4 should be catalyzing the reaction (Fig. 8). For the other genotypes, the effect of ketoconazole was approximately equal to or greater than that for CYP3cide. The differences, attributable to residual CYP3A5 metabolism, ranged from 11 to 23% for otenabant, 15 to 56% for midazolam, and 35 to 60% for tacrolimus (Fig. 8). The ability of CYP3cide to be used in an undiluted reaction to bolster the enzyme concentration for the use with low clearance compounds was also examined. This method was tested using midazolam. Although midazolam is a high clearance compound, it provided a direct comparison with the time-dependent inactivation method using a dilution step. Compared with ketoconazole inhibition, CYP3cide differences attributable to CYP3A5 ranged from 0.4 to 30%, which was somewhat lower than that found when the time-dependent inactivation method was used with a dilution step.

Impact of CYP3cide and ketoconazole on the consumption of midazolam (A), tacrolimus (B), and otenabant (C) in HLM from donors of genotyped CYP3A5 *1/*1 (red), *1/*3 (blue), and *3/*3 (yellow). Each representative genotype consisted of three individual donors, in which the mean inhibition of each genotype is indicated by each colored bar, and the error bars indicate the S.D. of the mean for each genotype.
Discussion
CYP3A plays a dominant role among enzymes of the P450 family in the metabolism of drugs. In reaction phenotyping studies conducted in vitro, using HLM, the role of CYP3A in the metabolism of a drug is typically determined by measuring the effect of ketoconazole. This has the shortcoming that ketoconazole inhibits both CYP3A4 and CYP3A5, so the relative contributions of these two enzymes cannot be discerned. The same is true for clinical drug-drug interaction studies. Ketoconazole, typically dosed at 200 to 400 mg/day, is coadministered with a second drug, and the decrease in clearance of the second drug is interpreted as reflecting the contribution of CYP3A to its clearance. However, this also does not identify the relative importance of CYP3A4 versus CYP3A5. Because CYP3A5 is subject to a genetic polymorphism such that individuals with the *3/*3 genotype lack expression of functional enzyme, it could be proposed that the contribution of CYP3A5 to drug clearance in vivo could be delineated by comparing the pharmacokinetics of the drug in *1/*1 homozygous subjects with that in *3/*3 homozygous subjects. However, this has not been as easily accomplished as it has been for measuring the impact of genetic polymorphism of other P450 enzymes (e.g., CYP2D6 and CYP2C19) because of the contribution of the closely related CYP3A4 to the metabolism of CYP3A substrates and its high variability in expression level. Most studies comparing CYP3A5 expressers and nonexpressers have only uncovered small differences, and many of the studies have low numbers of study subjects. The experimental drug otenabant was shown to have a 3.1-fold difference in exposure between CYP3A5 *1/*1 and *3/*3 subjects, which is the largest difference reported to date (Goosen et al., 2010).
Thus, developing improved tools for this work continues to be a valuable activity. CYP3cide should serve as an important in vitro tool, which when used side-by-side with ketoconazole to inhibit all CYP3A activity will permit distinction between the contribution of CYP3A4 to CYP3A5 (as well as CYP3A7 and maybe CYP3A43). Previous reports of compounds that show a preference for inhibiting CYP3A4 relative to CYP3A5 have been made. Khan et al. (2002) and Soars et al. (2006) described mifepristone as a selective CYP3A4 inactivator using recombinant enzyme; however, when we examined this using HLM and recombinant enzymes, mifepristone did not have suitable qualities to use in reaction phenotyping of CYP3A4 versus CYP3A5 (M. A. Zientek, manuscript in preparation). Raloxifene has also been described as inactivating CYP3A4 and not CYP3A5, and an elegant explanation for this selectivity was offered based on specific protein sequence differences (presence of a key nucleophilic cysteine in CYP3A4) (Pearson et al., 2007). However, further exploration of raloxifene for reaction phenotyping purposes was not undertaken to demonstrate its potential practical utility. An experimental compound 3-((quinolin-4-ylmethyl)amino)-N-(4-(trifluoromethoxy)phenyl)thiophene-2-carboxamide (OSI-930) was also shown to inactivate CYP3A4 and not CYP3A5 (Lin et al., 2011). However, with all three of these agents, testing the putative selective inactivator was not done using HLM from CYP3A5 expressers and nonexpressers, and for any compound to be used as a selective tool for phenotyping purposes, this must be done. In our initial efforts in searching for a selective inhibitor, we frequently encountered compounds showing promise of selectivity when examined in rCYP3A4 and rCYP3A5, only to have this not be the case when further examined in HLM. This may have to do with the “artificial nature” of the recombinant system with regard to ratios of POR to P450 and the lack of competition among the P450 enzymes for limiting oxidoreductase that would occur in liver microsomes. Thus, we extensively examined the properties of CYP3cide in HLM to ascertain acceptable performance characteristics in the in vitro system intended for its primary use.
In our efforts to provide a full characterization of CYP3cide, a few surprising observations were made. One particular observation occurred during the comparison of the activity data from the many genotyped microsomal donors. Insight was gained into pmol-to-pmol activity differences of CYP3A4 and CYP3A5 found throughout donors. Assuming the immunoquantitation of the CYP3A enzymes correctly identifies active enzyme abundance, one could extrapolate that CYP3A5 is approximately 4-fold more active in HLM than CYP3A4 on a pmol-to-pmol basis for midazolam hydroxylase activity. In addition, of significance is the correlation of CYP3A5 abundance with the activity remaining monitored via 1′-hydroxymidazolam formation. These data suggest a direct connection between the percentage of remaining activity and that of CYP3A5 abundance, thus further validating the use of CYP3cide as a CYP3A phenotyping tool.
Despite several efforts to uncover the mechanism by which CYP3cide inactivates CYP3A4, this remains undetermined. It is clear that the inactivation is irreversible, as shown through an inability to recover activity after extended dialysis. The enzyme structure is perturbed because it is incapable of forming a reduced CO-difference spectrum, indicating that the heme thiolate is disrupted. However, CYP3cide does not act by forming a metabolite-intermediate complex like other amines, and it does not act by forming an adduct to the porphyrin (Foti et al., 2011). Efforts to show covalent binding to the protein have been unsuccessful, although studies to detect drug-to-polypeptide or drug to heme via LC/MS will continue to be an area of effort in our laboratories. An examination of the metabolism of CYP3cide by CYP3A5 was made to glean insights into the possible mechanism of inactivation. Metabolism of the 3-piperdinylpyrrolidine to electrophilic iminium ions (as shown through α-cyanoamine adducts) as well as oxidation of the piperidine all the way to a 1° amine dominated the metabolism. Thus, it is possible that an iminium ion or downstream metabolite of the enamine form could be the inactivating electrophile that forms an adduct that is not stable enough to be analyzed. Similar challenges were encountered when determining the mechanism of inactivation caused by phencyclidine, and various electrophilic and radical intermediates have been proposed as the inactivating species (Hoag et al., 1987; Osawa and Coon, 1989; Crowley and Hollenberg, 1995; Sayre et al., 1995; Jushchyshyn et al., 2006; Shebley et al., 2006). Further work on delineating the mechanism of inactivation of CYP3A4 by CYP3cide is in progress. Although the mechanism of inactivation of CYP3A4 by CYP3cide could not be definitively determined, its kinetic behavior clearly meets the need for its use as a selective CYP3A4 mechanism-based inactivator.
The strategic use of CYP3cide in reaction phenotyping of a new chemical entity would be preceded by a standard P450 reaction phenotyping study using a combination of measurement of metabolism in recombinant human P450 enzymes and in HLM in the presence of P450 selective inhibitors. If after these two experiments it is shown that ketoconazole inhibits metabolism in HLM and that the chemical can be metabolized by rCYP3A5, then a follow-up experiment focused on the CYP3A4 versus CYP3A5 contribution using CYP3cide should be undertaken. Our investigation led us to define the range for optimal conditions of use of CYP3cide for delineating the relative contribution of CYP3A4 versus CYP3A5. Incubation of HLM with CYP3cide (0.25–2.5 μM) and NADPH for 3 to 5 min, followed by dilution into an incubation mixture containing the new compound, and comparing that with a control (no CYP3cide) as well as comparing it to an incubation containing ketoconazole at a standard concentration (usually ∼1 μM) should yield the best estimate of the contribution of CYP3A4 versus CYP3A5 to the hepatic metabolism. A comparison was also made of the time-dependent inactivation method with a method that did not use a dilution step. The method without the dilution step was proposed to investigate compounds that have extremely low microsomal clearance and thus need very high concentrations of enzyme to discern the CYP3A4 contribution to metabolism. This method tended to predict a lower contribution of CYP3A5 to the metabolism of midazolam but on average provided a similar contribution.
In conclusion, CYP3cide, a mechanism-based inactivator of CYP3A4 that is selective for this enzyme compared with CYP3A5, has been described. CYP3cide should be useful to investigators seeking to delineate the relative contribution of CYP3A4 versus CYP3A5 in the metabolism of compounds cleared by CYP3A. Future endeavors include the use of this tool across a wide range of drugs known to be metabolized by CYP3A as well as gaining greater insight into the underlying biochemical mechanism of its inactivation.
Authorship Contributions
Participated in research design: Walsky, Obach, Hyland, Kang, Zhou, West, Geoghegan, Goosen, and Zientek.
Conducted experiments: Walsky, Obach, Kang, Zhou, West, Geoghegan, Walker, and Zientek.
Contributed new reagents or analytic tools: Geoghegan and Helal.
Performed data analysis: Walsky, Obach, Hyland, Kang, Zhou, West, Geoghegan, Walker, and Zientek.
Wrote or contributed to the writing of the manuscript: Walsky, Obach, Hyland, Kang, Walker, and Zientek.
Acknowledgments
We thank James R. Halpert and Dmitri R. Davydov (University of California at San Diego) and Bill J. Smith (Pfizer, Inc.) for stimulating conversations, helpful suggestions on experimental design, and help in planning future experiments pertaining to CYP3cide.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- P450
- cytochrome P450
- CYP3cide
- 1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine
- HLM
- human liver microsomes
- POR
- P450 oxidoreductase
- r
- recombinant
- amu
- atomic mass units
- KCN
- potassium cyanide
- MS
- mass spectrometry
- LC
- liquid chromatography
- HPLC
- high-performance liquid chromatography
- ESI
- electrospray ionization
- NMR
- nuclear magnetic resonance
- DMSO
- dimethyl sulfoxide.

- Received March 6, 2012.
- Accepted May 29, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}