


Abstract
Compromised hepatic drug metabolism in response to proinflammatory cytokine release is primarily attributed to downregulation of cytochrome P450 (CYP) enzymes. However, whether inflammation also affects other phase I and phase II drug metabolizing enzymes (DMEs), such as the flavin monooxygenases (FMOs), carboxylesterases (CESs), and UDP glucuronosyltransferases (UGTs), remains unclear. This study aimed to decipher the impact of physiologically relevant concentrations of proinflammatory cytokines on expression and activity of phase I and phase II enzymes, to establish a hierarchy of their sensitivity as compared with the CYPs. Hereto, HepaRG cells were exposed to interleukin-6 and interleukin-1β to measure alterations in DME gene expression (24 h) and activity (72 h). Sensitivity of DMEs toward proinflammatory cytokines was evaluated by determining IC50 (potency) and Imax (maximal inhibition) values from the concentration-response curves. Proinflammatory cytokine treatment led to nearly complete downregulation of CYP3A4 (∼98%) but was generally less efficacious at reducing gene expression of the non-CYP DME families. Importantly, FMO, CES, and UGT family members were less sensitive toward interleukin-6 induced inhibition in terms of potency, with IC50 values that were 4.3- to 7.4-fold higher than CYP3A4. Similarly, 18- to 31-fold more interleukin-1β was required to achieve 50% of the maximal downregulation of FMO3, FMO4, CES1, UGT2B4, and UGT2B7 expression. The differential sensitivity persisted at enzyme activity level, highlighting that alterations in DME gene expression during inflammation are predictive for subsequent alterations in enzyme activity. In conclusion, this study has shown that FMOs, CESs, and UGTs enzymes are less impacted by IL-6 and IL-1β treatment as compared with CYP enzymes.
SIGNIFICANCE STATEMENT While the impact of proinflammatory cytokines on CYP expression is well established, their effects on non-CYP phase I and phase II drug metabolism remains underexplored, particularly regarding alterations in drug metabolizing enzyme (DME) activity. This study provides a quantitative understanding of the sensitivity differences to inflammation between DME family members, suggesting that non-CYP DMEs may become more important for the metabolism of drugs during inflammatory conditions due to their lower sensitivity as compared with the CYPs.
Introduction
Inflammation is increasingly recognized as a contributor to the regulation and variability of drug clearance in humans, presumably due to alterations in drug metabolism (Stanke-Labesque et al., 2020; Dunvald et al., 2022). More specifically, the widespread elevation of proinflammatory cytokines, such as interleukin (IL)-6 and IL-1β affects gene expression of drug metabolizing enzymes (DMEs) in hepatocytes (Aitken and Morgan, 2007; Dickmann et al., 2011, 2012; Klein and Zanger, 2013; Tanner et al., 2018), subsequently affecting hepatic drug clearance and efficacy or safety of drug treatments (Leung et al., 2014). Considering the high prevalence of both acute and chronic inflammatory diseases, it is crucial to take into account how hepatic drug metabolism of both novel and existing drugs can be affected by inflammation.
In vitro studies using human liver models have been instrumental in broadening our understanding of inflammation-induced alterations in drug metabolism and can facilitate in quantifying these effects. A promising approach to predict the subsequent impact of inflammation on drug clearance in vivo involves utilizing in vitro data coupled with physiologically based pharmacokinetic (PBPK) models. This approach has demonstrated its utility in predicting the influence of elevated IL-6 levels on drug clearance, particularly for substrates of the key DMEs cytochrome P450 (CYP) 3A4 and CYP2C19 (Machavaram et al., 2013; Xu et al., 2015; Jiang et al., 2016; Lenoir et al., 2022; Stader et al., 2022). Generating more physiologically relevant quantitative in vitro data will likely aid in utilizing PBPK models to predict the impact of inflammation on drug clearance for substrates of other CYP enzymes and non-CYP mediated pathways (Ozbey et al., 2023).
Importantly, it is estimated that clearance of ∼25% of the top 200 most prescribed small molecule drugs approved by the FDA is mainly dependent on non-CYP enzymes, with the UDP-glucuronosyltransferase (UGT) family contributing to biotransformation in 45% of the cases (Saravanakumar et al., 2019). However, whereas the impact of proinflammatory cytokines on CYP expression is well established, the potential impact on other DME families, including the UGTs, sulfotransferases, flavin-containing monooxygenases (FMOs), and carboxylesterases (CESs), has received considerably less attention. Yet, it remains unclear to what extent the activity of non-CYP metabolizing enzymes is affected by inflammation, and whether these enzymes exhibit a comparable sensitivity to the effects of inflammatory cytokines as compared with the CYP enzymes.
Another limitation of available in vitro data is that they have mostly focused on the impact of cytokines on the mRNA expression levels of DME enzymes rather than on their enzymatic activity. While significant changes in the expression of DME mRNA during inflammation have indeed prompted focus on transcription as the primary mechanism underlying changes in metabolic capacity, there is increasing acknowledgment of the influence of post-transcriptional mechanisms on DME activity (Stanke-Labesque et al., 2020). Consequently, a strong up- or downregulation of mRNA expression observed upon cytokine stimulation may not necessarily translate into similar alterations in enzyme activity. Furthermore, in vitro studies are often conducted using cytokine concentrations that surpass the physiological concentrations observed in patients, compromising clinical translation (de Jong et al., 2020). IL-6 levels typically range from 10 to 1000 pg/ml during inflammatory conditions, and IL-1β can reach up to 50 pg/ml (Jablonska et al., 2001; Machavaram et al., 2013; Coutant and Hall, 2018). However, most in vitro studies have exclusively examined the effects of 10 ng/ml IL-6 and 1 ng/ml IL-1β, concentrations that far exceed physiological levels. This underscores the necessity of investigating changes in enzymatic activity upon physiologically relevant concentrations of cytokines to generate reliable quantitative in vitro data.
In this study, we therefore investigated the concentration-dependent effects of IL-6 and IL-1β on both the mRNA expression and activity of CYP and non-CYP DMEs in a relevant human hepatocarcinoma cell line, i.e., in HepaRG cells. Quantifying the impact of inflammatory mediators across various DME families allowed us to establish a hierarchy of their sensitivity. By comparing the effects of IL-6 and IL-1β on transcription versus activity, we shed light on whether alterations in mRNA serve as a reliable predictor of corresponding changes in enzyme activity during inflammation. This information is essential for enhancing our understanding of the impact of inflammation on drug metabolism, and could be implemented in modeling tools aimed at optimizing drug dosing strategies for patients with inflammatory disease.
Material and Methods
Reagents and Chemicals
William’s E Medium with GlutaMAX Supplement and trypsin-EDTA (0.25%) were purchased from ThermoFisher (Waltham, MA, USA). Fetal bovine serum was obtained from Merck (Batch number: 0001663799), penicillin/streptomycin was obtained from Lonza (Basel, Switzerland). Hydrocortisone, DMSO, human insulin, and primers were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). Dulbecco’s phosphate-buffered saline (PBS) was obtained from Capricorn Scientific (Ebsdorfergrund, Hessen, Germany). SensiMix SYBR Lo-ROX kit and 10x NH4 Reaction Buffer for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) were purchased from Meridian BioScience (Cincinnati, Ohio, USA). Maxima H minus Reverse transcriptase and 5x RT buffer was purchased from Thermo Scientific (Waltham, MA, USA). Human recombinant IL-6 and human recombinant IL-1β were purchased from Peprotech (London, UK). All cytokines were reconstituted and stored as high concentration stocks according to the manufacturer’s instructions. S-mephenytoin, 4′-hydroxymephenytoin, 4′-hydroxymephenytoin-d3, diclofenac, 4′-hydroxydiclofac, 4′-hydroxydiclofenac-13C6, phenacetin, acetaminophen, benzydamine N-oxide, and benzydamine N-oxide-d6 were purchased from LGC (Wesel, Germany). Acetaminophen-d4 was purchased from Alsachim (Illkirch-Graffenstaden, France). Benzydamine was purchased from Sigma-Aldrich (St. Louis, MO, USA). 1’-Hydroxymidazolam was purchased from Cerilliant (Round Rock, Texas, USA) and 1’-hydroxymidazolam-d4 from Supelco (St. Louis, Missouri, USA). Midazolam hydrochloride, morphine, morphine-3-glucuronide, and morphine-3-gluronide-d3 were from Duchefa Farma (Haarlem, the Netherlands). Acetonitrile, methanol, water, and formic acid of LC-MS grade were obtained from Merk (Darmstadt, Germany).
HepaRG Culture and Treatment
HepaRG cells at passage 12 (batch HPR101067) were purchased from Biopredict International (Rennes, France) and expanded to set up a working bank according to the provider’s instructions. Cells plated in 96-wells plates at a density of 9000 cells/well were first grown in William’s E medium GlutaMAX supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, 5 μg/ml human insulin, and 20 μg/ml hydrocortisone for two weeks. Subsequently, cells were cultured for an additional two weeks in the same medium supplemented with 2% DMSO to get fully differentiated cells (Gripon et al., 2002). Cells were maintained at 37°C in 5% CO2 throughout the experiment.
The fetal bovine serum concentration in the DMSO-containing HepaRG medium was reduced to 1% at 24 hours before treatment with the cytokines IL-6 or IL-1β. Concentrations of IL-6 used for the experiments ranged from 0.0001 ng/ml to 10 ng/ml and from 0.001 pg/ml to 1 ng/ml for IL-1β, respectively. For gene expression analysis, cells were treated with IL-6 or IL-1β for 24 hours prior to lysis. For activity measurements, the cytokine-containing medium was renewed every 24 hours. After 72 hours, the medium was replaced by 2% DMSO-containing serum-free medium with a substrate specific to the DME of interest, as described in detail below. An CyQUANT LDH Cytotoxicity Assay (Thermo Scientific, Wilmington, US) was conducted after 72 hours to evaluate cytotoxicity at the highest concentrations of IL-6 and IL-1β, yielding cytotoxicity levels of 6% and 14%, respectively.
Human Liver Biopsies
Human liver biopsies were obtained from the gastroenterology biobank at the Leiden University Medical Center (LUMC, Leiden, the Netherlands), as described elsewhere (de Jong et al., 2023).
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from HepaRG cells or human liver biopsies following the acid guanidinium thiocyanate-phenol-chloroform extraction, as described elsewhere (Chomczynski and Sacchi, 1987). Concentration and purity of RNA was subsequently measured using a NanoDrop 3300 (Thermo Scientific, Wilmington, US). Synthesis of cDNA was performed with 0.5 μg RNA input using Maxima H Minus Reverse Transcriptase (Thermo scientific) according to the manufacturer’s instructions. RT-qPCR analysis was performed with a QuantStudio 6 Flex System using SYBR Green technology. RT-qPCR samples were run in duplicate. All PCR primers were designed in-house and subsequently checked for amplification efficiency (Supplemental Table 1). Relative mRNA levels were calculated using the comparative ΔΔCt method (Livak and Schmittgen, 2001).The expression in each HepaRG sample was normalized by subtracting the geometric mean Ct value of the endogenous control genes ribosomal protein lateral stalk subunit P0 (RPLP0), glyceraldehyde-3-phosphate dehydrogenase and β-actin (ACTB) from the target Ct value to obtain the ΔCt (eq. 1)

Subsequent relative gene expression levels were calculated as 2−ΔCt. Fold changes of treated cells as compared with PBS-control cells were calculated using Eq. 2 and 3.


Data are expressed as mean fold changes ± S.E.M. Basal gene expression in HepaRG cells and human liver biopsies presented in Fig. 1 and Supplemental Fig. 2 are exclusively normalized for RPLP0. This is due to the fact that RPLP0 was identified as a stable endogenous control in liver biopsies, unlike other housekeeping genes (de Jong et al., 2023). Statistical analyses were carried out on ΔCT values due to the considerably skewed symmetry of up- and downregulation in the linear fold change.

Basal mRNA expression levels of phase I and phase II drug metabolizing enzymes in HepaRG cells and in human livers. mRNA expression of the gene of interest was normalized to the housekeeping gene RPLP0 and presented as a fold change compared with basal CYP3A4 expression of either HepaRG cells or human livers. All values are means + S.E.M. from eight independent experiments (HepaRG) or from biopsies of 40 human livers.
DME Activities in HepaRG Cells
Determination of DME activity was based on the metabolic conversion of probe substrates, i.e., midazolam for CYP3A4, phenacetin for CYP1A2, diclofenac for CYP2C9, S-mephenytoin for CYP2C19, benzydamine for FMO3, and morphine for UGT2B7 using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). CYP2D6 activity could not be determined since HepaRG cells are derived from a CYP2D6 poor metabolizer patient and was thus excluded from our analysis (Guillouzo et al., 2007). Cells were exposed to 5 μM midazolam for 30 minutes, 50 μM phenacetin for 2 hours, 10 μM diclofenac for 2 hours, 100 μM S-mephenytoin for 2 hours, 10 μM benzydamine for 4 hours, or 100 μM morphine for 4 hours in serum-free William’s E medium supplemented with 2% DMSO. Substrate concentrations were selected below the Michaelis-Menten constant to achieve selective metabolic conversion by the specific DME isoform (Störmer et al., 2000; Court et al., 2003; Spaggiari et al., 2014). Afterward, cell medium samples containing the probe substrates and their metabolites were collected and mixed with 250 mM formic acid, and immediately frozen at -20 degrees. Notably, UGT2B7 activity samples were mixed with 1 M sodium carbonate and then frozen. For quantification of the metabolites 1’hydroxymidazolam (CYP3A4), acetaminophen (CYP1A2), 4’hydroxydiclofenac (CYP2C9), 4’hydroxymephentoin (CYP2C19), benzydamine-N-oxide (FMO3), or morphine-3-glucuronide (UGT2B7) samples were subjected to LC-MS/MS based analysis. A detailed description of the LC-MS/MS analysis can be found in the Supplemental Methods ‘LC-MS/MS method to quantify CYP activity’ or ‘LC-MS/MS method to quantify FMO3 and UGT2B7 activity’, where MS-specific parameters are listed in Supplemental Tables 2 and 3. CES1 activity was not determined due to the absence of a probe-based analytical detection method. Enzyme activity data were normalized to the amount of cells per well and presented as the rate of metabolite formation in picomole/min/million cells as compared with untreated cells.
Statistical Analysis
Results were generated from at least four independent experiments. Relative IC50 of IL-6 and IL-1β for DME expression and activity were determined using GraphPad Prism 9.2.0 software (GraphPad Software, La Jolla, CA, USA) through nonlinear regression on the basis of the four-parameter logistic function (Sebaugh, 2011). In case the concentration-response curve did not reach the lower asymptote upon the highest cytokine stimulation, IC50 values were determined by directly interpolating from the studied concentration-response curve, without extrapolation for higher cytokine concentrations beyond the range of observed data points. Percentual maximal inhibition (Imax) values were calculated based upon the upper and lower asymptotes of the concentration-response curves. Statistical significance in IC50 and Imax values between DME isoforms was determined by the parametric one-way ANOVA test assuming normal distribution of data and applying the Dunnet’s post hoc test for comparison with CYP3A4 in GraphPad Prism 9. Statistical significance between IC50 and Imax values on mRNA and activity was done using an unpaired t test. The criterion was based on the P values and indicated with * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001 and NS, not significant.
Results
Basal mRNA Expression of DMEs in HepaRG is Comparable to Human Livers
The mRNA expression levels of four CYP enzymes (CYP3A4, CYP2C9, CYP2C19, CYP1A2), five other phase I enzymes (FMO1, FMO3, FMO4, CES1, CES2), and four phase II enzymes (UGT1A4, UGT2B4, UGT2B7 and UGT2B15) were analyzed by RT-qPCR in HepaRG cells and biopsies of human livers (Fig. 1). Rank order of P450 expression was CYP3A4 > CYP2C9 > CYP2C19 > CYP1A2 in HepaRG cells and CYP2C9 > CYP3A4 > CYP2C19 > CYP1A2 in human livers. CYP1A2 expression was relatively low in HepaRG as compared with human livers, consistent with previous characterization studies (Guillouzo et al., 2007). The rank order of other phase I enzymes expression was FMO3 > FMO4 > FMO1 and CES1 > CES2. For the included phase II enzymes, the expression order was UGT2B4 > UGT2B15 > UGT1A4 > UGT2B7. This pattern was consistent in both HepaRG cells and human livers, aligning with previous research (Hines, 2006; Izukawa et al., 2009). Thus, the rank order within DME families exhibited strong similarity between human livers and the HepaRG cell model, suggesting that the HepaRG cell model is not only suitable for providing translation input regarding CYP enzymes but also for other DME families.
Impact of Proinflammatory Cytokine Treatment on CYP Expression and Activity
The effect of inflammation on the gene expression and enzyme activity of selected phase I and phase II DMEs was evaluated by determining the IC50 (potency) and Imax (efficacy) values of IL-6 and IL-1β on individual isoforms.
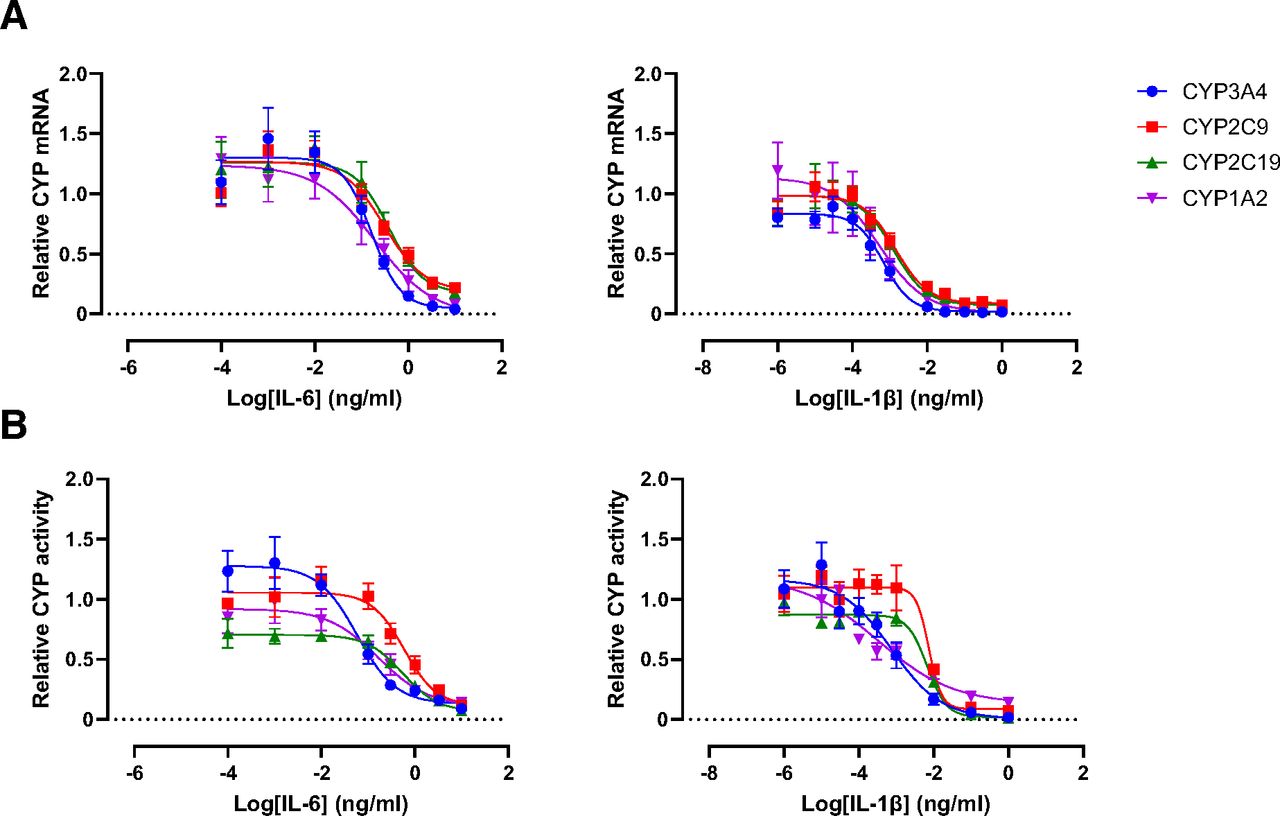
A concentration-dependent decrease in the relative mRNA expression of all CYP isoforms was observed following treatment with both IL-6 and IL-1β. Among the CYP family members, no substantial differences were noted in the isoform-specific response to cytokine treatment, as evident from the comparable potency and efficacy values (Fig. 2A, Table 1). Comparison of IC50 values and maximum suppression values for IL-1β and IL-6 indicated that in general, IL-1β is much more potent than IL-6 in suppressing DME gene expression and enzyme activity. This finding corroborates previous research in HepaRG cells (Klein et al., 2015). We next examined whether the alterations at the DME gene expression level were retained at the enzyme activity level. Indeed, a concentration-dependent decrease was observed for CYP activity of all isoforms (Fig. 2B, Table 2). In contrast to the similar potencies of IL-6 and IL-1β in modulating expression levels of different CYP isoforms, there was a distinct potency difference (∼10-fold) between the impact of inflammation on CYP2C19 and CYP2C9 enzyme activities as compared with CYP3A4 activity, which was reflected by a higher sensitivity of CYP3A4 activity toward IL-6 and IL-1β.

Cytokine concentration-response curves for regulation of CYP isoforms CYP3A4, CYP2C9, CYP2C19, and CYP1A2 on expression (A) and activity level (B). Cells were treated with concentrations of 0.0001 ng/ml to 10 ng/mL (IL-6) or 0.001 pg/ml to 1 ng/ml (IL-1β) for 24 hours to analyze gene expression alterations via RT-qPCR or for 72 hours to analyze activity alterations via probe substrate metabolism with LC-MS/MS. mRNA and activity data are expressed as fold change of levels found in untreated control cells, arbitrarily set to 1.0. Each data point represents the average ± S.E.M. of at least four independent experiments. Data were fit with a nonlinear regression model.
Quantified IC50 and Imax values for DME mRNA expression levels obtained from fitting a nonlinear regression model on the concentration-effect curves after treatment with IL-6 or IL-1β for 24 h
The IC50 values are reported in ng/ml for IL-6 treatment and in pg/ml for IL-1β treatment. One-way ANOVA and Dunnett’s post hoc test with comparison with CYP3A4 was done to investigate differences in potency and maximal effect between DME families, for both IL-6 and IL-1β treatment. * P < 0.05, ** P < 0.01, *** P < 0.001
Quantified IC50 and Imax values for DME activity obtained from fitting a nonlinear regression model on the concentration-effect curves after treatment with IL-6 or IL-1β for 72 h
The IC50 values are reported in ng/ml for IL-6 treatment and in pg/ml for IL-1β treatment. One-way ANOVA and Dunnett’s post hoc test with comparison with CYP3A4 was done to investigate differences in potency and efficacy between DME families, for both IL-6 and IL-1β. * P < 0.05, ** P < 0.01, *** P < 0.001
Non-CYP Isoforms Are Differentially Affected by Cytokine Treatment as Compared with CYP Isoforms
We next examined the impact of IL-6 and IL-1β treatment on the different members of the most important non-CYP DME families. Sensitivity differences in response to cytokine treatment among DME families were defined by benchmarking potency and efficacy values against CYP3A4, which is recognized as the most important DME in humans because of its clinical importance and high expression (Zanger and Schwab, 2013). Interestingly, gene expression of FMO3, FMO4, CES1, CES2, UGT1A4, UGT2B4, and UGT2B7 was in terms of potency less sensitive toward the effects of IL-6 as compared with CYP3A4, with IC50 values that were 4- to 9-fold higher than for CYP3A4 (Fig. 3A, Table 1). Additionally, while IL-6 elicited a maximal downregulation of only 55 ± 9% for FMO3, 57 ± 4% for FMO4, 39 ± 15% for CES1, and 48 ± 13% for CES2, it led to a nearly complete downregulation of 97 ± 1% for CYP3A4 expression. This difference in efficacy of IL-6 was similarly observed across all members of the UGT family, where maximal downregulation ranged from 60 ± 12% to 73 ± 7%.

Cytokine concentration-response curves for regulation of CYP3A4, FMO3, UGT2B7, and CES1 on expression (A) and activity level (B). Cells were treated with concentrations of 0.0001 ng/ml to 10 ng/ml (IL-6) or 0.001 pg/ml to 1 ng/ml (IL-1β) for 24 hours to analyze gene expression alterations via RT-qPCR or for 72 hours to analyze activity alterations via probe substrate metabolism with LC-MS/MS. mRNA and activity data are expressed as fold changes of levels found in untreated control cells, arbitrarily set to 1.0. Each data point represents the average ± S.E.M. of at least four independent experiments. Data were fit with a nonlinear regression model.
Similar patterns were observed for the impact of IL-1β on non-CYP DME isoforms. FMO3, FMO4, CES2, UGT2B4, and UGT2B7 exhibited a significantly lower sensitivity to IL-1β as compared with CYP3A4, indicating that a, respectively, 18-, 28-, 30-, 9-, and 14-fold higher concentration of IL-1β was needed to exert 50% of the maximal downregulation by this cytokine. Interestingly, IL-1β did not impact CES1 expression across all concentrations tested. In addition, the maximal inhibitory effect of IL-1β on gene expression levels of FMO3, FMO4, CES2, UGT1A4, and UGT2B7 ranged from 80 ± 3% to 84 ± 17%, which was less as compared with the observed near-complete downregulation of 99 ± 2% of CYP3A4.
Importantly, the differential potency and maximal inhibitory impact of inflammatory mediators on different members of the DME families could be confirmed at the enzyme activity level (Fig. 3B, Table 2). Compared with CYP3A4 activity, FMO3 activity was less sensitive toward the effects of IL-6, as evident by a 26-fold difference in potency. UGT2B7 activity was even less sensitive toward IL-6, with a 35-fold difference in IC50 value as compared with CYP3A4 activity. In addition, maximal inhibition by IL-6 was only 29 ± 5% for FMO3, and 69 ± 7% for UGT2B7, significantly less than the maximal inhibition of 93 ± 2% that was observed for CYP3A4 activity. The maximal downregulation of FMO3 activity following IL-1β treatment was 54 ± 5%, which was also less than observed for the CYP3A4 activity (98 ± 1%). However, IL-1β showed comparable potency toward FMO3 activity inhibition as compared with CYP3A4 activity inhibition, highlighting that the efficacy of IL-1β rather than the sensitivity to IL-1β differed between FMO3 and CYP3A4 activities. UGT2B7 activity displayed lower sensitivity toward IL-1β, which was reflected by a 31-fold difference in IC50 value as compared with CYP3A4.
Transcriptional Regulation is the Main Driver of the Cytokine-Mediated Inhibition of DMEs
Several studies have suggested that inflammation-related post-transcriptional mechanisms may modulate CYP activity, which would theoretically result in a mismatch in the overall impact of inflammatory mediators in altering DME gene expression versus enzyme activity. To investigate whether post-transcriptional modifications induced by inflammation are indeed critical to the effect, acquired IC50 and Imax values for DME gene expression and enzyme activity were compared (Fig. 4). Overall, there was a strong linear relationship between the potency of IL-6 and IL-1β on DME expression and DME activity (P < 0.0001) (Fig. 4A). Importantly, 90% of the variability in DME activity could be explained by changes in transcription (R2 = 0.9), highlighting the strong association between alterations in gene expression and enzyme activity during inflammation. We next compared individual expression versus activity IC50 values for CYP3A4, CYP2C19, CYP2C9, CYP1A2, FMO3, and UGT2B7, visually presented in Supplemental Fig. 1. CYP3A4 activity was more sensitive toward IL-6 induced downregulation compared with CYP3A4 expression, and this was similarly seen for FMO3 activity upon IL-1β treatment. In contrast, CYP2C19 and CYP2C9 expression was more sensitive toward IL-1β treatment as compared with CYP2C19 and CYP2C9 activity. For other isoforms, similar IC50 values on expression and activity level were found. The maximal impact of IL-6 and IL-1β on expression and activity of the DMEs was highly similar, except for the mismatches observed for FMO3 (Fig. 4B).

Simple linear regression analysis to investigate the relationship between the impact of IL-6 and IL-1β treatment on DME mRNA expression vs. activity for LogIC50 values (A) and Imax values (B). The regression line represents the best-fit line calculated from the data, and the dotted lines indicate the 95% confidence interval. Blue dots represent data obtained from IL-6 treated cells, and brown dots represent data obtained from IL-1β treated cells.
Comparison of IC50 Values for Cytokine-Induced CYP Changes in HepaRG Cells versus Two-Dimensional (2D) and Three-Dimensional (3D) Primary Human Hepatocyte (PHH) Models
To further highlight the translational value of the HepaRG cell line as in vitro liver model, we compared our quantitative cytokine-induced changes to what has been reported before in 2D and 3D PHH models (Dickmann et al., 2011, 2012; Klöditz et al., 2023). Comparing our HepaRG IL-6 IC50 values with those previously determined for CYP isoforms in 2D/3D PHHs showed good agreement between the results (Table 3). The potency of IL-6 in inducing transcriptional alterations in CYPs in 3D PHH spheroids was almost identical as compared with the potency found in HepaRG cells. The IC50 data acquired in a 2D PHH model were also comparable. However, it should be noted that basal CYP expression rapidly declines in 2D cultures of PHH, even in the absence of a proinflammatory stimulus (Kiamehr et al., 2019). The correspondence of our HepaRG IC50 data does not hold so well for comparing the potency of IL-1β on CYP expression and activity in PHHs. Although we found the most pronounced effects on CYP3A4, similarly to the results in 3D PHHs, IL-1β was much more potent in HepaRG cells as compared with PHHs. This might in part be due to the morphological heterogeneity of HepaRG cells, where biliary-like cells release additional proinflammatory cytokines, amplifying the IL-1β response (Pinto et al., 2018). Indeed, aggravation of the IL-1β, but not the IL-6 response has been demonstrated in hepatocyte coculture models as compared with hepatocytes alone, where a sensitivity increase up to 50-fold was observed for CYP3A4 (Nguyen et al., 2015). Taken together, these findings demonstrate that HepaRG cells exhibit comparable sensitivity to IL-6-induced transcriptional changes in CYP enzymes as observed in 2D and 3D PHH models.
Quantified IC50 values in HepaRG cells from this study as compared with reported values in 2D and 3D PHH models
Data from 3D PHH models was extracted from the publication by Klöditz et al. (2023) and represents the average of four independent donors. Data from 2D PHHs was extracted from the studies by Dickmann et al. (2011, 2012) and represents the average of five independent donors unless stated otherwise.
Cytokine Specific Effects on Nuclear Receptors and Transcription Factors Regulating the DMEs
Our data indicates that transcriptional alterations in DME are the primary mechanism underlying inflammation-related changes in CYP enzyme activity in vitro. To gain mechanistic insight into the differential regulation of hepatic gene expression by cytokines, we investigated the effects of IL-6 and IL-1β on a selection of nuclear receptors and transcription factors generally considered to be involved in DME gene expression regulation (Fig. 5). Pregnane X receptor (PXR) and constitutive androstane receptor (CAR) are identified as key transcriptional regulators of the CYP enzymes, with confirmed binding sites in the response elements of human CYP3A4/5, CYP2C9, CYP2C19, and CYP1A2 (Xie et al., 2000; Ferguson et al., 2002; Chen et al., 2003). Nuclear factor Y (NFY) and upstream transcription factor 1 (USF1) are essential for constitutive FMO3 transcription via promoter binding (Klick and Hines, 2007), while liver X receptor α (LXRα) has recently been identified as regulator of human CES (Collins et al., 2022). UGT family regulation is isoform-specific, with the aryl hydrocarbon receptor (AhR) and hepatocyte nuclear factor (HNF) 1α implicated in UGT1A4 regulation, farnesoid X receptor (FXR) and peroxisome proliferator activated receptor α (PPARα) in UGT2B4 regulation, and nuclear factor E2-related factor 2 (Nrf2), FXR, HNF4α, HNF1, vitamin D receptor (VDR), and forkhead box protein A1 (FOXA1) in the regulation of UGT2B7 and UGT2B15 (Hu et al., 2014). Basal gene expression of these regulators in HepaRG cells was confirmed with RT-qPCR (Supplemental Fig. 2). PXR and CAR expression was most strongly downregulated, i.e., >60% by IL-6 treatment and >90% by IL-1β treatment. IL-1β also downregulated PXR and CAR's binding partner retinoid X receptor α (RXRα) (∼60%), LXRα (∼80%), HNF1α (∼80%), AhR (∼50%), Nrf2 (∼70%), and PPARα (∼80%), which was not seen after IL-6 treatment. Expression of HNF4α was downregulated by ∼70% following IL-1β treatment and ∼40% by IL-6 treatment. The other regulators FXR, NFY, and USF1 were unaffected by both IL-6 and IL-1β. Sensitivity toward IL-6 and IL-1β was evaluated for PXR and CAR, as these regulators were most affected by cytokine treatment. The IC50 values for IL-6 treatment were 0.86 ± 0.46 ng/ml for PXR and 0.38 ± 0.56 ng/ml for CAR, while for IL-1β treatment, the IC50 values were 4.37 ± 3.68 pg/ml for PXR and 2.50 ± 7.41 pg/ml for CAR. Concentration-response curves for PXR and CAR as compared with one of the key genes they regulate, CYP3A4, is presented in Supplemental Fig. 3.

The impact of IL-6 or IL-1β on transcription factors and nuclear receptors that regulate the various DMEs families. Cells were treated with 10 ng/ml IL-6 or 1 ng/ml IL-1β for 24 hours to analyze gene expression alterations via RT-qPCR. Data are expressed as the mean fold change ± S.E.M. of mRNA compared with untreated control cells of 6 independent experiments. One way ANOVA with Dunnett post hoc test was performed for every gene separately. ** P < 0.01, *** P < 0.001.
Discussion
Proinflammatory cytokine release during inflammatory conditions is associated with compromised metabolism of drugs in the liver. The impact of proinflammatory cytokines on in vitro CYP expression is well-characterized (de Jong et al., 2020). However, less attention has been credited to the effects on non-CYP phase I and phase II drug metabolism, and especially data on the effects of inflammation on DME activity is lacking. Our results demonstrate that members of the non-CYP families FMOs, CESs, and UGTs were less sensitive toward the effects of IL-6 and IL-1β as compared with the CYP family. This differential sensitivity was evident at both the DME gene expression and DME enzyme activity level, highlighting that alterations in transcription during inflammation are highly predictive for subsequent alterations in enzyme activity.
Our concentration-response experiments defined differences in both the potency and efficacy of cytokines in inducing downregulation of expression and activity of individual DME family members. While results from previous in vitro studies at supraphysiological concentrations of IL-6 have hinted toward a more limited impact on UGT isoforms as compared with CYP isoforms (Klein and Zanger, 2013; Keller et al., 2016; Gramignoli et al., 2022), this study is the first to directly compare multiple DME families on both expression and activity. Rank ordering of DME sensitivity highlighted that CYP isoforms exhibited the highest sensitivity to the modulatory effects of IL-6 and IL-1β, whereas members from the FMO, CES, and UGT families consistently showed a lower sensitivity. Importantly, this differential sensitivity was observed for both IL-6 and IL-1β treatment, even though IL-6 and IL-1β induce different inflammatory signaling pathways (Weber et al., 2010; Schaper and Rose-John, 2015) and exert different effects on transcriptional regulators (Klein et al., 2015).
The mechanisms underlying this differential sensitivity could stem from the differential impact of cytokines on the regulators of the DMEs. IL-6 and IL-1β stimulation of HepaRG cells profoundly and significantly suppress mRNA expression of PXR and CAR by >60%, whereas presumed transcriptional regulators of UGT and CES enzymes are less impacted, and FMO regulators are not at all impacted by cytokine treatment. Nuclear receptors and transcription factors implicated in DME transcriptional modulation are thus transcriptionally differentially regulated by cytokines, which might underlie the differential sensitivity to inflammation observed for various DME families. In addition to inflammation-induced alterations in gene expression of regulators, a loss of nuclear localization or alterations in the phosphorylation status of regulators has also been proposed, i.e., for the dimerization partner RXRα (Ghose et al., 2004; Keller et al., 2016). This might explain the observed mismatch between the sensitivity toward proinflammatory cytokines for CYP3A4 expression as compared with expression of the key regulators PXR and CAR. Future studies should aim to investigate whether the transcriptional downregulation concordantly leads to lower transcriptional activation of DME regulators.
Post-transcriptional mechanism related to inflammation may, alongside transcriptional changes, further affect CYP activity (Stanke-Labesque et al., 2020). For instance, nitric oxide-dependent ubiquitination leading to enhanced proteasomal degradation, or the release of inflammation-related miRNAs, have been implicated in this post-transcriptional regulatory process (Ferrari et al., 2001; Lee et al., 2009; Kugler et al., 2020). To investigate the importance of post-transcriptional mechanisms in modulating CYP activity under inflammatory conditions, we analyzed the correlation between the impact of IL-6 and IL-1β on DME expression versus DME activity. We found that, in HepaRG cells, alterations in gene expression are highly predictive for alterations in enzyme activity, providing limited evidence for inflammation-associated post-transcriptional modifications of DMEs. Previous studies suggesting the importance of post-transcriptional modifications on CYP activity mainly stem from observed mismatches between mRNA and protein levels in PHHs (Aitken and Morgan, 2007) or from animal studies (Stanke-Labesque et al., 2020). The time kinetics of alterations in expression versus protein/activity levels could partially account for the observed mismatches, and future studies should therefore evaluate the temporal dynamics of DME expression and activity alterations in response to inflammation. We conducted our activity measurements after 72 hours, in accordance with other studies and considering the reported half-life of CYP3A4, which is approximately 37 hours (Willmann et al., 2003). However, half-life of CYP2C9 is reported to be 104 hours (Willmann et al., 2003), which could explain why we found a stronger effect of inflammation on CYP2C9 expression compared with its activity. This finding is thus likely unrelated to post-transcriptional modifications but rather an effect of the protein’s half-life. All in all, our results have highlighted that the transcriptional alterations in DME expression are the main driver of the alterations in enzyme activity observed in vitro.
PBPK modeling is increasingly exploited to predict the impact of inflammation or inflammatory diseases on drug clearance. A major advantage of PBPK modeling combined with in vitro to in vivo extrapolation (IVIVE) is the ability to translate in vitro data into biologically relevant parameters for model input to predict clinical inflammation-related alterations in pharmacokinetics. Specifically, IC50 and Imax values obtained in vitro can be used to model CYP enzyme dynamics under inflammatory conditions, and this approach has been shown successful for the prediction of disease–drug interactions with CYP substrates in, for example, patients with rheumatoid arthritis, leukemia, or surgical trauma (Machavaram et al., 2013; Xu et al., 2015; Jiang et al., 2016; Machavaram et al., 2019; Lenoir et al., 2022). Despite the growing interest in PBPK modeling for non-CYP enzymes, current models predominantly focus on predicting drug–drug interactions rather than the impact of inflammation on non-CYP mediated drug clearance (Ozbey et al., 2023). This limitation arises partly due to the scarcity of physiologically relevant quantitative in vitro data on the effects of cytokines on non-CYP enzymes (Kenny et al., 2013; Liu et al., 2023). To address this gap, we provided IC50 and Imax values for non-CYP enzymes, which can serve as critical inputs for PBPK modeling to better predict inflammation-related changes in non-CYP mediated drug metabolism. Importantly, comparing our HepaRG IL-6 IC50 values with those previously determined for CYP isoforms in 2D/3D PHHs showed good agreement between the results, enhancing our confidence in the validity of HepaRG data as input for PBPK modeling approaches. Also, our reported IC50 data are within the physiological range of serum IL-1β and IL-6 in patients experiencing inflammation-related diseases (Coutant and Hall, 2018). Ultimately, PBPK models, when integrated with robust in vitro data, could serve as a powerful tool for optimizing drug dosing strategies and enhancing therapeutic outcomes in the presence of inflammation.
In the clinic, a differential impact of inflammation on DME family members has been observed, for example in nonalcoholic fatty liver disease (NAFLD) patients, where hepatic inflammation is an important contributor to disease progression (Song et al., 2023). Protein levels of CYPs were lower in diseased patients, but non-CYP enzyme levels remained relatively unchanged, except for select UGTs (Murphy et al., 2023). This was confirmed in other studies which showed CYP2C19 to be most impacted by NAFLD, whereas other DMEs were less affected (Powell et al., 2023; Govaere et al., 2024). For antifungal agents, a differential impact of inflammation has been demonstrated based on the metabolic route of the drug. Exposure of posaconazole, which is mainly metabolized by UGT1A4, was not influenced by inflammation as assessed by C-reactive protein (CRP) levels (Märtson et al., 2019). Conversely, different studies have demonstrated that trough levels of voriconazole, a substrate for CYP2C19/3A4, are increased during inflammation (Van Wanrooy et al., 2014; Veringa et al., 2017). As such, patients with inflammatory conditions may experience variation in pharmacokinetics of concomitant medication depending on the specific DME engaged in the drug’s metabolic pathway. Our study suggests that drugs utilizing secondary or alternative routes via non-CYP clearance may be less susceptible to the effects of inflammation as compared with drugs fully metabolized by CYP enzymes.
In conclusion, our study has shown that UGT, FMO, and CES enzymes are less sensitive toward the effects of proinflammatory cytokines IL-6 and IL-1β as compared with the CYP enzymes. Additionally, the findings highlight that transcriptional alterations in the DME expression are highly predictive for the alterations in enzyme activity, arguing against inflammation-related post-transcriptional modifications. Patients suffering from acute or chronic inflammatory diseases may thus be at risk for alterations in their drug metabolism, where the magnitude of the alteration likely depends on the DME family members involved in the clearance route of the drug.
Acknowledgments
The authors would like to thank and acknowledge Johan van der Reijden and Luuk Hawinkels (Department of Gastroenterology-Hepatology, Leiden University Medical Center) for their excellent support with acquiring and processing the human liver biopsies from the biobank.
Data Availability
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Material.
Authorship Contributions
Participated in research design: de Jong, Harpal, Manson.
Conducted experiments: de Jong, Harpal, van den Berg, Peter.
Performed data analysis: de Jong, Harpal, van den Berg, Hoekstra, Peter, Manson.
Wrote or contributed to the writing of the manuscript: de Jong, Harpal, Hoekstra, Rissmann, Swen, Manson.
Footnotes
- Received July 2, 2024.
- Accepted September 10, 2024.
No funding was received for this work.
The authors declare that there are no conflicts of interest.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ACTB
- β-actin
- AhR
- aryl hydrocarbon receptor
- CAR
- constitutive androstane receptor
- CES
- carboxylesterase
- CYP
- cytochrome P450
- 2D
- two-dimensional
- 3D
- three-dimensional
- DME
- drug metabolizing enzyme
- FMOs
- flavin monooxygenases
- FXR
- farnesoid X receptor
- HNF
- hepatocyte nuclear factor
- IL
- interleukin
- Imax
- maximal inhibition
- LC-MS/MS
- liquid chromatography-mass spectrometry
- PBPK model
- physiologically based pharmacokinetic model
- PHH
- primary human hepatocyte
- PXR
- pregnane X receptor
- RPLP0
- ribosomal protein lateral stalk subunit P0
- RT-qPCR
- reverse transcription-quantitative polymerase chain reaction
- UGT
- UDP glucuronosyltransferase
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}