


Abstract
Endoxifen (ENDX) is an active metabolite of tamoxifen (TAM), a drug commonly used for the treatment of estrogen receptor–positive breast cancer and metabolized by CYP2D6. Genetic or drug-induced reductions in CYP2D6 activity decrease plasma ENDX concentrations and TAM efficacy. It was proposed that direct oral administration of ENDX would circumvent the issues related to metabolic activation of TAM by CYP2D6 and increase patient response. Here, we characterized the pharmacokinetics and oral bioavailability of ENDX in female rats and dogs. Additionally, ENDX exposure was compared following equivalent doses of ENDX and TAM. ENDX exposure was 100-fold and 10-fold greater in rats and dogs, respectively, with ENDX administration compared with an equivalent dose of TAM. In single-dose administration studies, the terminal elimination half-life and plasma clearance values were 6.3 hours and 2.4 L/h per kg in rats given 2 mg/kg i.v. ENDX and 9.2 hours and 0.4 L/h/kg in dogs given 0.5 mg/kg i.v. ENDX, respectively. Plasma concentrations above 0.1 µM and 1 µM ENDX were achieved with 20-mg/kg and 200-mg/kg doses in rats, and concentrations above 1 µM and 10 µM were achieved with 15-mg/kg and 100-mg/kg doses in dogs. Oral absorption of ENDX was linear in rats and dogs, with bioavailability greater than 67% in rats and greater than 50% in dogs. In repeated-dose administration studies, ENDX peak plasma concentrations reached 9 µM in rats and 20 µM in dogs following four daily doses of 200 mg/kg or 30 mg/kg ENDX, respectively. The results indicate that ENDX has high oral bioavailability, and therapeutic concentrations were maintained after repeated dosing. Oral dosing of ENDX resulted in substantially higher ENDX concentrations than a similar dose of TAM. These data support the ongoing development of ENDX to overcome the limitations associated with CYP2D6-mediated metabolism of TAM in humans.
SIGNIFICANCE STATEMENT This study presents for the first time the pharmacokinetics and bioavailability of endoxifen and three key tamoxifen metabolites following repeated oral dosing in female rats and dogs. This study reports that endoxifen has high oral bioavailability, and therapeutic concentrations were maintained after repeated dosing. On the basis of these data, Z-endoxifen (Z-ENDX) was developed as a drug based upon the hypothesis that oral administration of Z-ENDX would overcome the limitations of CYP2D6 metabolism required for full metabolic activation of tamoxifen.
Introduction
The selective estrogen receptor modifier tamoxifen (TAM, Fig. 1A) is a prodrug widely used in the treatment of women diagnosed with estrogen receptor (ER)-positive breast cancer. Cytochrome P450–catalyzed oxidation plays a crucial role in TAM biotransformation and yields several metabolites that contribute to its antitumor activity (Stearns et al., 2003; Murdter et al., 2011). Endoxifen (ENDX, Fig. 1A) and 4-hydroxy tamoxifen (4HT) are the most potent active metabolites of TAM, with 100-fold greater binding affinity of the estrogen receptor and 30-fold to 100-fold greater suppression of estrogen-dependent cell proliferation compared with TAM (Lim et al., 2005; Wu et al., 2009; Murdter et al., 2011). Tamoxifen is metabolized to ENDX and 4HT through activity of cytochrome P450 enzymes, and CYP2D6 is the main enzyme responsible for the conversion of TAM to 4HT and the primary tamoxifen metabolite, N-desmethyltamoxifen, to ENDX (Stearns et al., 2003). Previous studies indicate genetic variation in CYP2D6 influences TAM effectiveness in women with ER+ breast cancer (Madlensky et al., 2011; Goetz et al., 2013; Karle et al., 2013), and ENDX concentrations are associated with TAM efficacy (Stearns et al., 2003). TAM-treated ER-positive breast cancer patients with genetic or drug-induced reductions in CYP2D6 metabolism have a higher risk of recurrence in the adjuvant settings. Furthermore, ENDX concentrations display considerable variability (20–180 nM), and TAM-treated women with reduced or absent CYP2D6 enzyme activity have significantly lower ENDX plasma concentrations (Jin et al., 2005; Borges et al., 2006; Goetz et al., 2018). The impact of CYP2D6 genotype status on TAM efficacy resulted in clinical practice guidelines regarding use of CYP2D6 genotype for TAM dosing (Goetz et al., 2018; Drögemöller et al., 2019). On the basis of these data, Z-endoxifen (Z-ENDX) was developed as a drug based upon the hypothesis that oral administration of Z-ENDX would overcome the limitations of CYP2D6 metabolism required for full metabolic activation of tamoxifen.

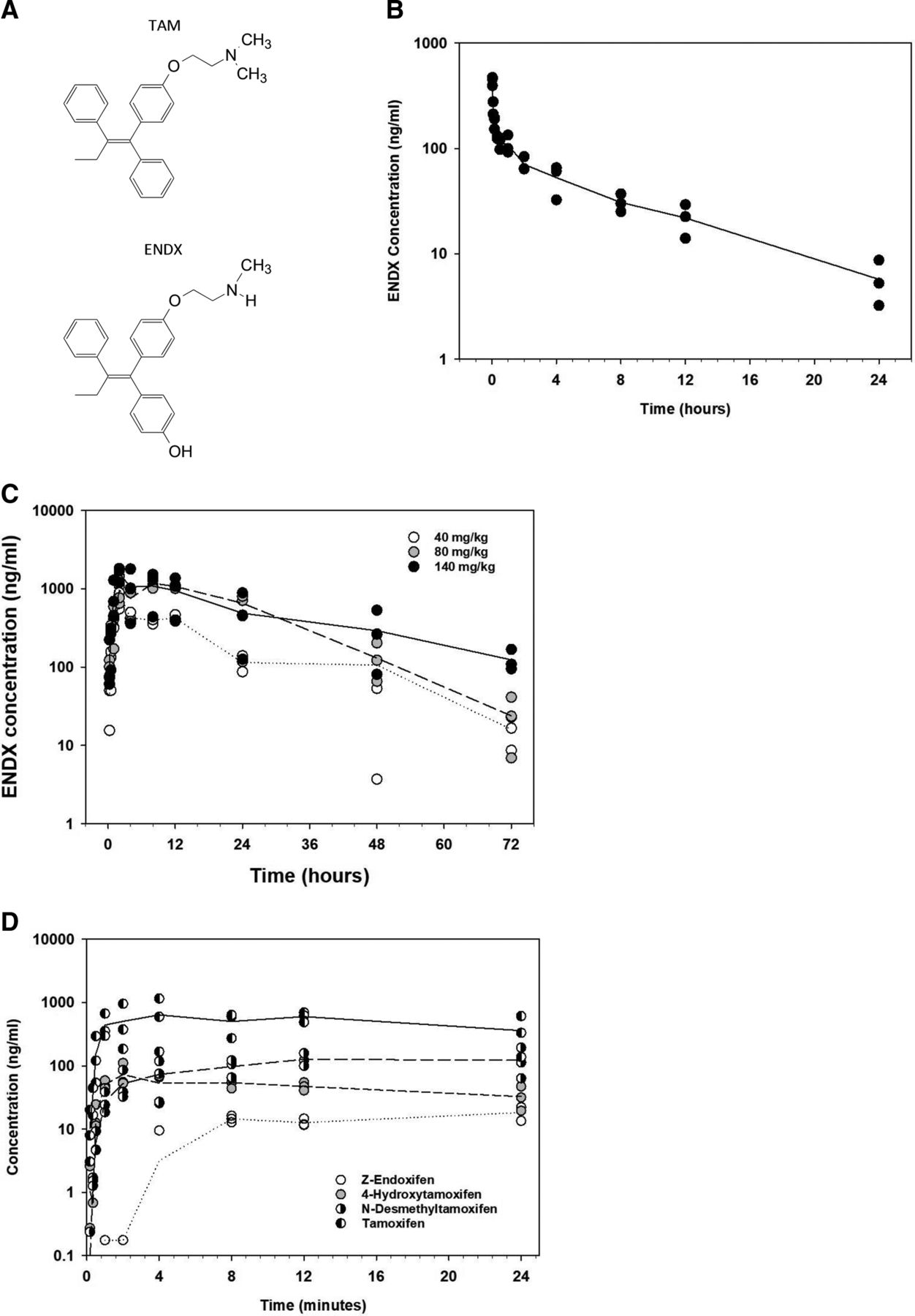
(A) Chemical structures of TAM and ENDX. Plasma profiles for (B) ENDX following intravenous administration of 2 mg/kg ENDX·HCl, (C) ENDX following oral administration of 40, 80, and 140 mg/kg ENDX·HCl, or (D) TAM and its major metabolites following oral administration of 80 mg/kg TAM to female Sprague-Dawley rats (n = 3 per dose group).
ENDX displays superior antitumor activity to TAM in preclinical model systems, ENDX has high oral bioavailability in mice, and plasma concentrations of ENDX following an oral dose of ENDX are eightfold higher than those achieved following a comparable dose of TAM (Ahmad et al., 2010; Reid et al., 2014; Jayaraman et al., 2020). These data led to two phase I studies in which Z-ENDX was delivered in escalating doses to women with hormone-refractory cancer (#NCT01327781 and #NCT01273168) and a phase II study in endocrine-refractory ER+/HER2− breast cancer (#NCT02311933). Substantial ENDX plasma concentrations (>1900 ng/mL) were achieved with ENDX clearance unaffected by CYP2D6 genotype, and encouraging antitumor activity in endocrine-refractory metastatic breast cancer was observed (Goetz et al., 2017; Takebe et al., 2021).
Here, we characterized the pharmacokinetics and bioavailability of ENDX in female rats and dogs. The results of this work support the ongoing development of ENDX for ER+ breast cancer and future clinical trials.
Materials and Methods
Chemicals and Reagents
Z-ENDX free base and Z-ENDX hydrochloride (ENDX·HCl) were provided by the Developmental Therapeutics Program, National Cancer Institute. TAM, 4HT, N-desmethyltamoxifen (NDMT), and toremifene (a selective ER modulator used as the internal standard) were purchased from Sigma Chemical Company (St. Louis, MO). The primary stock solutions of ENDX (2 mg/ml), 4HT (1 mg/ml), NDMT (1 mg/ml), TAM (1 mg/ml), and toremifene (1 mg/ml) were prepared in ethanol and stored at −20°C. Working standard solutions were prepared in ethanol and stored at −20°C.
High Performance Liquid Chromatography Assay
Plasma concentrations of TAM, ENDX, 4HT, and NDMT were measured using a modification of a previously validated and published high performance liquid chromatography assay with fluorescence detection (Lee et al., 2003). In brief, TAM, ENDX, 4HT, NDMT, and toremifene (internal standard) were isolated from plasma samples using liquid-liquid extraction. Dried extracts were reconstituted in 100 μl of 55:45 (v/v) ACN:20 mM KH2PO4, pH 3.0, transferred to an amber autosampler vial with glass insert, and placed in an autosampler maintained at 4°C. Compounds were separated on a Genesis C18 (250 mm × 3.0 mm, 3 m particle size) analytical column protected by a Genesis C18 (10 mm × 3.0 mm, 3-µ particle size) precolumn using a concave gradient elution profile from 43:57 (v/v) ACN:20 mM KH2PO4, pH 3.0, to 75:25 (v/v) ACN:20 mM KH2PO4, pH 3.0 (flow rate: 0.5 mL/min; injection volume: 50 μL). Following the separation, the effluent was passed through a postcolumn photochemical reactor (PHRED-8, Aura Industries), and the fluorescent products were quantified (excitation wavelength: 250 nm; emission wavelength: 370 nm). All procedures were performed in the dark under minimum exposure to light. This assay has been validated in our laboratory according to Food and Drug Administration guidance. The lower limit of quantification and linear range were 1 ng/ml and 1–500 ng/ml, respectively.
Animals
All animal studies were performed under protocols and guidelines reviewed and approved by the Institutional Animal Care and Use Committee of Southern Research Institute, which is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. Additionally, all animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. Female Sprague-Dawley rats with an indwelling jugular vein cannula were purchased from Charles River Laboratories (Raleigh, NC). Purebred beagle dogs were obtained from Ridglan Farms (Mt. Horeb, WI) for the 28-day pharmacokinetic (PK) study and Marshall BioResources (North Rose, NY) for the 4-day PK study. The animals were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum. Rats were acclimated for at least 5 days and dogs for 14 days prior to study start.
Single-Dose Pharmacokinetics and Bioavailability in Sprague-Dawley Rats
Female Sprague-Dawley rats (200–250 g, 7 to 8 weeks old, n = 3 per dose group) with an indwelling jugular vein cannula were used in the study. For intravenous and oral (p.o.) dosing of the salt form of ENDX (ENDX·HCl), ENDX·HCl was dissolved in 0.1 M HCl and then adjusted to a pH of approximately 4.5–5.0 using 0.1 N sodium hydroxide for the 2-mg/kg i.v. dose and 20-, 40-, 80-, 140-, and 200-mg/kg p.o. doses. For p.o. administration of the ENDX free base, ENDX free base was suspended in 0.5% carboxymethyl cellulose in sterile water for an 80-mg/kg dose. TAM citrate salt was dissolved in 0.5% carboxymethyl cellulose in sterile water for the 80-mg/kg p.o. dose. Six rats were assigned to each treatment group and three rats to one untreated group. Rats were further divided into subgroups of three for blood sample collection. Blood samples (approximately 0.7 mL) were collected from the jugular vein catheter into tubes containing EDTA prior to dosing and approximately 2 (intravenous groups only), 5 (intravenous groups only), 10, 20, and 30 minutes and 1, 2, 4, 8, 12, 24, 48 (40-, 80-, and 140-mg/kg ENDX·HCl groups only), and 72 (40-, 80-, and 140-mg/kg ENDX·HCl groups only) hours after dosing. Upon collection, each blood sample was placed on ice and subsequently centrifuged (9000g, 5 minute) to separate plasma. Plasma was then transferred to a separate tube and immediately frozen (at or below –70°C) until analysis.
Multiple-Dose (4-Day) Pharmacokinetics in Sprague-Dawley Rats
Female Sprague-Dawley rats (200–250 g, 7 to 8 weeks old, n = 3 per dose group) with an indwelling jugular vein cannula were used in this study. ENDX·HCl (20, 40, 60, 100, or 200 mg/kg) was suspended in 0.9% sterile saline and administered once daily for 4 consecutive days. Three rats were assigned to each treatment group. Blood samples were collected on days 1 and 4 approximately 4 hours after dose administration. For blood collection, rats were anesthetized with CO2/O2, and blood samples were collected from the retro-orbital plexus into a tube containing EDTA. The blood sample was mixed by gentle inversion, placed on ice, and subsequently centrifuged in a refrigerated centrifuge to separate plasma. Plasma was then transferred to a separate tube and immediately frozen (at or below –70°C) until analysis.
Multiple-Dose (28-Day) Pharmacokinetics in Sprague-Dawley Rats
Female Sprague-Dawley rats (200–250 g, 7 to 8 weeks old, n = 3 per dose group) with an indwelling jugular vein cannula were used in this study. ENDX·HCl (2, 20, 80, and 200 mg/kg) was suspended in 0.9% sterile saline and administered once daily for 28 consecutive days. Three rats were assigned to each treatment group. Blood samples were collected from the jugular vein catheter into tubes containing EDTA prior to dosing and approximately 1, 2, 4, 8, 12, 24, 48 (day 28 only), and 72 (day 28 only) hours postdosing on day 1 and day 28. Upon collection, each blood sample was placed on ice and subsequently centrifuged in a refrigerated centrifuge to separate plasma. Plasma was then transferred to a separate tube and immediately frozen (at or below –70°C) until analysis.
Multiple-Dose (4-Day) Pharmacokinetics in Beagle Dogs
Female beagle dogs (7–10 kg, 12-15 months, n = 2 per dose group) were used in this study. Individual doses of ENDX (15, 30, or 100 mg/kg per day) and TAM (30 mg/kg per day) were placed in gelatin capsules from Torpac, Inc. (Fairfield, NJ). Capsules were filled within 4 days of use for dosing and maintained at room temperature from the time of filling through the period of use. The amount of ENDX added to each capsule was based on the most recent individual animal body weight. ENDX for intravenous administration (0.5 mg/kg per day) was dissolved in 0.9% sterile saline. ENDX and TAM were administered by p.o. or intravenously once daily for 4 consecutive days. Blood samples (approximately 2.0 mL) were collected from the peripheral vein of each dog into tubes containing EDTA prior to dosing and approximately 2 (intravenous only), 5 (intravenous only), 10, 20, and 30 minutes and 1, 1.5, 2, 4, 8, 12, and 24 hours after dosing on days 1 and 4. Upon collection, each blood sample was mixed by gentle inversion, placed on ice, and subsequently centrifuged in a refrigerated centrifuge to separate plasma. Plasma was then transferred to a separate tube and immediately frozen (at or below –70°C) until analysis.
Multiple-Dose (28-Day) Pharmacokinetics in Beagle Dogs
Male and female purebred beagle dogs (7–14 kg, 12–16 months, n = 2 males and 2 females per dose group) were used in this study. Individual doses of ENDX (5, 15, or 30 mg/kg per day) were placed in gelatin capsules. ENDX was administered orally by capsule daily for 28 consecutive days. Dogs in the control group received empty capsules daily for 28 consecutive days. Blood samples were drawn from dogs receiving ENDX on day 1 immediately before dosing, 24 hours after dosing, on day 28 immediately before dosing, and 1, 2, 4, 8, 24, 48, and 72 hours after dosing. Blood samples (approximately 2.0 mL) were mixed with EDTA and centrifuged to separate plasma. Plasma was then transferred to a separate tube and immediately frozen at −20°C until analysis.
Data Analysis
Half-life, area under the curve (AUC), clearance, and volume of distribution values were estimated by standard noncompartmental analysis methods using WinNonlin (Professional Version 3.0; Pharsight Corp; Mountain View, CA). The apparent terminal elimination rate constants (λz) were determined by linear least-squares regression through the linear terminal portion of the graph of the log plasma concentration versus time data. The apparent elimination half-life (t1/2) was calculated as 0.693/λz. Area under the plasma concentration-time curve (AUC0-T) was determined using the linear trapezoidal rule from time zero to the time of the last detectable sample. AUC0-T through infinite time was calculated by adding CT/λz to AUC0-T. The CLp was calculated as dose/AUC0-T through infinite time. Bioavailability was calculated as follows: (AUCpo/AUCiv) ・ (Doseiv per dosepo) ・ 100%. Half-life values following multiple dosing were predicted by rearrangement of the following equation:
 where R is the accumulation ratio based on comparing the day-4 versus day-1 plasma concentrations, t1/2 is the elimination half-life, and τ is the dosing interval (24 hours).
where R is the accumulation ratio based on comparing the day-4 versus day-1 plasma concentrations, t1/2 is the elimination half-life, and τ is the dosing interval (24 hours).
Results
Single-Dose Pharmacokinetic and Bioavailability Study in Rats
The pharmacokinetics of ENDX and TAM were characterized in female Sprague-Dawley rats administered an intravenous dose of 2.0 mg/kg, oral doses of 20, 40, 80, 140, and 200 mg/kg Z-ENDX·HCl, an oral dose of 80 mg/kg ENDX free base, or an oral dose of 80 mg/kg TAM. The pharmacokinetic data are summarized in Table 1. Plasma profiles for intravenous and oral administration of ENDX (Fig. 1, B and C) and oral administration of TAM (Fig. 1D) are shown in Fig. 1.
Single-dose pharmacokinetics of endoxifen and tamoxifen in rats
After rapid distribution following intravenous administration, the plasma concentration declined with a terminal elimination half-life of 6 hours and a plasma clearance value of 2.39 L/h per kg. The peak plasma concentration was 0.4 µg/mL. After oral administration of ENDX·HCl, peak plasma concentrations ranging from 0.3 to 1.7 µg/mL were achieved within 3 to 4 hours. A sustained concentration of ENDX approaching 1000 ng/mL (3.7 µM) was maintained for longer than 24 hours after administration of a dose ≥80 mg/kg (Fig. 1C). ENDX pharmacokinetics were linear over the 20–80-mg/kg dose range, with evidence of saturable pharmacokinetics at 140 mg/kg and above. ENDX free base and hydrochloride salt forms appear to be bioequivalent as plasma concentrations of 1.4 µg/mL and 1.2 µg/mL were achieved 3 to 4 hours after administration of an 80-mg/kg dose of ENDX free base or hydrochloride salt, respectively. Plasma concentrations with both formulations were maintained near 1.0 µg/mL for 24 hours after administration of the oral dose. The absolute bioavailability of ENDX was calculated by comparing the AUC for the oral dose with the AUC for the 2-mg/kg i.v. dose and demonstrated high oral bioavailability (>67%).
TAM absorption was rapid, and peak concentrations of TAM and its metabolites NDMT and 4HT were achieved 1 to 2 hours following oral TAM administration. These peak concentrations were maintained for 24 hours post–oral dosing. ENDX was also measured in plasma as a metabolite of TAM. Appearance of ENDX was delayed, and a peak concentration of ENDX of 20 ng/mL (54 nM) was not achieved until 6 hours after the oral TAM dose. This peak concentration of ENDX was maintained for 18 hours post–TAM oral dosing. The relative bioavailability of ENDX following TAM administration was calculated by comparing the area under the concentration time curve from 0-24 hours (AUC0-24h) for the 80-mg/kg oral TAM dose with the AUC0-24h for the 80-mg/kg oral ENDX dose and demonstrated extremely low relative ENDX bioavailability (1.1%) following an oral dose of TAM.
Repeated-Dose 4-Day Pharmacokinetic Study in Rats
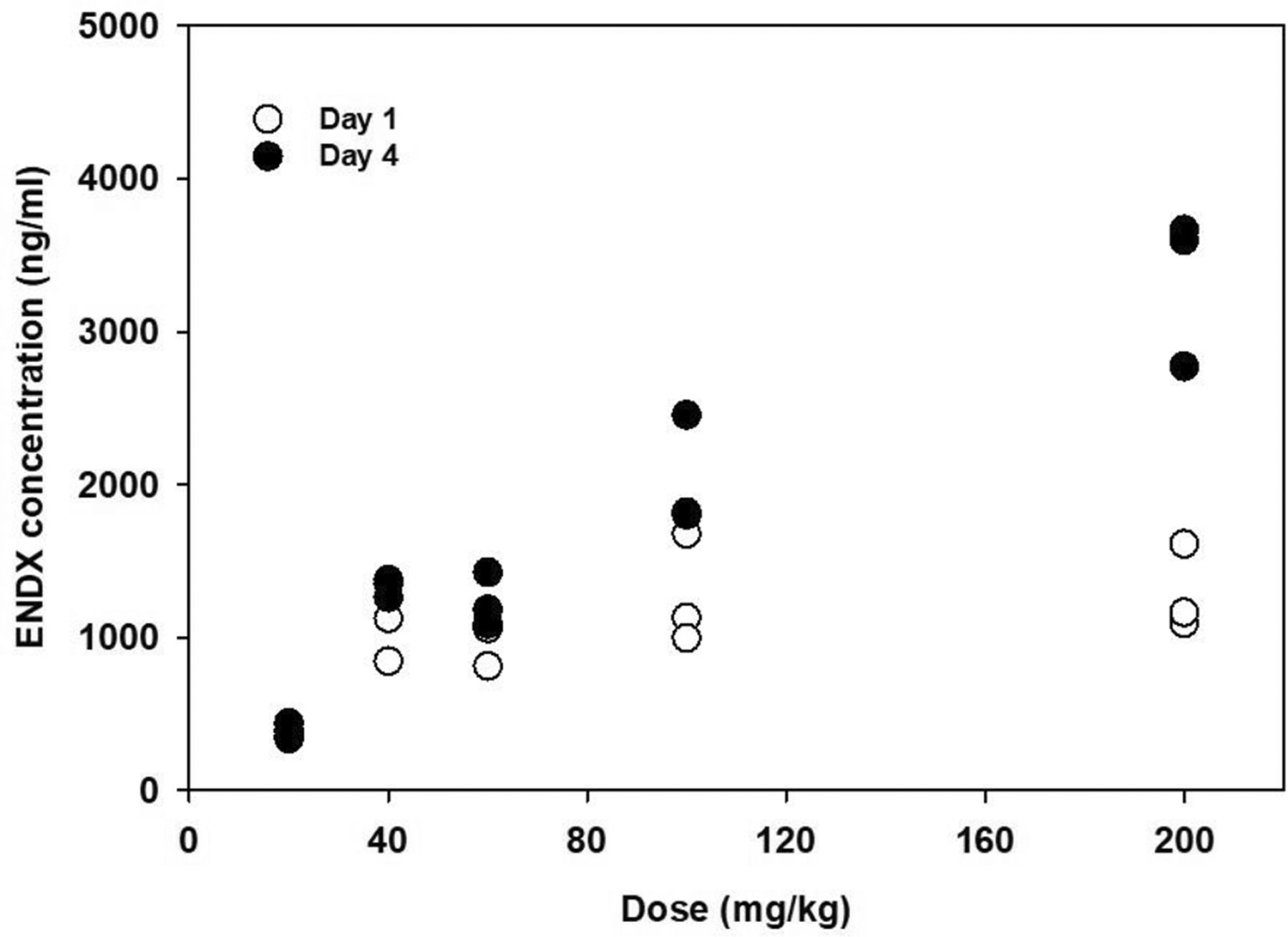
Oral ENDX·HCl doses of 20, 40, 60, 100, and 200 mg/kg were selected for a 4-day dose range–finding study. ENDX plasma concentrations were measured in plasma samples 4 hours after the daily dose on day 1 and on day 4 following 4 consecutive days of dosing. The plasma profile of ENDX is illustrated in Fig. 2, and pharmacokinetics are described in Supplemental Table 1. ENDX plasma concentrations appeared to reach a plateau in the 40–60-mg/kg dose range on day 1. On day 4, ENDX concentrations increased in proportion to dose, up to the 100-mg/kg dose. Plasma accumulation was calculated as the ratio of the day-4 to day-1 plasma concentrations and was found to increase with dose. These data suggest that the terminal elimination half-life of ENDX is ≥34 hours, and high concentrations are maintained with 4 days of dosing.

ENDX plasma concentrations 4 hours after oral administration of the specified doses to female Sprague-Dawley rats (n = 3 per dose group).
Repeated-Dose 28-Day Pharmacokinetic Study in Rats
The pharmacokinetics of ENDX were characterized in female Sprague-Dawley rats administered daily oral doses of 2, 20, 80, and 200 mg/kg ENDX·HCl. Blood samples were obtained 4 hours after drug administration on day 1 and 28. Plasma concentration-time data for each treatment group are illustrated graphically in Fig. 3, and pharmacokinetic data are summarized in Table 2. After oral administration, ENDX Cmax was achieved within 8 hours, and oral absorption appeared saturable above the 80-mg/kg dose. The bioavailability estimate for each dose was determined by comparison of the AUC0-24h estimate for the oral doses with the AUC0-24h estimate from the 2-mg/kg i.v. dose from the single-dose ENDX PK study. From this calculation, oral bioavailability was determined to be greater than 50%.
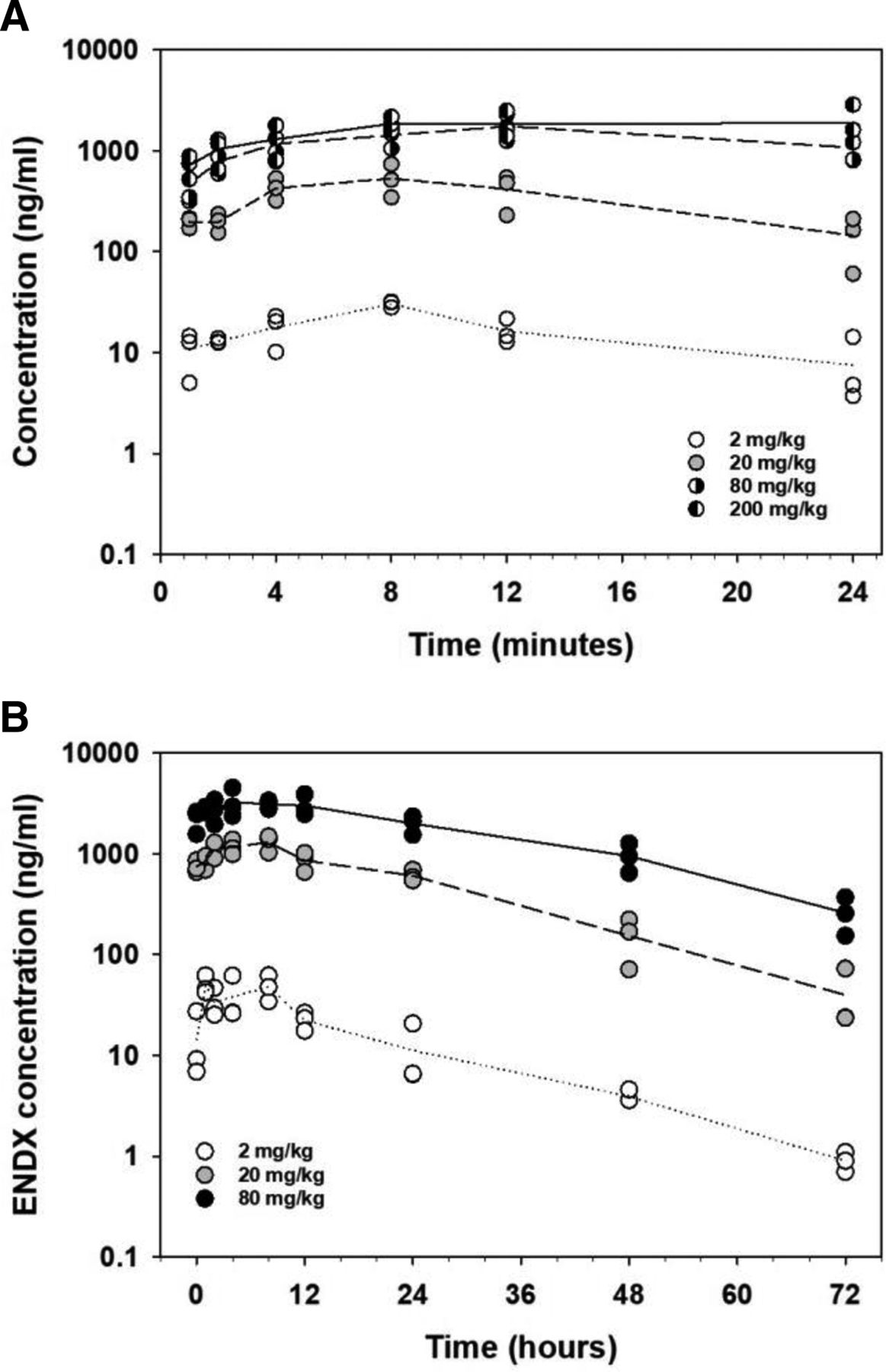
Twenty-eight–day multiple-dose pharmacokinetics of endoxifen in rats

Plasma profiles for ENDX following oral administration of 2, 20, 80, and 200 mg/kg/kg ENDX·HCl to female Sprague-Dawley rats (n = 3 per dose group) on (A) day 1 and (B) day 28 of a 28-day daily dosing schedule.
ENDX pharmacokinetics on day 28 did not differ greatly from those observed on day 1 as peak plasma concentrations achieved within 4–8 hours after the day-28 dose. Cmax and AUC0-24h increased in proportion to dose, and an increase of approximately twofold in the Cmax, AUC0-24h, and steady-state trough concentration (C0h and C24h on day 28) values was consistent with an apparent elimination half-life under 24 hours. Due to the saturable absorption noted at the 80-mg/kg dose and above, the day-28 pharmacokinetics were not determined for the 200-mg/kg dose.
Repeated-Dose 4-Day Pharmacokinetic Study in Beagle Dogs
A 4-day repeated-dose pharmacokinetic study with ENDX and TAM was also carried out in fasted female beagle dogs using an intravenous dose of 0.5 mg/kg of ENDX·HCl, oral doses of 15, 30, and 100 mg/kg of ENDX·HCl, and an oral dose of 30 mg/kg TAM citrate (two dogs per group). ENDX and TAM pharmacokinetics were characterized on day 1 and day 4 and are summarized in Tables 3 and 4, respectively. ENDX and TAM concentration-time profiles are illustrated graphically in Fig. 4.
Four-day multiple-dose pharmacokinetics of endoxifen in fasted and fed female beagle dogs
Four-day multiple-dose pharmacokinetics of tamoxifen in fasted female beagle dogs

Plasma profiles for (A) ENDX following intravenous administration of 0.5 mg/kg ENDX·HCl to fasted female beagle dogs, (B) ENDX following oral administration of 15, 30, and 100 mg/kg ENDX·HCl (day 1 on left, day 4 on right) to fasted female beagle dogs, (C) ENDX following oral administration of 100 mg/kg ENDX·HCl to fed female beagle dogs, or (D) TAM and ENDX following oral administration of 30 mg/kg TAM to fasted female beagle dogs (day 1 on left, day 4 on right). Dogs received daily oral administration of ENDX or TAM for 4 days (n = 2 per dose group).
A peak concentration of 0.76 µg/mL ENDX was achieved after the first intravenous dose of 0.5 mg/kg. After a rapid distribution phase, the plasma concentration declined with a terminal elimination half-life of 9.2 hours and a clearance of 0.40 L/h per kg. ENDX did not appear to accumulate in plasma after four intravenous doses. One of the dogs (5F9841) had similar pharmacokinetics on day 1 and day 4, whereas the other dog (5F9845) had lower ENDX plasma concentrations on day 4 compared with day 1, which resulted in higher apparent clearance.
Following oral administration of ENDX·HCl, ENDX peak plasma concentrations were achieved within 1–8 hours of dosing. While Cmax and AUC0-24h values appeared to increase with increasing dose, it is not clear if absorption was saturable as there was a large difference in the peak plasma concentration and AUC0-24h values at the 100-mg/kg dose. There was some evidence that ENDX accumulates in plasma after oral administration as the day-4 AUC0-24h values were higher than the day-1 values for both dogs administered the 30-mg/kg dose and one of the two dogs administered the 15-mg/kg dose. Unfortunately, day-4 plasma samples were not available for fasted dogs administered the 100-mg/kg dose as the study had to be stopped for this group due to vomiting. Because of this, plasma samples were only collected over 24 hours after the day-1 dose administration. The 100-mg/kg dose was then repeated in two fed female dogs, and ENDX concentrations were measured in plasma samples collected for 24 hours after oral drug administration. Peak plasma concentrations greater than 8 µM were achieved 4–8 hours after the oral dose was given, and plasma ENDX concentrations remained above 1.0 µM for the 24-hour study period. The AUC0-24h estimate determined from the oral dose was compared with the AUC0-24h estimate from the 0.5-mg/kg i.v. dose data to define oral bioavailability. Based on this calculation, ENDX has approximately 50% oral bioavailability in beagle dogs.
Following the oral TAM dose, peak concentrations of TAM, NDMT, and 4HT were achieved after 1–8 hours, and these concentrations were sustained for 24 hours after the initial oral dose. Appearance of ENDX in plasma was delayed, and metabolite concentrations of ENDX did not reach their peak of 0.07 and 0.38 µg/mL until 12 and 4 hours, respectively, post–TAM oral administration. These concentrations were maintained through the 24-hour sample collection period. The ENDX metabolite ratios for the two dogs treated with 30 mg/kg TAM were 0.08 and 0.295. Additionally, the ENDX metabolite AUC0-24h values of dogs administered 30 mg/kg TAM were 3%–20% that of the ENDX AUC0-24h values for dogs treated with 30 mg/kg ENDX.
Repeated-Dose 28-Day Pharmacokinetic Study in Dogs
The pharmacokinetics of ENDX were characterized on day 1 and day 28 in male and female beagle dogs administered daily oral doses of 5, 15, or 30 mg/kg ENDX·HCl for 28 days. The pharmacokinetic data are summarized in Table 5. ENDX plasma profiles are illustrated in Fig. 5. Peak plasma concentrations were achieved 2–4 hours after oral administration. Following the 5- and 15-mg/kg doses, plasma concentrations greater than 40 ng/mL (100 nM) were maintained for 8 and 24 hours, respectively. Plasma concentrations greater than 400 ng/mL (1000 nM) were maintained for longer than 24 hours in three of the four dogs administered the 30-mg/kg dose. In terms of gender, AUC values were higher in female dogs. Male dogs displayed a dose-proportional increase in AUC over the 5–30-mg/kg range. Female dogs displayed a dose-proportional increase in AUC from 5 to 15 mg/kg. However, AUC increased threefold in female dogs when the dose was increased from 15 to 30 mg/kg. The reason for this threefold increase in AUC is currently unknown, but similar results were observed in mice.
Pharmacokinetics of endoxifen in male and female beagle dogs on day 28 following 28 days of daily dosing

Plasma profiles for ENDX following oral administration of (A) 5 mg/kg, (B) 15 mg/kg, and (C) 30 mg/kg ENDX·HCl to male and female beagle dogs on day 28 of a 28-day daily dosing schedule (n = 2 males and 2 females per dose group).
Discussion
TAM is widely used as a treatment option for ER+ breast cancer. Cytochrome P450–catalyzed oxidation of TAM produces 4HT and ENDX, which display greater antiestrogenic activity than TAM (Wu et al., 2009; Murdter et al., 2011). ENDX plasma concentrations exceed those of 4HT but vary widely (3–26 ng/mL) due to CYP2D6 polymorphisms and concomitant use of CYP2D6 inhibitors (Murdter et al., 2011). This led to the development of ENDX as a therapy for ER+ breast cancer to avoid the genetic and environmental factors that affect TAM metabolism and efficacy. Preclinical pharmacology and toxicology studies in rats and dogs are crucial to the development of ENDX and comparison with TAM.
We first sought to characterize the pharmacokinetics and bioavailability of TAM and its metabolites in rats and dogs following oral administration. For rats, our TAM and 4HT results compare well with several single-dose pharmacokinetic studies (Shin et al., 2006; Choi and Kang, 2008; Shin et al., 2008; Shin and Choi, 2009; Kim et al., 2010; Li et al., 2011) but are substantially lower than reports in two other studies (Robinson et al., 1991; Yang et al., 2010). We observed high concentrations and systemic exposure of TAM and NDMT and low concentrations and systemic exposure of 4HT. However, extrapolation of our data to the 200-mg/kg TAM dose used by Robinson et al. (1991) yielded values that were approximately twofold lower than those reported in that study. These differences may be due to variation in the strain, age, and metabolic status of rats used or analytical methodology. Additionally, Robinson et al. (1991) reported variable presence of an unknown peak eluting before 4HT. This peak was later identified as ENDX and measured in our study. For dogs, the published pharmacokinetics of TAM is limited to a single-canine phase I trial of TAM with doxorubicin (Waddle et al., 1999). In that study, dogs were administered 150, 300, and 600 mg/m2 (equivalent to 7.5, 15, and 30 mg/kg for a 10-kg beagle dog) TAM orally every 12 hours for 7 days. The combined plasma concentration of TAM and NDMT was 6–11 µM at the 600-mg/m2 dose level. This is similar to the combined concentrations of 1830 and 2970 ng/ml (4.9 and 8.0 µM, respectively) for beagle dogs in our study.
There are few published multiple-dose reports of TAM and its metabolites in rats. Lien et al. (1991) reported tissue distribution of TAM and its metabolites in female rats and showed that ENDX was not detectable in serum following administration of three daily doses of 1 mg/kg TAM but reached ∼0.5–1 ng/ml after 14 days of treatment. Interestingly, 4HT concentrations were higher than NDMT on day 3, whereas the relative concentrations of TAM and each metabolite were TAM>NDMT>4HT>ENDX on day 14. Other reports show greater accumulation of NDMT compared with TAM but little accumulation of 4HT during prolonged TAM treatment (Robinson et al., 1991; Kisanga et al., 2003). Interestingly, when 10 mg/kg TAM was administered intraperitoneally, NDMT plasma concentrations were lower than TAM. These concentrations were approximately two- to threefold higher than 4HT and ENDX (Gamboa da Costa et al., 2007). We previously reported relative concentrations of TAM and metabolites as TAM>NDMT>4HT>ENDX on day 3 following 3 days of treatment with 20 mg/kg or 200 mg/kg oral TAM in ovariectomized Sprague-Dawley rats (Schweikart et al., 2014). The order of metabolite concentrations in our study differed from Lien et al. (1991) but was similar to other reports. Although the TAM concentration was 10-fold greater in the 200-mg/kg treatment group compared with the 20-mg/kg treatment group, the NDMT, 4HT, and ENDX concentrations were only seven-, six-, and twofold greater, respectively. These differences may reflect dose-dependent metabolism of TAM and/or alteration of CYP3A-catalyzed metabolism.
No prior studies in rats and dogs report detailed pharmacokinetics of the three TAM metabolites that are crucial to clinical use of TAM and development of ENDX. Here, we found that pharmacokinetics of TAM, NDMT, 4HT, and ENDX in rats and dogs are similar to those reported for mice (Reid et al., 2014). TAM has good oral bioavailability in all three species, high concentrations are achieved and maintained after dosing, drug accumulation is slow due to the long half-life, and concentrations of NDMT are higher than 4HT and ENDX but lower than TAM. In mice and rats, 4HT plasma concentrations are higher than ENDX. In dogs, ENDX concentrations are half of 4HT following a single 30-mg/kg TAM dose but are twofold higher than 4HT after 4 days of daily dosing. Concentrations of both active TAM metabolites are in the range of demonstrated in vitro and in vivo activity. This is in contrast to humans, where, following TAM administration, 4HT plasma concentrations are low (<5 ng/ml), whereas ENDX concentrations are 10-fold higher (∼27 ng/mL) (Murdter et al., 2011).
The observation that 4HT levels are higher than ENDX in rats and dogs, but not humans, may be due to species differences in cytochrome P450 activity and expression. The rat CYP3A family consists of five members: CYP3A1, CYP3A2, CYP3A9, CYP3A18, and CYP3A23. Gender differences for CYP3A exist, with male rats displaying greater CYP3A isoform expression and activity compared with females (Mahnke et al., 1997). CYP3A2 and CYP3A18 are present constitutively in male rats, whereas CYP3A1 and CYP3A9 are predominantly found in female rats (Debri et al., 1995; Mahnke et al., 1997; Jan et al., 2006). The human CYP3A enzyme family consists of four members: CYP3A4, CYP3A5, CYP3A7, and CYP3A43. Like rats, expression of CYP3A isoforms is often sex-dependent and varies based on isoform. Expression of CYP3A4 is higher in women than men due to differences in circulating growth hormone (Dhir et al., 2006). CYP3A5 expression is also elevated in women compared with men (Thangavel et al., 2013). Metabolic discrepancies also exist as rat CYP3A isoforms are not orthologous with human CYP3A4. For example, nifedipine is a prototypical substrate of the human CYP3A enzyme but is not metabolized by rat CYP3A1 (Zuber et al., 2002). For CYP2D, rats have six CYP2D enzymes, whereas humans only have one (CYP2D6). Human CYP2D isoforms display high overall sequence identity with the rat CYP2D isoforms but lower sequence identity in the active region (Edmund et al., 2013). In dogs, CYP2D15 and CYP3A12 are the orthologs of human CYP2D6 and CYP3A4, respectively (Roussel et al., 1998; Court, 2013). The canine CYP2D15 displays enzymatic activity close to human CYP2D6 but is affected by the inhibitors quinine and quinidine in a way that is reminiscent of rat CYP2D1 (Roussel et al., 1998; Court, 2013). Few substrates of canine CYP3A12 are known, and the overlap between human CYP3A4 and canine CYP3A12 substrates is not clear. Thus, the oral disposition of TAM in rats and dogs is not representative of human oral TAM disposition. These preclinical species provide inaccurate models for oral TAM disposition and antitumor activity in humans.
We also sought to compare the pharmacokinetics of ENDX in rats and dogs with the pharmacokinetics of ENDX in mice and following TAM administration. High concentrations of ENDX are achieved and maintained throughout the dose range examined in our studies, and drug accumulation is slow due to the long half-life. A single intravenous dose of 2 mg/kg ENDX·HCl to rats achieved a peak concentration that was eightfold higher than that associated with anticancer activity in women following TAM treatment (Madlensky et al., 2011). A single oral dose of 20 mg/kg ENDX·HCl in rats and 15 mg/kg ENDX·HCl in dogs reached peak concentrations associated with maximal activity in vitro (Wu et al., 2009). Concentrations greater than 1 µM (370 ng/mL) were sustained for 24 hours with a single dose of 80 mg/kg ENDX· HCl in rats and 15 mg/kg ENDX· HCl in dogs. ENDX displayed good oral bioavailability in all three species. When ENDX concentrations and exposure in rats and dogs were compared for equivalent doses of TAM, the results were similar to those previously reported by us and others (Ahmad et al., 2010; Reid et al., 2014). The systemic exposure of ENDX in rats was extremely low (2.5%) after an oral dose of 80 mg/kg TAM compared with an equivalent 80-mg/kg ENDX·HCl dose. Similarly, the systemic exposure of ENDX in beagle dogs was low (5% and 19%) after oral doses of 30 mg/kg TAM in comparison with an equivalent ENDX·HCl dose.
In conclusion, this is the first study to evaluate and compare tamoxifen and Z-endoxifen pharmacokinetics in different animal systems. These data demonstrate the substantial differences in the concentrations of tamoxifen and its metabolites across these different models (including our prior published murine data) and demonstrate the substantial differences in Z-endoxifen concentrations with tam administration compared with humans, where CYP2D6 metabolism is rate limiting. These data support the hypothesis that administration of ENDX, the putative active metabolite of TAM, will yield higher, more reproducible concentrations of ENDX than a comparable dose of TAM. These data further support the ongoing clinical development of ENDX as a hormonal therapy for ER+ breast cancer.
Acknowledgements
We thank Drs. Karen Schweikart, Joe Covey, and Myrtle Davis at the National Cancer Institute for their assistance and guidance during planning and completion of this work.
Authorship Contributions
Participated in research design: Jia, Goetz, Ames, Reid.
Conducted experiments: Buhrow, Safgren.
Performed data analysis: Koubek, Goetz, Reid.
Wrote or contributed to the writing of the manuscript: Koubek, Buhrow, Safgren, Jia, Goetz, Ames, Reid.
Footnotes
- Received April 21, 2022.
- Accepted September 16, 2022.
↵This project was funded in part with federal funds from National Institutes of Health National Cancer Institute [Contracts HHSN261200800001E and N01-CM52206] and the Mayo Clinic Cancer Center Support [Grant P30 CA15083].
M.P.G. is the Erivan K. Haub Family Professor of Cancer Research Honoring Richard F. Emslander, M.D., and reports personal fees CME activities from Research to Practice and Clinical Education Alliance; consulting fees to Mayo Clinic from Eagle Pharmaceuticals, Lilly, Biovica, Novartis, Pfizer, Sermonix, AstraZeneca, Blueprint Medicines, and Biotheranostics; and grant funding to Mayo Clinic from Pfizer, Lilly, and Sermonix. The other authors have nothing to disclose.
1Current Affiliation: College of Materials and Chemical Engineering, Minjiang University, Fuzhou, China
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- 4HT
- 4-hydroxy tamoxifen
- AUC
- area under the concentration time curve from 0-24 hours, AUC0-24h
- AUC0-T
- area under the plasma concentration-time curve
- ENDX
- endoxifen
- ENDX·HCl
- Z-ENDX hydrochloride
- ER
- estrogen receptor
- NDMT
- N-desmethyltamoxifen
- PK
- pharmacokinetics
- p.o.
- oral
- t1/2
- apparent elimination half-life
- TAM
- tamoxifen
- Z-ENDX
- Z-endoxifen
- λz
- terminal elimination rate constants
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}