


Abstract
The distribution of a drug within the body should be considered as involving movement of unbound drug between the various aqueous spaces of the body. At true steady state, even for a compound of restricted lipoidal permeability, unbound concentrations in all aqueous compartments (blood, extracellular, and intracellular) are considered identical, unless a compartment has a clearance/transport process. In contrast, total drug concentrations may differ greatly, reflecting binding or partitioning into constituents of each compartment. For most highly lipid permeable drugs, this uniform unbound concentration is expected to apply. However, many compounds have restricted lipoidal permeability and are subjected to transport/clearance processes causing a gradient between intracellular and extracellular unbound concentrations even at steady state. Additional concerns arise where the drug target resides in a site of limited vascularity. Many misleading assumptions about drug concentrations and access to drug targets are based on total drug. Correction, if made, is usually by measuring tissue binding, but this is limited by the lack of homogenicity of the organ or compartment. Rather than looking for technology to measure the unbound concentration it may be better to focus on designing high lipoidal permeable molecules with a high chance of achieving a uniform unbound drug concentration. It is hoped this paper will stimulate greater understanding of the path from circulation to cell interior, and thereby in part avoid or minimize the need to provide the experimentally very determining, and sometimes still questionable, answer to this problem.
Introduction
It is recognized that unbound drug is in equilibrium with on and off targets that define the pharmacology and toxicology of a drug. The concentration of unbound drug could potentially differ from that present in the circulation where a distribution barrier such as a cell membrane needs to be crossed. Most methods measuring the distribution of drug rely completely or partially on assay of the total drug. Reliance on these methods may be widely misleading in understanding drug action.
Access to the external surface of cells is generally available to most drug types due to the relative leakiness of the vascular endothelium. This leakiness is provided by the junctions between cells, which can be viewed as aqueous pores. Certain organs such as the brain and testes have vasculature with much tighter junctions that restrict the diffusion of even relatively small molecules via these aqueous pores. Rapid passive transfer into these organs can only occur by crossing the cells of the endothelium of the vasculature by lipoidal diffusion. Because this transfer involves passage into and out of cells, measurements of drug penetration into organs such as the brain can be related to other cell types to understand intracellular concentrations, even if the measurement is not actually intracellular [e.g., drug concentration in cerebrospinal fluid (CSF)]. Although the external cell surface provides a rich source of drug targets (G-protein–coupled receptors, ion channels, etc.) that can be accessed from the interstitial fluid bathing the cells, many drug targets, off targets, and enzymes are located intracellularly (nuclear hormone, cytochrome P450, etc.) or have a key binding site accessed from the cytosol (tyrosine kinases). Understanding what concentrations of drugs are present inside cells, and more importantly the unbound concentrations, would provide enormous clarity to drug effects on these types of targets. This mini-review examines the state of knowledge and common understanding around intracellular drug concentration and whether technology can provide relevant answers.
Plasma Protein Binding and Unbound Drug In Vivo.
A key driver of intracellular concentration in vivo must be the unbound drug present in the circulation. Unfortunately, this subject is greatly misunderstood in the scientific community. Prestige publications in high-impact international journals still carry a message that plasma protein binding determines the unbound concentration in the circulation. Anecdotal collection of publications and presentations would indicate that over one-half of the drug research scientific community has been misled. Statements highlighting this problem include “Plasma proteins, by virtue of their high concentration, control the free drug concentration in plasma and in compartments in equilibrium with plasma, thereby, effectively attenuating drug potency in vivo” (Trainor, 2007). The truth is otherwise: following repetitive oral dosing of drugs, for a given rate of entry, the therapeutically important average unbound plasma concentration at steady state, or the area under the unbound drug plasma concentration curve (AUCu), is determined by intrinsic clearance, the unbound intracellular parameter that controls the rate of elimination at the site(s) of elimination, with plasma protein binding playing no part (Benet and Hoener, 2002; Smith et al., 2010). In the simplest case of oral drugs cleared purely by metabolism the AUCu is defined only by the fraction absorbed (Fabs), the fraction escaping gut first pass metabolism (Fgut), the dose, and the intrinsic clearance (Clint): AUCu = Fabs. Fgut. Dose/Clint. Supporting this expectation, for example, no drug interaction purely due to displacement of protein binding of an orally administered drug has been found to cause either an elevation of average unbound plasma concentration or a sustained increase in clinical effect (Benet and Hoener, 2002), with the rare exception of high extraction ratio drugs given parentally (Benet and Hoener, 2002).
Total Drug and Unbound Drug in Cells: What Can Be Learned from In Vitro Studies?
Mateus et al. (2013) studied the intracellular concentration of drugs in vitro using a homogenization method and human embryonic kidney (HEK) 293 cells. Tissue partioning (the tissue/medium concentration ratio) correlated with molecular charge, increasing from a mean of 7 for the negatively charged compounds, to 47 for the neutrals, and 74 for the compounds with a positive charge (relative to that at pH 7.4). These values reflected large differences in binding to cell constituents. When unbound concentration was considered a mean tissue water/medium concentration ratio [i.e., unbound drug in brain/unbound drug in plasma partition coefficient (Kpuu)] of 1.2 was established for the uncharged compounds, as expected for passive diffusion of unbound drug. The negatively charged compounds (acids) had a mean Kpuu value slightly lower (0.8), while a higher mean Kpuu value was observed for the positively ionizable (bases) compounds (4.0). The higher value of Kpuu for the positively charged compounds is consistent with trapping of charged species in acidic subcellular compartments, such as endosomes and lysosomes (pH approximately 5), while the lower-than-unity value for acids is expected from the pH gradient across the plasma membrane (pH 7.4 in the extracellular medium and pH 7.1 in the cytosol of (HEK) 293 cells). The process of lysosomal trapping (Kaufmann and Krise, 2007) is due to the increased ionization of the drug and may be considered as unbound drug. However, the higher concentrations are sequestered within the lysosome and are not representative of other parts of the cell interior, such as the cytosol. Moreover, increased ionization (or decreased zwitterionic character) may move the drug to a less active form, as exemplified by macrolide and fluoroquinolone antibiotics (Van Bambeke and Tulkens, 2001).
In Vivo Considerations around Volume of Distribution, Unbound Intracellular Concentration, and Target Access.
Like plasma protein binding, there is confusion around the term volume of distribution. Drugs of low volume are assumed to achieve restricted intracellular concentrations, or even have difficulty in cellular access, in contrast to high-volume drugs. General statements such as poor efficacy related to a low volume or toxicity related to a high volume can be heard at many drug discovery project reviews. Statements such as this also are incorporated into publications. Much of what has been described previously in Total Drug and Unbound Drug in Cells: What Can Be Learned from In Vitro Studies? for in vitro also relates to vivo (Rodgers and Rowland, 2007). If a drug moves across membranes purely by passive diffusion, then the results detailed previously for (HEK) 293 cells can be extrapolated to in vivo volume of distribution.
Table 1 details the volume of distribution at steady state (Vss) for a number of drugs. The major influence on Vss for most of the drugs is binding. Gillette (1971) defined Vss in terms of plasma volume (Vp), tissue volume (Vt), and fraction of unbound drug in tissues (fut) and unbound drug plasma (fu): Vss = Vp + (fu/fut) × Vt, which ignores transport effects. Being physical spaces, plasma volume and tissue volume are both essentially constant in any one species and the major influence on Vss is, therefore, the ratio fu/fut. Classification of the drugs in Table 1 is based on their pKa, lipophilicity, and whether a significant proportion is ionized at physiologic pH. Thus, drugs such as diazepam (basic pKa 3.4) and fluconazole (basic pKa 2.6, acidic pKa 12.7) are effectively totally unionized at physiologic pH, and so are classed as neutral. The neutral less lipophilic drugs, such as fluconazole, pyrazinamide, and isoniazid, exhibit little binding in both plasma and tissues, and thus have a Vss value close to total body water (TBW). More lipophilic neutral drugs, such as diazepam, bind to both serum and tissue proteins, and thus the Vss value reflects a comparable balance between fu and fut, whereas even more lipophilic neutral drugs, such as cyclosporine, partition extensively into fat and the lipid portion of membranes with a resultant higher Vss value (Rodgers and Rowland, 2006). The predominantly ionized acidic drugs indomethacin and ketoprofen have a large proportion bound in plasma (low fu) and low affinity for cellular constituents (high fut), and hence have a low Vss value (Rodgers and Rowland, 2006). Ionized basic drugs bind to acidic phospholipids within membranes, a binding of relatively high affinity due to a combination of hydrophobic and ion-pair interactions (Rodgers et al., 2005a,b). These drugs, such as chlorpheniramine, fluoxetine, and hydroxyzine now have a low fut value and all have a high Vss value (Table 1). The actual total drug concentration of an ionized basic drug varies widely across tissues, but this merely reflects variation in the tissue distribution of the acidic phospholipids rather than actual differences in drug properties (Rodgers et al., 2005a,b).
The pKa, log D, TPSA, and Vss values for a number of acidic, neutral, and basic drugs
These binding differences of neutral, acidic, and basic drugs produce a large range of Vss values, but have no effect on intracellular or extracellular unbound drug concentrations that are theoretically the same (or very similar) throughout body water spaces (apart from possible lysosomal accumulation for bases, as previously described). Most of the drugs listed in Table 1 have relatively high lipoidal permeability, as can be judged by their positive lipophilicity and low topological polar surface area (TPSA), but it should be noted that pyrazinamide and isoniazid (log D values of −0.7) achieve Vss of TBW. Also, despite the large range of Vss values, the same unbound drug concentrations occur across TBW spaces (identical unbound extracellular and intracellular concentrations). Proof of this is provided by analysis of CSF [e.g., indomethacin (Bannwarth et al., 1990), pyrazinamide (Phuapradit et al., 1990), and isoniazid (Holdiness, 1985)], synovial fluid [e.g., ketoprofen (Netter et al., 1987)], and vaginal fluid and saliva [e.g., fluconazole (Grant and Clissold, 1990)], in addition to plasma. Positron emission tomography scanning and displacement of probe drugs at concentrations identical to that predicted from in vitro affinity and potency measurement is available for some molecules [e.g., chlorpheniramine, Tagawa et al. (2001)]. Thus, the unbound concentrations, which interact with proteins and trigger pharmacodynamic effects, are identical to unbound drug concentrations in the circulation (see Plasma Protein Binding and Unbound Drug In Vivo). Although, as shown previously, volume of distribution plays little part in understanding drug penetration to the target, it is a key component of understanding the pharmacokinetics of a drug. Together with systemic clearance it is the determinant of drug half-life.
Impact of Steady State on Intracellular Unbound Drug Concentrations.
As discussed previously, for a lipoidally permeable drug the steady-state unbound concentrations in any aqueous compartment (blood, extracellular, and intracellular) should be identical, unless that compartment has a clearance or transport process. The total drug concentration reflects unbound concentrations plus the bound drug; however, it should be noted that the unbound drug controls the concentration of bound drug and not the other way round (Rodgers and Rowland, 2007).
Unionized drug is the prevalent form that diffuses across membranes; therefore, when considering rates of transfer, correction needs to be made for ionization influenced by different pH environments within organs and compartments (as described in vitro in Total Drug and Unbound Drug in Cells: What Can Be Learned from In Vitro Studies?). Where lipoidal permeability per se—without any other processes, such as transporters—limits a molecule’s passage into cells or across barriers is not well defined. Clearly, passage will be slower but at steady state, the authors believe a low permeability drug, subject only to passive diffusion with no other influence, should achieve unity between extracellular and intracellular concentrations. In support of this, pyrazinamide and isoniazid with log D values of −0.7 have Vss values of TBW and identical CSF and plasma concentrations in patients. This gray area of lipoidal permeability and intracellular concentration allows questionable assumptions to be made: an example is sleep disorders with beta-adrenoceptor antagonists. Lipophilic drugs such as propranolol are nonselective (interacting with β1, β2, and 5-HT receptors), whereas para-substituted (and often more hydrophilic) drugs such as atenolol are highly selective (β1). The lower incidence of sleep disorders with drugs such as atenolol is often assumed to be exclusively due to limited brain penetration; however, no significant correlations are observed between CSF concentration/β1 receptor occupancy and sleep disorders. In contrast, highly significant relationships have been observed between central and peripheral β2/central 5-HT receptor occupancies and sleep disorders (Yamada et al., 1995).
Permeability is a measure of the velocity of movement of a compound through a membrane, thus the extent of tissue distribution is time dependent. A revealing in vivo study on the balance between permeation and time was reported by Abrahamsson et al. (1989). Metoprolol log D0.1 and atenolol log D−2.0 were given to Beagle dogs as single and multiple doses The concentration of atenolol in CSF compared with plasma had a delayed Cmax and a slower decline, indicative of its intrinsic low permeability. The CSF/plasma concentration ratio increased during repeated drug administration from 0.48 ± 0.12 on day 1 to 0.83 ± 0.14 on day 7. It is likely that the ratio will never reach unity, due to a combination of efflux by P-glycoprotein (P-gp) at the blood-brain barrier (BBB) and CSF flow acting as a clearance pathway (Iliff et al., 2012). In contrast, the CSF concentration of the more lipophilic analog metoprolol was the same as the unbound concentration of the drug in circulating plasma at every time point throughout the study. Although this study concerns penetration into the CSF it applies to other cell membrane systems, such that over time (steady state) most small molecules of even modest lipophilicity and lipoidal diffusion are expected to penetrate into cells by passive diffusion to achieve identical extracellular and intracellular unbound unionized concentrations. This is rendered more complex since transporters tend to yield the most effect on the disposition of moderate or low lipoidally permeable drugs; therefore, finding examples of low permeability, nontransported drugs is difficult. In the case of atenolol (normally considered passive to transporters) the relatively low penetration observed in rat into the central nervous system (CNS) (albeit over a short infusion regimen) has been explained by possible transporter efflux (Chen et al., 2017).
Unbound Drug in Cytosol and Target Exposure: Role of the Membrane.
A number of different drug targets and proteins involved in drug disposition are quoted as being accessed from the cell membrane rather than the cytosol. If this was the general case, then thoughts on intracellular or intraorgan unbound drug concentrations would need to be modified. P-gp is a quoted example with statements appearing in the literature such as “It is known that P-gp binds its substrates in the cytoplasmic membrane leaflet of apical membranes.” These often reference a landmark study by Dey et al. (1997), which in fact stated “The On-site is closer to the cytosolic phase of the membrane to recruit drug molecules from the cytosol or from the inner leaflet of the lipid bilayer. Movement of the drug substrate from the ON-site to the OFF-site is unfavorable and rate-limiting for the drug to be translocated, but can be driven by the large free energy change that occurs during ATP hydrolysis.” Studies including X-ray crystallography (Aller et al., 2009) show a chamber (with multiple binding sites) open to the cytoplasm, in substrate binding mode, and the inner leaflet of the cell membrane. Regardless of the actual route of active site access, an equilibrium concentration will be reached between the inner leaflet and the cytoplasm that ultimately is controlled by unbound concentrations in the circulation. The open nature of the binding site and ready cytoplasmic access is demonstrated by quaternary substrates and the use of inside-out vesicles; which measure accumulation in the cell via P-gp or similar transport proteins of the substrate. Examples of these include quaternary derivatives of substrate drugs such as propafenone, quinidine, and quinine, which accumulate by eliminating the passive membrane partitioning and diffusion due to the permanent positive charge. These substrates accumulated in P-gp containing inside-out vesicles and the accumulation could be inhibited by cyclosporine A. Moreover, there was a lack of accumulation in inside-out vesicles in the absence of ATP. Clearly, the active site in these models was assessed via the solvent and not the membrane (Schmid et al., 1999; Hooiveld et al., 2002). Membrane affinity and possible passage to the active site of the pharmacological target have also been suggested for some drugs, such as amlodipine, with an intrinsic long duration of action (slow receptor off rate). Again, quaternary derivatives supply strong evidence of solvent rather than membrane access to the protein target: quaternary amlodipine cannot penetrate the membrane and access to the binding site of the calcium channel is restricted to the aqueous channel pore. Quaternary amlodipine shows the same slow off-rate kinetics as amlodipine (Kwan et al., 1995), indicative of a pure receptor effect and one discrete from membrane affinity. It can be assumed in most cases that the active site of drug targets, and systems that change the disposition of drugs (metabolizing enzymes and transporters), are either accessed directly from the aqueous media or from a membrane location in equilibrium with it.
Unbound Drug in Homogenous Organs and Cells.
Probably the most studied organ for unbound drug concentration content is the brain (Hammarlund-Udenaes, 2010). The BBB is formed by the very tight junctions in the vascular endothelium, meaning that the passage of a drug molecule into the brain is determined by pure lipoidal and transporter influences. Compounds that rapidly cross into the CNS obey a general rule of having positive lipophilicity and <75 Å2 TPSA (Pajouhesh and Lenz, 2005). Clearly, the brain is a complex organ with different cell types, but for the purpose of this review it is relatively discrete and can be easily homogenized to a fairly consistent degree. When the homogenization approach is compared with other methods (Table 2) there is generally a high consistency of results. Adding to the definition homogenous is that the BBB represents a fairly severe test of lipoidal diffusion, and that unbound drug distribution across the cells (both the vasculature and brain) should be fairly uniform. This allows readily crude techniques such as homogenization followed by dialysis to yield consistent estimates of the unbound concentration of drug in brain. The most used preclinical methods of microdialysis, homogenization and dialysis, and CSF sampling show reasonable agreement and tend to cross validate each other (Liu et al., 2009). They, of course, make the reasonable assumption that passage across the BBB equates to passage across the membranes of different cell types in the brain.
Methods and techniques used to study unbound drug concentration in the brain
Although physical chemical parameters can largely predict brain penetration (Rankovic, 2015), the interplay between intrinsic permeability and transporters will still show diversity and difficulty in predicting across a chemical series as to where the exact boundaries on H bonding and lipophilicity lie in determining free passage across the BBB (reflected by Kpuu close to unity). This is illustrated by a study of seven opioids in mouse (Kalvass et al., 2007b), in which loperamide and alfentanil showed relative exclusion from the brain, as judged by their low partitioning value and higher unbound brain EC50 values, compared with their in vitro binding against the opioid receptor. Inspection of their physicochemical properties indicated that alfentanil has a TPSA above 75 Å2 and six H-bond acceptors. Loperamide also possesses an H-bond donor group (see Table 3). Morphine with a negative log D value has associated poorer intrinsic lipoidal permeability, which translates into a slower approach to equilibrium (as discussed in Impact of Steady State on Intracellular Unbound Drug Concentrations) between plasma and brain. In the study, total plasma, total brain, unbound plasma, and unbound brain EC50 estimates were used to express opioid potency, and they were evaluated as potential surrogates for biophase EC50. Unbound EC50 values were calculated by multiplying the total EC50 by the appropriate unbound fraction value determined from equilibrium dialysis. Of these metrics, those between unbound brain EC50 and receptor binding showed the strongest relationship among the compounds. Importantly the misuse of the brain plasma partition coefficient (Kpbrain) in isolation as a measure of CNS exposure, under the assumption that larger values of Kpbrain equate with higher CNS exposure, was highlighted by the authors. The investigators pointed out that CNS drug discovery researchers had devoted much effort and resources to predicting and maximizing the Kpbrain value of drug candidates and stressed the fallacy of pursing this strategy. Even though Kpbrain values differed by more than 50-fold among the opioids examined, there was no correlation between Kpbrain and any relevant pharmacodynamic parameter. When correction was made for brain and plasma binding (Kpuu), sufentanil, fentanyl, and morphine showed that passive diffusion was the predominant driver of the equilibrium of unbound drug between brain and plasma. Loperamide, alfentanil, and to a much lesser extent methadone show restricted access, indicative of transporter influence, and confirmed by studies in cell lines and knockout mice (Kalvass et al., 2007a,c; Mercer and Coop, 2011).
Physicochemical properties of opioid agonists with different brain penetrations
Brain penetration data were obtained from Kalvass et al. (2007a,b,c).
Clearly, physicochemical properties are a very useful guide to understanding brain penetration. However, even in molecules with apparent favorable properties for lipoidal diffusion, transporter effects cannot entirely be discounted in understanding intracellular flux or concentration, as illustrated by loperamide.
The Problem with Nonhomogenous Tissues: Does the Drug Get There or Is It Ineffective When It Does?
The problem is graphically illustrated around tuberculosis (TB) drug discovery programs and even treatment. The replicating bacteria (Mycobacterium tuberculosis) are largely systemic and relatively easy to treat. However, the bacteria can reside in sanctuary sites in a nonreplicating state. The nonreplicating bacteria are resident in poorly perfused casea, the result of pathologic damage and the body’s response to this damage. These nonreplicating bacteria are largely resistant to drug therapy, hence the severe and prolonged dosage regimens used. The resistance can be ascribed to two mechanisms: persistent bacilli (resistance per se to therapy) and persistent disease due to the sanctuary of the caseum restricting access of some drugs to the bacteria (Horsburgh et al., 2015).
Attempts to measure drug in the caseum and surrounding tissue, while sophisticated, have relied on total drug measurements (Dartois, 2014). Such methods are difficult to interpret, since numerous complications will occur when comparing a caseum largely full of low protein aqueous fluid to the blood supply to cells and their content surrounding it. Because of the cell damage and invading bacteria the caseum is surrounded by macrophages, i.e., cells rich in lysosomes. Figure 1 illustrates the total drug distribution expected from the physicochemical and protein binding data of a number of drugs used in tuberculosis. Drugs that are basic at acidic pH values will concentrate in the lysosomes of macrophages surrounding the caseum, suggesting it is a barrier to free diffusion. The concept that more water soluble, neutral drugs may surmount this barrier is easily made from total drug measurements since they will appear to have uniform distribution, but this is potentially misleading. In the absence of a reliable method, an assumption that unbound drug is uniform between plasma and the aqueous content of the caseum seems valid regardless of the drug class. Different culture techniques have provided support for the persistent bacilli theory rather than the persistent disease mechanism. Under culture conditions close to the nonreplicating caseum environment (but without any barrier, thus mimicking the persistent bacilli rather than the persistent disease scenario), bacteria are at least 10-fold more resistant to drugs (rifampin, isoniazid, moxifloxacin, linezolid, bedaquiline, rifapentine, and rifabutin) than when cultured in a replicating assay (Sarathy et al., 2018).
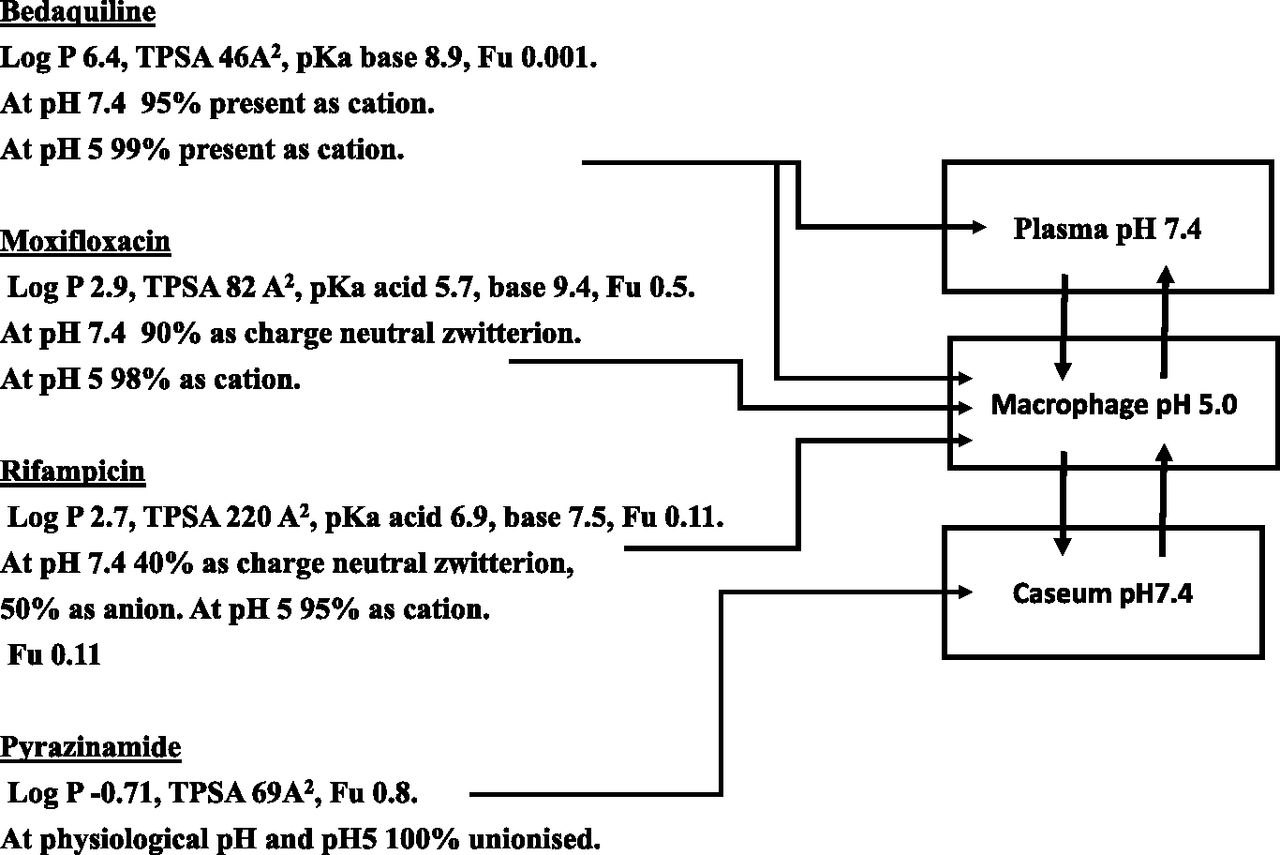
Physicochemical properties (obtained from the DrugBank database; https://www.drugbank.ca) and plasma protein binding (from the same database) dictate major locations of total drug in a tuberculosis drug program, which may not relate to unbound drug. Bedaquiline is likely to have high general tissue concentrations due to its basic nature. Bedaquiline, moxifloxacin, and rifampicin will concentrate in macrophages (lysosomes rich) since all are basic at low pH. Only pyrinzamide will appear to have reasonable concentrations in the caseum relative to its surroundings. However, unbound drug concentrations would be expected to be identical at any sampling site.
An analogous situation is seen in oncology. The blood supply of tumors is not regular when compared with normal tissue (Nagy et al., 2009). Normal vasculature is organized in evenly spaced, well-differentiated arteries, arterioles, capillaries, venules, and veins. In contrast, the tumor vasculature is unevenly distributed and chaotic, branching irregularly and forming arterio-venous shunts. Tumor blood vessels are more abundant at the interface between the host and the tumor. The amount and size of blood vessels decrease as tumors grow, leading to zones of ischemia and necrosis. Blood vessels formed by tumors have very leaky vasculature with large tight junctions. Drug delivery to tumors has been studied using the drug doxorubicin, which can be followed semi-quantitatively via its fluorescence. There is a marked fall off in fluorescence intensity in mouse breast adenocarcinoma cross section as a function of distance from the nearest blood vessel. Modeling of these data suggests that the tumor cell populations furthest from a blood vessel are the most refractory to treatment (Trédan et al., 2007). An additional factor in tumor drug access is the high interstitial pressure creating a net fluid flow from the tumor. Therefore, for drugs of limited permeability, concentration gradients could arise by this mechanism. These experiments probably do not mirror the typical steady-state situation in oncology treatment. Doxorubicin, used frequently in these experiments because of its fluorescence, is probably a drug of lower permeability with a low log P/D7.4 of 1.4/0 and high TPSA of 206 Å2. The log D value may be typical for normal tissue but the pH falls with distance from the nearest blood vessel, thus a negative log D may be more appropriate deeper into the tumor. Of high importance is that many of these types of studies, including the one highlighted, are single dose experiments with sampling over a relatively short time period (not steady state).
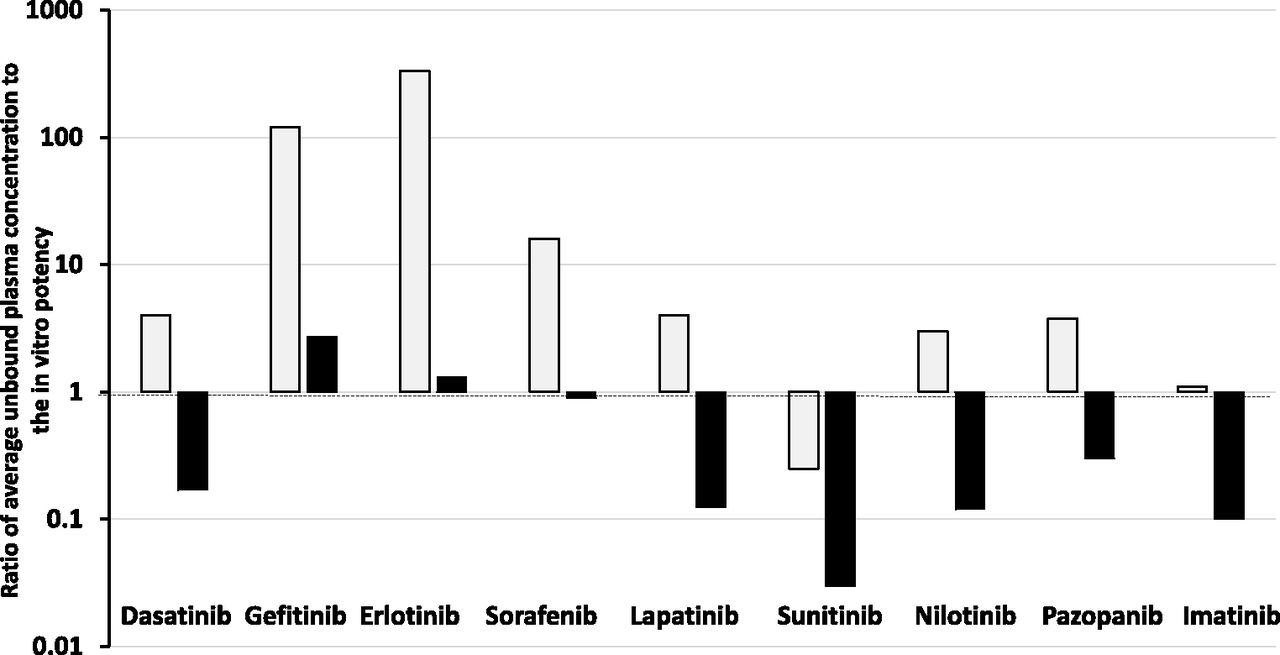
To examine if oncology drugs at steady state are more penetrant is difficult without the fluorescent properties. An attempt has been made by examination of data published by Kitagawa et al. (2013) and Hoshino-Yoshino et al. (2011) to explore if the clinical concentrations in the circulation of tyrosine kinase inhibitors (which are probably also of higher lipoidal permeability that doxorubicin) at the registered efficacious clinical doses are much higher than expected from in vitro measurement of potency (inhibition) to compensate for poor penetration into the tumor sites (non-CNS). Surprisingly, the data displayed in Fig. 2 show that in the majority of cases (66%) at the doses approved clinically the unbound average steady-state concentrations [calculated by daily plasma AUCu/24 hours corrected for fu and validated by unbound Cmax and unbound Cmin comparisons published by Kitagawa et al. (2013) and Hoshino-Yoshino et al. (2011)] were less than four times higher than the Ki or IC50 values measured in a biochemical assay, with an ATP concentration set at the Km value of the kinase. When a more physiologic ATP concentration of 1 mM was used, the majority of compounds achieved unbound plasma concentrations below the Ki or IC50 value.
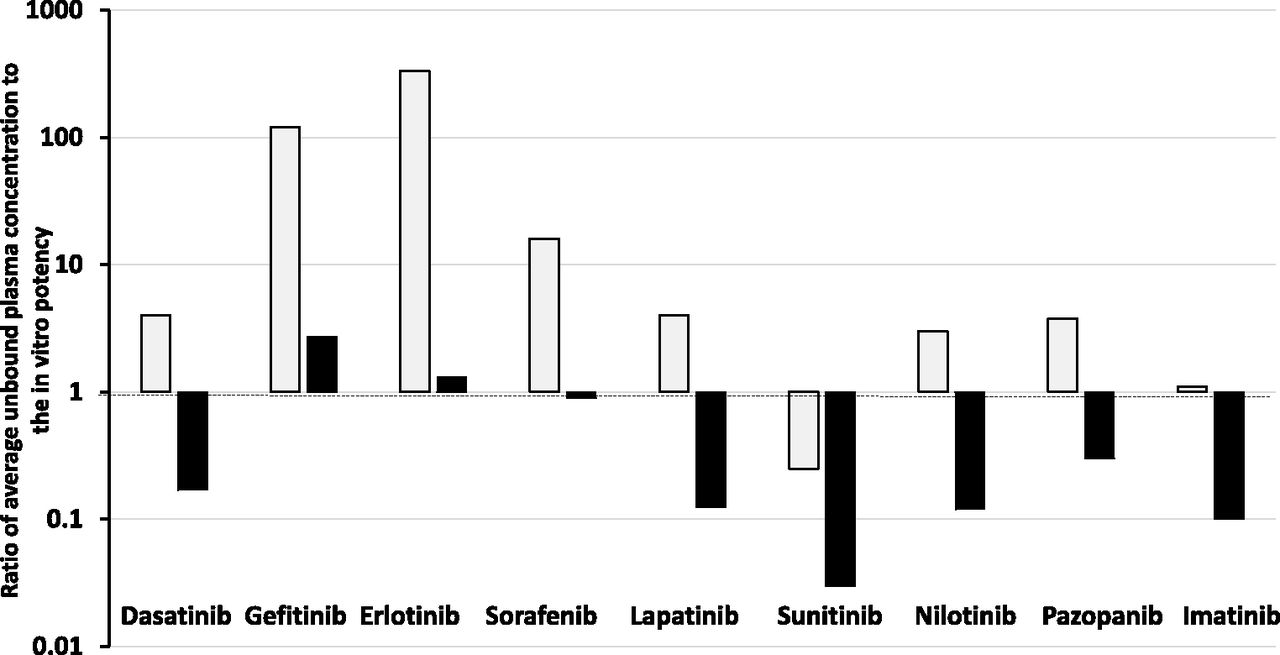
Ratio of average unbound plasma concentration at the therapeutic dose of various kinase inhibitors to their unbound in vitro potency (IC50) against the kinase at Km (open bars) and physiologic (1 mM, shaded bars) concentrations of ATP.
Many factors impact on kinase inhibitors, including the activity state of the kinase and the possibility of active metabolites, but the data discussed give no indication that oncology drugs such as kinase inhibitors need to be dosed to achieve concentrations well above in vitro estimates to compensate for tumor access in vivo. The dosage regimens approved for use could be limited by toxicity—and, therefore, are suboptimal in terms of therapeutic effect—or they could achieve and maintain concentrations of the kinase inhibitors (with greater lipoidal permeability than doxorubicin) that provide excellent drug-tumor tissue access and ultimately a uniform unbound drug concentration in circulation and tissues (TBW). As discussed previously, permeability is a measure of velocity and at steady state poorly perfused tumor water should still achieve the same unbound concentration as that in total body water (in the absence of local clearance or transport). Importantly, cell-based assays (normally considered more robust than biochemical assays) may be limited, in extrapolation to in vivo, by cell binding and/or not achieving equilibrium between the added drug and the cell interior due to the short time course of the study.
Cancer Makes the Brain Nonhomogeneous.
The BBB is an organized lipoidal barrier between the blood and the brain interstitial fluid. Disruption of this barrier happens when tumors form in the brain and trigger the growth of new blood vessels. Therefore, the blood-brain/tumor barrier (BBTB) encompasses existing and newly formed blood vessels. The high metabolic demands of high-grade glioma tumors, for instance, create hypoxic areas that trigger increased expression of vascular endothelial growth factor and angiogenesis, leading to the formation of abnormal vessels and a dysfunctional BBTB.
The BBTB is considered leaky in the core part of glioblastomas (as shown for tumor blood vessels in The Problem with Nonhomogenous Tissues: Does the Drug Get There or Is It Ineffective when It Does?). In large parts of glioblastomas, and even more so in lower grade diffuse gliomas, the BBTB more closely resembles the intact BBB (see Unbound Drug in Homogenous Organs and Cells) and prevents efficient passage of cancer therapeutics, including small molecules and antibodies. Thus, drugs can still be blocked from reaching the many infiltrative glioblastoma cells that demonstrate within-organ metastasis away from the core part of brain areas displaying a more organized and less leaky BBTB (van Tellingen et al., 2015).
Brain is a major sanctuary site for the metastases of systemic tumors. New drugs are needed in cases where the original primary cancer is cured with drugs with low CNS access but at which metastases occur subsequently in the brain. For example, patients with HER2-positive metastatic breast cancer have experienced a dramatic improvement in overall survival with HER2-targeted therapy, such as the antibody trastuzumab. The advances in systemic treatments for these patients directly lead to an increase in the rate of brain metastases. Breast cancer is a common cause of brain metastases, with these metastases occurring in at least 10%–16% of patients (Leone and Leone, 2015). To be highly effective it seems that drugs with good lipoidal permeability are necessary to access all the tumor cells. Lapatinib, a small molecule inhibitor of HER2, which clearly has more potential to cross the BBB (log D = 5.82; TPSA = 106 Å2) than an antibody, has been extensively tested in the treatment of HER2-positive brain metastases. As a single agent, lapatinib has shown response rates in the brain ranging from 2.6% to 6% in pretreated patients. When added to capecitabine, response rates increase to 20%–33%. When studied by positron emission tomography imaging 11C-lapatinib concentrations were higher in cerebral metastases than in normal brain tissue (Taskar et al., 2012), which shows some access; however, it is reasonable to question the nonuniformity as evidence of suboptimal BBB penetration. Alternatively, it could be due to different binding of the drug in brain versus tumor tissue. The different concentrations illustrate the nonhomogeneity of the brain in the disease state. When CSF was examined, lapatinib concentrations were around 0.1% of plasma concentrations, which must be considered against a plasma protein binding of 99.9% (Gori et al., 2014). Again, these data could be highly supportive of good BBB penetration, but small variations in plasma protein binding and the resultant error in estimating the unbound drug concentration could change the interpretation considerably. Similar metastatic events occur with lung cancer, with the same restrictions posed by the BBB (Preusser et al., 2018). Focusing on anaplastic lymphoma kinase positive non-small-cell carcinoma, early anaplastic lymphoma kinase inhibitors such as crizonib (log D = 3.6; TPSA = 78 Å2) are poor with highly variable activity against brain metastases (Metro et al., 2015). A large factor in this case is due to very low penetration (CSF/Cpu = 0.03, where Cpu is the unbound drug concentration in plasma). Newer generation compounds such as alectinib (log D = 5.5; TPSA = 72 Å2) with much better brain penetration (CSF/Cpu = 0.7–0.9) show promising activity in early trials (Gainor et al., 2015).
The brain represents a formidable barrier to achieving uniform unbound plasma concentrations between the organ and the circulation. It represents a lipoidal membrane barrier with high expression of efflux proteins together with a clearance pathway provided by brain fluid flow. Even when more permeable vasculature is formed by growing tumors, access to the total tumor burden will be significantly compromised by low lipoidal permeability in drugs.
Where Should New Technology Go?
The example of brain and cancer potentially can be interpreted two ways: 1) we need breakthrough new technology that can measure unbound drug in discrete locations of a population of cells of mixed function and even origin; or 2) we should just focus on basics and make the assumption that we need high lipoidal permeability in drugs and an absence (resultant) of transporter effects to ensure uniform exposure at steady state of all aqueous compartments equal to unbound drug in the circulation. The problem of new technology is revealed by work on individual cancer cells, which shows different expressions of P-gp, different intracellular concentrations, and different responses (Bithi and Vanapalli, 2017). This work used doxorubicin fluorescence to examine the single cells. Such experiments could be repeated with other drugs using single-cell technology, but the problem is how to estimate the intracellular unbound concentration. Even the latest single-cell technology that combines nondestructive and quantitative withdrawal of intracellular fluid with subpicoliter resolution using fluidic force microscopy and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry still only gives total concentration of drug and metabolites (Guillaume-Gentil et al., 2017). As soon as the actual cells being targeted are not uniform and offer individually different access to intracellular targets, the idea of technology helping to provide a global solution becomes almost fanciful. The problem becomes getting the drug to and into the worst case examples and leads to a natural conclusion: achieve the highest lipoidal permeability possible, and ensure the drug is dosed to achieve the closest to steady state as possible. With these objectives, we conjecture that in the majority of cases unbound plasma concentration will be a reasonable surrogate of the intracellular unbound concentration acting on the target. The strategy of seeking high lipoidal permeability in oncology is beginning to be embedded in the process (Zeng et al., 2015). The discovery of AZD3759 (an epidermal growth factor receptor kinase inhibitor) focused on achieving high passive permeability (29.5 × 10−6 cm/s), on not being a substrate of the efflux transporters P-gp or breast cancer resistance protein, and having a value of Kpuu > 0.5 for brain and CSF in preclinical species. Early clinical data established the concept of a brain penetrant epidermal growth factor receptor inhibitor and resultant encouraging efficacy (Yang et al., 2016; Ahn et al., 2017).
Conclusion: Mastering Complexity through Simplification
Intracellular drug concentrations to link to intracellular targets seem a desirable aspiration, but the more we ask the more complex it becomes. NMR, NMR imaging, magnetic resonance imaging, and other techniques may offer hope, but in the search for new technology remember what we take for granted is still not perfect. We struggle to measure protein binding and free fraction accurately in simple plasma once we are above 98%. The biggest problem perhaps is not the need for new technology and the perceived absence of data, but the widespread availability and misuse of total drug concentration information. Total drug measures, even if providing organ compartment values may be wildly misleading when trying to understand, for example, the concentration of drug acting on proteins (drug targets and off targets). Probably biomarker pharmacodynamics is the only (surrogate) measure that does not rely on extrapolation from homogenates. In the absence of an answer, our suggestion is simple first principles: we can be highly guided by lipoidal permeability and existing global measures such as CSF or brain Kpuu. Highly lipoidal permeable drugs at steady state will not usually have large concentration gradients across cells (apart from known pH effects); therefore, unbound drug in plasma is a reliable indicator of all aqueous unbound concentrations. Increasingly, the problem of understanding intracellular concentrations is being partly solved by in silico approaches, often physiologically based pharmacokinetic modeling in various forms. Of course, the correct principles and, where available, data sets need to be adopted to be successful. Helping achieve this is the encouraging formation of expert groups to combine expertise and share best practice and correct science (Chu et al., 2013; Guo et al., 2018; Yamamoto et al., 2018). The further challenge is to communicate this work to the wider community of scientists involved in drug research.
Acknowledgments
D.A.S. thanks ISSX for inviting and supporting him to give a presentation on the topic of this paper at the 22nd North American Meeting. The talk and the many stimulating discussions at the meeting and afterward with ISSX members, and the encouragement of Dr. Grover Paul Miller led to this final paper coauthored by M.R.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Smith, Rowland.
Footnotes
- Received December 18, 2018.
- Accepted March 22, 2019.
Abbreviations
- AUCu
- area under the unbound drug plasma concentration curve
- BBB
- blood-brain barrier
- BBTB
- blood-brain/tumor barrier
- CNS
- central nervous system
- CSF
- cerebrospinal fluid
- fu
- unbound drug in plasma
- fut
- unbound drug in tissues
- Kpbrain
- brain plasma partition coefficient
- Kpuu
- unbound drug in brain/unbound drug in plasma partition coefficient
- P-gp
- P-glycoprotein
- TBW
- total body water
- TPSA
- topological polar surface area
- Vss
- volume of distribution at steady state
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}