


Kobayashi and associates recently reported that CYP2E1 is the major low-affinity phenacetin O-deethylase in human liver microsomes using a combination of chemical inhibition and correlation studies on human liver microsomes, and kinetic studies on recombinant human cytochromes, heterologously expressed in insect cells using a baculovirus expression system (Kobayashi et al., 1999). We previously identified and kinetically characterized five distinct cytochrome P-450 (CYP)1isoforms (2A6: Km 4098 μM; 2C9:Km 566 μM; 2C19: Km 656 μM; 2D6: Km 1021 μM; 2E1:Km 1257 μM) as low-affinity phenacetinO-deethylases, all of which have an affinity more than an order of magnitude lower than that of CYP1A2 (Km31 μM) (Venkatakrishnan et al., 1998). Simulation of the relative contribution of each isoform to the net human liver microsomal rate of phenacetin O-deethylation after correction for intrahepatic enzyme abundance suggests that CYPs 2C9 and 1A2 may play a major role at high substrate concentrations with added minor contributions of CYP2E1, 2C19, 2D6, and 2A6. The importance of CYP2C9 at higher substrate concentrations was further verified by chemical inhibition studies using the CYP2C9-selective inhibitor sulfaphenazole (Venkatakrishnan et al., 1998). This report provides further evidence for the involvement of CYP2C9 as an important human liver microsomal low-affinity phenacetin O-deethylase. We also offer an explanation for the identification of CYP2E1 by Kobayashi and associates as the major low-affinity phenacetin O-deethylase and the failure to identify a role for CYP2C9.
Although heterologously expressed forms of other CYP isoforms showed measurable phenacetin O-deethylase activity,Kobayashi et al. (1999) characterized only 1A1, 1A2, and 2E1. The stated reason was that plots of substrate concentration versus reaction velocity were linear. This finding is explained by the use of 1 mM as the highest concentration of substrate, which is less than 2-fold higher than the Km values of CYPs 2C19 and 2C9, and below the Km for CYPs 2A6 and 2D6, based on our previously published study (Venkatakrishnan et al., 1998).
Although fluvoxamine is a potent inhibitor of human liver microsomal CYP1A2 (Brøsen et al., 1993; von Moltke et al., 1996a), fluvoxamine is not by any means a specific inhibitor of this isoform. Fluvoxamine is an equipotent inhibitor of CYP2C19 [IC50 value of 0.24 μM versus S-mephenytoin 4′-hydroxylation (von Moltke et al., 1999) and Ki value of 0.7 μM versus formation of cycloguanil from proguanil (Rasmussen et al., 1998)] and can cause clinically significant drug interactions with substrates that are primarily metabolized by this isoform. In addition, fluvoxamine also inhibits CYPs 2C9 (Ki value of 6 μM versus phenytoin p-hydroxylation (Schmider et al., 1997) and 13 μM versus tolbutamide 4′-hydroxylation and (S)-warfarin 7-hydroxylation (Hemeryck et al., 1999) and 3A4 [Ki values ranging from 5 to 20 μM for the parallel 1- and 4-hydroxylation pathways of alprazolam and triazolam biotransformation (von Moltke et al., 1995, 1996b)], both of which are high-abundance human liver microsomal isoforms, and causes clinically significant alterations in the pharmacokinetics of co-administered substrates of these isoforms. Another example is the effect of fluvoxamine on fluoxetine metabolism (von Moltke et al., 1997). Fluoxetine N-demethylation is not catalyzed by CYP1A2, as evident from the lack of metabolite formation by cDNA-expressed CYP1A2 and the lack of inhibition by both α-naphthoflavone and furafylline (von Moltke et al., 1997). Interestingly, fluvoxamine at a concentration of 10 μM inhibited the reaction by 40% in human liver microsomes, with a Ki value of 5 μM (von Moltke et al., 1997).
It is thus likely that the rates of phenacetinO-deethylation measured in the presence of 10 μM fluvoxamine do not accurately reflect the low-affinity (non-1A2) component of this reaction, because fluvoxamine inhibits CYPs 2C19 and 2C9 as well, both of which are phenacetin O-deethylases with affinities greater than that of CYP2E1. We tested this hypothesis using heterologously expressed forms of all the phenacetinO-deethylases. At a substrate concentration of 500 μM and inhibitor concentration of 10 μM, fluvoxamine almost completely inhibited CYP1A2- and 2C19-mediated phenacetinO-deethylation, and also significantly inhibited CYP2C9 (60% inhibition), confirming the lack of specificity of fluvoxamine towards CYP1A2 (Fig. 1). Interestingly, 2D6 and 2E1 were the only isoforms that were not inhibited by fluvoxamine. von Moltke et al. (1995) reported a fluvoxamineKi of 17 μM versus desipramine hydroxylation in vitro. At a substrate concentration of 500 μM, simulation analyses predict only a minor role for CYPs 2D6 and 2A6 in phenacetinO-deethylation (Venkatakrishnan et al., 1998). It is thus not surprising that the residual activity in the presence of fluvoxamine was aniline inhibitable, correlated well with measures of CYP2E1 activity, and did not correlate with measures of CYP2C9 activity.

Inhibitory effects of 10 μM fluvoxamine (FVX) and 2 μM α-naphthoflavone (ANF) on phenacetin O-deethylation by lymphoblast-expressed human CYP isoforms.
The substrate concentration used was 500 μM for fluvoxamine inhibition and 1000 μM for inhibition by α-naphthoflavone.
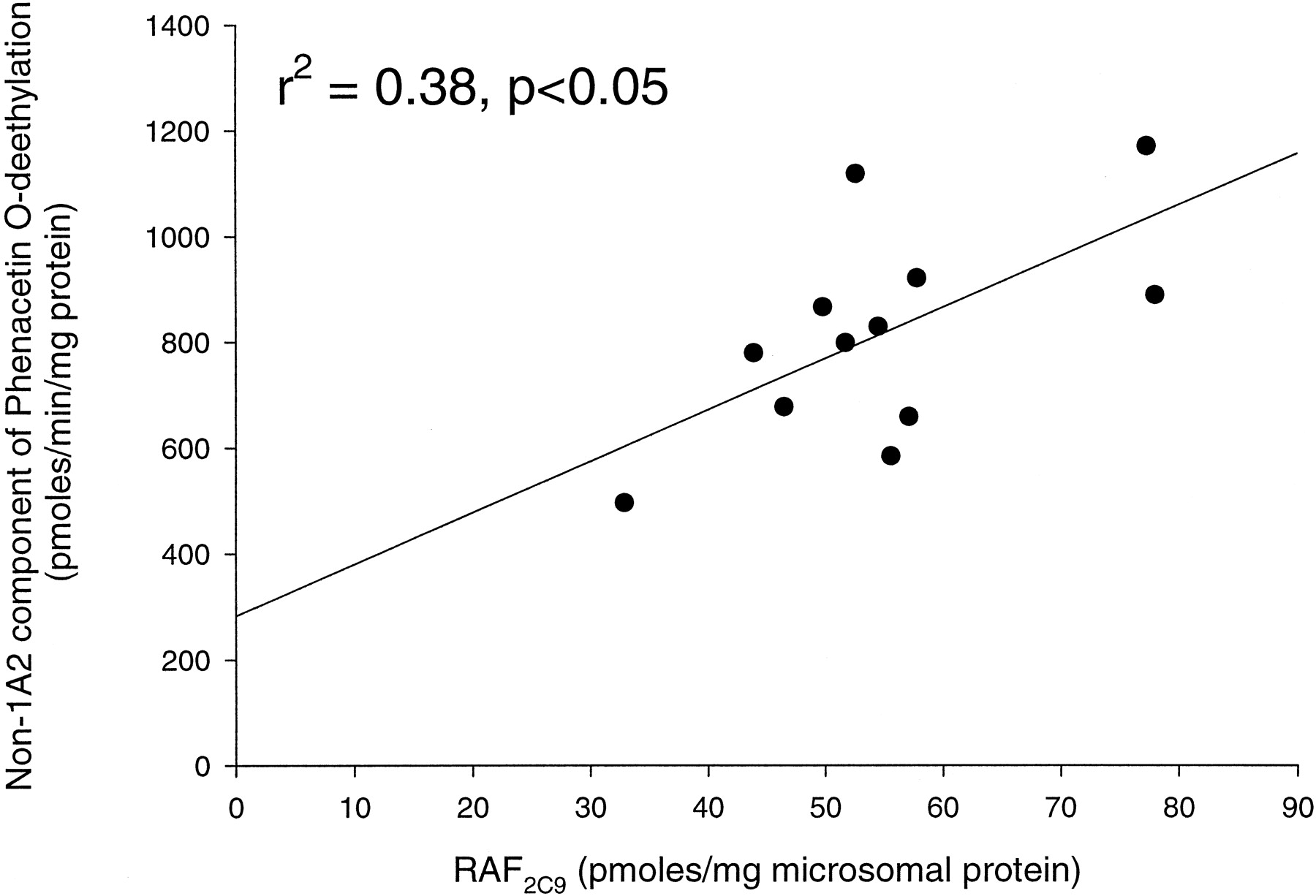
At a concentration of 2 μM, α-naphthoflavone, had no effect on the rates of phenacetin O-deethylation by CYPs 2A6, 2C9, 2C19, 2D6, and 2E1, but almost completely inhibited CYP1A2 (IC500.16 μM), validating its use as a potent CYP1A2-specific inhibitor. We have isolated the low-affinity component of phenacetinO-deethylation as the α-naphthoflavone-inhibitable component (using a substrate concentration of 1000 μM and inhibitor concentration of 2 μM) in a panel of 12 human liver microsomes. A significant correlation (r2 = 0.38,p < .05) was observed between the low-affinity component of phenacetin O-deethylation and the relative activity factor for CYP2C9 determined using flurbiprofen 4′-hydroxylation as the index reaction (Fig.2). This further emphasizes the importance of CYP2C9 as a low-affinity human liver microsomal phenacetin O-deethylase.

Correlation of the α-naphthoflavone-resistant (non-CYP1A2) component of phenacetin O-deethylation with CYP2C9 activity (relative activity factor determined using flurbiprofen 4′-hydroxylation) in a panel of 12 human liver microsomal preparations.
Although diethyldithiocarbamate (DDC) is a relatively specific CYP2E1 inhibitor, it has been shown to inhibit CYPs 2C9 and 3A as well, at the concentrations used in the study by Kobayashi and associates (Eagling et al., 1998). Inhibition by 100 to 1000 μM DDC is nonspecific and does not imply a role for CYP2E1, especially when viewed in the light of the lack of inhibition by an inhibitory CYP2E1 antibody. In fact, Eagling et al. (1998) have shown that testosterone 6β-hydroxylation (an index of CYP3A activity) is inhibited to an extent similar to that observed by Kobayashi et al. (1999) at the concentrations of DDC used in their study.
When used at a concentration under 100 μM, phenacetinO-deethylation is a valid index of CYP1A2 activity in vitro. Fluvoxamine cannot be used as a CYP1A2-specific inhibitor in human liver microsomes. The use of nonspecific chemical inhibitors can lead to the misidentification of the major CYP isoforms catalyzing a reaction, especially when the pathway of interest is catalyzed by multiple enzymes. For some CYP isoforms, specific chemical inhibitors are not available, CYP2B6 being an example. In such cases, it has been necessary to resort to nonspecific inhibitors such as orphenadrine, until the recent introduction of a CYP2B6-specific inhibitory monoclonal antibody by Gentest corporation. Fortunately, two highly specific inhibitors of CYP1A2 are available, namely α-naphthoflavone and the mechanism-based inhibitor furafylline. Both inhibitors have been validated for their specificity and potency in several studies independently (Newton et al., 1995; Bourrié et al., 1996; Ono et al., 1996; von Moltke et al., 1996a). The lack of specificity of fluvoxamine towards CYP1A2 has also been demonstrated by several laboratories, and is evident from the data presented here. Fluvoxamine is an equipotent CYP2C19 inhibitor, and also a moderate inhibitor of CYPs 2C9 and 3A4.
Footnotes
- Abbreviations used are::
- CYP
- cytochrome P-450
- DDC
- diethyldithiocarbamate




{kind=link}
{kind=link}