


Abstract
Icenticaftor (QBW251) is a potentiator of the cystic fibrosis transmembrane conductance regulator protein and is currently in clinical development for the treatment of chronic obstructive pulmonary disease and chronic bronchitis. An absorption, distribution, metabolism, and excretion study was performed at steady state to determine the pharmacokinetics, mass balance, and metabolite profiles of icenticaftor in humans. In this open-label study, six healthy men were treated with unlabeled oral icenticaftor (400 mg b.i.d.) for 4 days. A single oral dose of [14C]icenticaftor was administered on Day 5, and unlabeled icenticaftor was administered twice daily from the evening of Day 5 to Day 12. Unchanged icenticaftor accounted for 18.5% of plasma radioactivity. Moderate to rapid absorption of icenticaftor was observed (median time to reach peak or maximum concentration: 4 hours), with 93.4% of the dose absorbed. It exhibited moderate distribution (Vz/F: 335 L) and was extensively metabolized, principally through N-glucuronidation, O-glucuronidation, and/or O-demethylation. The metabolites M8 and M9, formed by N-glucuronidation and O-glucuronidation of icenticaftor, respectively, represented the main entities detected in plasma (35.3% and 14.5%, respectively) in addition to unchanged icenticaftor (18.5%). The apparent mean terminal half-life of icenticaftor was 15.4 hours in blood and 20.6 hours in plasma. Icenticaftor was eliminated from the body mainly through metabolism followed by renal excretion, and excretion of radioactivity was complete after 9 days. In vitro phenotyping of icenticaftor showed that cytochrome P450 and uridine diphosphate glucuronosyltransferase were responsible for 31% and 69% of the total icenticaftor metabolism in human liver microsomes, respectively. This study provided invaluable insights into the disposition of icenticaftor.
SIGNIFICANCE STATEMENT The absorption, distribution, metabolism, and excretion of a single radioactive oral dose of icenticaftor was evaluated at steady state to investigate the nonlinear pharmacokinetics observed previously with icenticaftor. [14C]Icenticaftor demonstrated good systemic availability after oral administration and was extensively metabolized and moderately distributed to peripheral tissues. The most abundant metabolites, M8 and M9, were formed by N-glucuronidation and O-glucuronidation of icenticaftor, respectively. Phenotyping demonstrated that [14C]icenticaftor was metabolized predominantly by UGT1A9 with a remarkably low Km value.
Introduction
Cystic fibrosis (CF) is a recessive genetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (Hanssens et al., 2021). The CFTR protein is an adenosine triphosphate–binding transporter that plays a vital role in the transport of chloride and bicarbonate anions across the epithelial cell apical membrane of the lungs, pancreas, intestine, reproductive tract, and sweat glands, among others; it regulates salt absorption and is essential for the osmotic balance of mucus and its viscosity (Grand et al., 2021; Hanssens et al., 2021). CF and chronic obstructive pulmonary disease (COPD) are considered to be distinct diseases of unrelated origins; however, CFTR protein dysfunction commonly occurs in both conditions (Shi et al., 2018).
The flow of ions through activated CFTR channels can be increased by CFTR potentiators that enable effective opening of these channels (Van Goor et al., 2009; Grand et al., 2021). The CFTR potentiator ivacaftor has been shown to provide clinical and physiological improvements in patients with CF and is approved for the treatment of patients with CF who have specific CFTR mutations (Ramsey et al., 2011; US Food and Drug Administration, 2012; European Medicines Agency, 2012a; Rowe et al., 2014; Moss et al., 2015). Icenticaftor (QBW251) is a new, orally bioavailable, low-molecular-weight CFTR potentiator of both the wild-type and mutated/defective forms of the CFTR protein that has been investigated in patients with CF and COPD (Rowe et al., 2020; Kazani et al., 2021; Mall et al., 2023; Martinez et al., 2023). Icenticaftor up to 750 mg twice daily was well tolerated in the first-in-human study of healthy subjects and up to 450 mg twice daily in patients with CF (Kazani et al., 2021). Moderate to rapid absorption of icenticaftor (time to reach peak or maximum concentration [Tmax], 0.8–4 hours) was observed, and the overall terminal half-life (T1/2) was 10–13 hours in healthy subjects. The mean maximum observed concentration (Cmax) in patients with CF ranged from 419 ng/ml (150 mg b.i.d., Day 1) to 4080 ng/ml (450 mg b.i.d., Day 14). Icenticaftor is currently in development as an adjunctive therapy for the treatment of COPD and chronic bronchitis. In a Phase 2 proof-of-concept study conducted in patients with COPD, improvements in respiratory function (pre- and postbronchodilator forced expiratory volume in 1 second [FEV1]) were observed over 4 weeks of icenticaftor treatment (Rowe et al., 2020). In addition, the results of a Phase 2b dose-finding study with icenticaftor demonstrated potentially clinically relevant benefits for patients with COPD and chronic bronchitis, including improvement in trough FEV1, reduction in cough, sputum, and rescue medication use, and a decline in fibrinogen levels at 24 weeks (Martinez et al., 2023).
The metabolic characteristics of icenticaftor have not been reported previously. Human absorption, distribution, metabolism, and excretion (ADME) studies are essential for understanding the pharmacokinetic (PK) properties of pharmaceutical drug candidates within the body, including the processing of a drug and its metabolites and their impact on drug safety (Coppola et al., 2019; Lindmark et al., 2023).
This study was designed to evaluate the ADME properties of icenticaftor, including PK parameters, in healthy volunteers. In addition, it was supplemented with an in vivo rat ADME study and in vitro assays with mouse, rat, monkey, and human hepatocytes for better elucidation of the clearance pathways and enzymes involved in the metabolism of icenticaftor.
Materials and Methods
Study Rationale
This single-center, open-label study investigated the ADME of icenticaftor, which has been shown to have nonlinear exposure with increasing dose after both single- and multiple-dose administration (Supplemental Fig. 1). The European Medicines Agency guidelines assert that a single-dose study is sufficient to investigate the ADME properties of a drug if there is no dose or time dependency in the first-pass metabolism or elimination of the drug (European Medicines Agency, 2012b). These guidelines also state that if the elimination is nonlinear, the design and degree of saturation should mimic the therapeutic situation for nonlinear elimination, that is, for drugs with dose-dependent elimination and significant accumulation under multiple dosing, a single dose of the radiolabeled drug should be administered when steady state has been reached with nonradiolabeled drug (European Medicines Agency, 2012b). Similarly, the United States (US) Food and Drug Administration guidelines state that a single-dose mass balance study is generally sufficient (US Food and Drug Administration, 2024). Under certain instances, a multiple-dose study can be considered, in which subjects would receive a single dose of radiolabeled drug after reaching steady state conditions with the nonradiolabeled drug; however, this approach evaluates only the clearance pathway of the radiolabeled drug, whereas bioanalysis of the nonradiolabeled moieties at steady state can be useful for interpreting the results (US Food and Drug Administration, 2024). Therefore, to satisfy agency guidelines, the present human ADME study was conducted at steady state with a single, oral, radiolabeled dose of icenticaftor.
In vitro investigations were also conducted to identify the metabolizing enzymes involved in the clearance of icenticaftor. Early biotransformation data indicated the involvement of glucuronidation and oxidative metabolism; therefore, human cytochrome P450 (CYP) and uridine diphosphate glucuronosyltransferase (UGT) phenotyping was performed.
Study Design and Subjects
This Phase 1, single-center, open-label study was conducted in six healthy male volunteers. Subjects were 18–55 years old, with a body mass index of 18.0–30.0 kg/m2 and body weight of 55–120 kg. Other key inclusion and exclusion criteria are detailed in the Supplemental Material.
The study included a 28-day screening period (including baseline period of 1 day) followed by a 12-day treatment period (see Supplemental Fig. 2). Nonradiolabeled icenticaftor (400 mg) was administered twice daily for the first 4 days (Days 1–4). On Day 5, at steady state, a single dose of [14C]icenticaftor 3.7 MBq (100 μCi) 400 mg was administered in the morning followed by a single dose of nonradiolabeled icenticaftor 400 mg in the evening. Further doses of nonradiolabeled icenticaftor (400 mg b.i.d.) were given on Days 6–12. All treatment administrations occurred approximately 30 minutes after consumption of a standardized meal.
Ethics Approval
The study was conducted in accordance with the ethical principles consistent with the International Conference on Harmonization Good Clinical Practice guidance E6, and the protocol was reviewed and approved by the independent ethics committee at the study site. All subjects provided written informed consent.
Study Objectives
The primary objectives of the study were to (1) determine the rates and routes of excretion of [14C]icenticaftor-related radioactivity, including mass balance of total drug-related radioactivity in urine and feces following a single 400 mg oral dose of [14C]icenticaftor at steady state; (2) determine the PK of total radioactivity in blood and plasma; and (3) characterize the plasma PK of icenticaftor. The secondary objective was to assess the safety and tolerability of multiple oral doses of 400 mg of icenticaftor. Exploratory objectives included the identification and semiquantification of icenticaftor and its metabolites in plasma and excreta (urine and feces) to elucidate key biotransformation pathways and clearance mechanisms in humans and to characterize the plasma PK of icenticaftor and its key metabolite(s), based on radiometry data.
Study Treatments
The 400 mg dose containing 3.7 MBq [14C]-radiolabeled icenticaftor was chosen to provide sufficiently high drug and metabolite levels to meet the objectives of this study without safety concerns. The radiation exposure for a subject had been estimated according to the International Commission on Radiological Protection and was considered to be acceptable (<1 mSv). Nonradiolabeled icenticaftor was provided as 100 mg hard gelatin capsules (Novartis Pharma AG, Basel, Switzerland), and the molecular weight of the nonradiolabeled, free base compound was 361.24 g/mol. [14C]Icenticaftor was supplied as 100 mg hard gelatin capsules (Almac Sciences, Ltd., Craigavon, UK). Radiochemical purity was 99.7%. The actual specific radioactivity of the solid drug substance was 9.15 kBq/mg (free base) and was measured at Almac Sciences, Ltd. (Craigavon, UK). The bottles containing the capsules and outer packaging were individually labeled with details of the investigational agent. The chemical structure of the compound, including the position of the radiolabel, is shown in Fig. 1.

Chemical structure of the radiolabeled icenticaftor.
Chemicals and Standards
Nonradiolabeled icenticaftor was synthesized and provided by Novartis Pharma AG (Basel, Switzerland). Radiolabeling of icenticaftor ([14C]icenticaftor) was performed by Almac Sciences Ltd. (Craigavon, UK) and [13CD3] icenticaftor (internal standard) was supplied by Tjaden Biosciences (Iowa, USA). Reference standards for the metabolites M8, M9, and M14 were synthesized and provided by Novartis Pharma AG (Basel, Switzerland), and the standard for M5 was provided by Hypha Discovery Ltd. (Abingdon, UK), as detailed in the Supplemental Materials, Section 6. Other chemicals and solvents were obtained from commercial sources and were of analytical grade.
Sample Collection and Aliquoting
Blood and plasma samples
Blood samples were collected predose on Days −1 and 5 and then at certain time points (0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168, 192, and 216 hours) after the administration of the [14C]icenticaftor dose on Day 5. All blood samples (8 ml or 18 ml) were collected either by direct venipuncture or using an indwelling cannula inserted in a forearm vein. Aliquots were collected for radioactivity determination and metabolite analysis.
Urine samples
Predose (blank) samples were collected from each subject on Day −1. After the administration of the radiolabeled dose on Day 5, all urine samples were collected during the time intervals 0–6, 6–12, and 12–24 hours and, thereafter, in 24-hour fractions up to 216 hours. To stabilize the acid-labile N-glucuronide in the individual urine fractions, the pH was adjusted to neutral pH values. Aliquots were collected for radioactivity determination (two aliquots of 1 ml) and metabolite analysis (two aliquots of 20 ml). The urine samples were frozen and stored below −60°C until analysis.
Fecal samples
Predose (blank) samples were collected from each subject on Day −1. Following the administration of the radiolabeled dose on Day 5, all fecal samples were collected and pooled per 24 hours, if needed, during the postdose sample collection period of 216 hours. Each sample was diluted with one to two volumes of water containing a suitable suspension stabilizer and then homogenized. Aliquots were collected for radioactivity determination and metabolite analysis.
Full details of the sample collection schedule, storage conditions and processing/analytical facilities are provided in the Supplemental Material.
Analysis of Total Radioactivity
Total [14C] concentrations of icenticaftor and its metabolites were determined using liquid scintillation counting or combustion with subsequent liquid scintillation counting (Supplemental Material).
Determination of Plasma Icenticaftor Concentrations
Plasma concentrations of unchanged icenticaftor were measured using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) assay (Supplemental Material).
Determination of Metabolite Profiles in Plasma, Urine, and Feces
Metabolite profiles in plasma, urine, and feces were investigated for up to 48, 96, and 120 hours, respectively. Plasma samples were pooled by combining identical aliquots of the same timepoints (time pools). Extracts of plasma samples were obtained by protein precipitation with acetonitrile. The final extracts were evaporated, reconstituted, and analyzed using high-performance liquid chromatography mass spectrometry (HPLC-MS).
Urine samples collected from each subject and pooled across 0–96 hours were prepared by combining identical volume percentages of the different urine fractions. A 150 μl aliquot was used for HPLC-MS analysis (Supplemental Material).
Fecal samples collected from each subject were pooled across 0–120 hours, and a homogenate pool prepared by combining identical volume percentages of the different homogenate fractions. Aliquots of pooled feces reconstituted sample extracts (100 μl) were analyzed by HPLC-MS using the Agilent model 1200 HPLC system (Agilent Technologies, Waldbronn, Germany). The detailed methods are described in the Supplemental Material.
Structural Characterization of Metabolites
Structures of metabolites in plasma and excreta were characterized using LC-MS/MS analysis. Offline radioactivity detection was used to correlate peaks with mass spectral data. Product ion mass spectra, exact mass measurement, and hydrogen/deuterium exchange experiments were used to derive metabolite structures, which were compared with those of synthetic standards where possible. Synthetic reference standards were prepared using biosynthetic methods (Supplemental Material, Section 6) and characterized by nuclear magnetic resonance spectroscopy (Supplemental Fig. 3).
Data Analysis
Multiple-dose plasma PK parameters Cmax, Tmax, area under the concentration-time curve calculated from time zero to the last measured time point [AUClast], apparent volume of distribution during the terminal elimination phase following extravascular administration [Vz/F], apparent systemic [or total body] clearance from plasma following extravascular administration at steady state [CLss/F]) for icenticaftor and its metabolites at steady state were derived from plasma concentration versus time data. Single-dose blood and plasma PK parameters (Cmax, Tmax, T1/2, AUClast, and area under the concentration-time curve from time zero to infinity [AUCinf]) for total radioactivity were derived from plasma and blood concentration versus time data for total radioactivity.
PK calculations were based on the recorded time of sample collection, and PK parameters were calculated using noncompartmental analysis (WinNonlin version 6.4; Certara, Princeton, NJ, USA). The terminal rate constant (λz) was determined from linear regression of at least the last three, nonzero measurements in the terminal phase of the log-transformed concentration-time profile (WinNonlin algorithm). The extent of icenticaftor and metabolite excretion in urine and feces was expressed as a percentage of the total radioactive dose administered.
The fraction of radioactivity in plasma was calculated: Fraction of plasma radioactivity (%) = (plasma radioactivity concentration/blood radioactivity concentration) × (1 – hematocrit) × 100.
In Vitro Phenotyping of Icenticaftor Metabolites
In vitro phenotyping was conducted to quantitatively evaluate the contribution of human CYP and UGT enzymes in the metabolism of [14C]icenticaftor in human liver microsomes (HLMs) and recombinant enzymes (Supplemental Material).
In Vitro Biotransformation in Mouse, Rat, Monkey, and Human Hepatocytes
[14C]Icenticaftor was incubated with hepatocytes derived from male ICR/CD-1 mice, male Sprague Dawley rats, male cynomolgus monkeys, and humans (both males and for up to 24 hours). Metabolites were extracted and analyzed by HPLC, with offline radioactivity detection. Metabolite structures were characterized using liquid chromatography mass spectrometry with accurate mass or LC-MS/MS and deuterium exchange. The detailed methods are described in the Supplemental Material.
Rat ADME Study
The ADME of icenticaftor in male Wistar rats (Han:WIST, albino) was assessed after the administration of a single intravenous dose (3 mg/kg) or oral administration (10 mg/kg) of [14C]icenticaftor. The detailed methods are described in the Supplemental Material.
Assessment of Safety
All adverse events (AEs), serious AEs, vital signs, laboratory evaluations (hematology, clinical chemistry, and urinalysis), and electrocardiogram were monitored and recorded.
Statistical Analysis
No inferential statistical analysis was performed. Descriptive statistics are provided.
Results
Subject Demographics
All six male subjects completed the study. Their median age was 32.0 years (range: 23–55 years), and the median body mass index was 26.34 kg/m2 (range: 21.39–29.41 kg/m2).
Absorption and Concentration of Radioactivity and Icenticaftor in Plasma or Blood
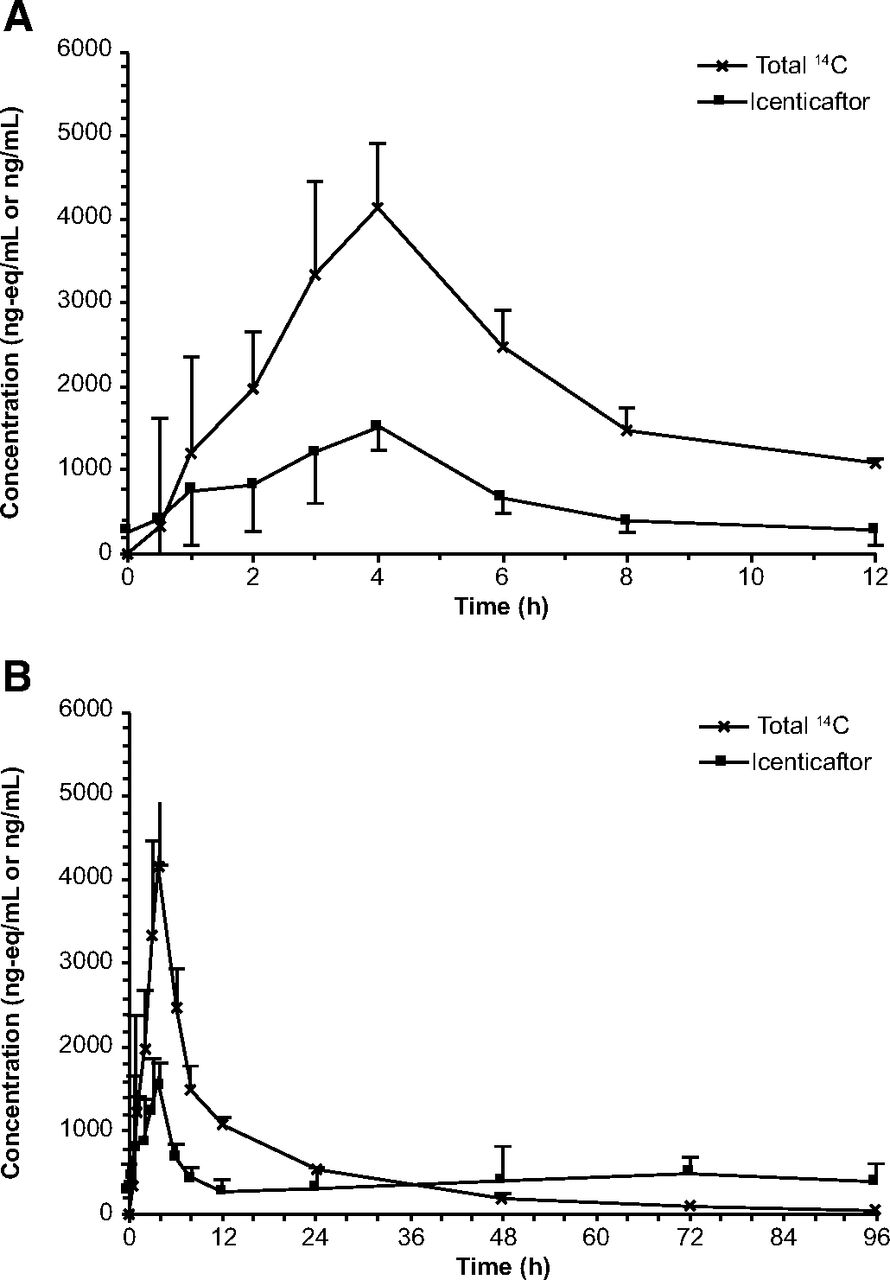
Icenticaftor demonstrated moderate to rapid absorption following oral administration, with a median Tmax of 4 hours (range: 1–4 hours) and 93.4% of the oral dose absorbed. Exposure to icenticaftor relative to total radioactivity was 16.6% of radioactivity AUCinf (Table 1). Arithmetic mean plasma concentrations of total radioactivity and icenticaftor are shown in Fig. 2. After oral administration of [14C]icenticaftor at steady state, radioactivity was detected in blood and plasma for up to 48 hours and 96 hours postdose, respectively; thereafter, radioactivity levels were below the limit of quantification (Supplemental Fig. 4).
Primary pharmacokinetic parameters of radioactivity and icenticaftor in blood and plasma

Arithmetic mean (S.D.) plasma concentrations of total radioactivity and icenticaftor. (A) Linear view 0–12 hours after the administration of the [14C]icenticaftor dose. (B) Linear view 0–96 hours after the [14C]icenticaftor dose. The figures show a combination of radioactive (liquid scintillation counting) and nonradioactive (LC-MS/MS) methods. The radiolabeled drug-related compounds (total 14C) display single-dose PK, whereas the icenticaftor concentrations reflect steady state PK.
The PK parameters of icenticaftor in plasma at steady state (Table 1) showed moderate intersubject variability for Cmax (33.8%) and AUClast (40%). The mean values of Cmax and AUClast were 1680 ng/ml and 8170 ng*h/ml, respectively. The individual Cmax and AUClast (0–12 hours) values ranged from 912 to 2360 ng/ml and from 4240 to 12900 ng*h/ml, respectively.
Distribution of Icenticaftor
The mean Vz/F of icenticaftor in plasma was 335 L (Table 1), with a mean coefficient of variation percentage of 48.1%.
The mean blood/plasma AUCinf ratio of radioactivity was 0.579 (range: 0.542–0.620). Additionally, icenticaftor and/or its metabolites did not exhibit any special affinity to erythrocytes as indicated by the similar ratio and decline of compound-related radioactivity in blood and plasma in the concentration-time curves (Supplemental Fig. 4). Compared with blood, the mean ± S.D. fraction of total radioactivity in plasma was 93.3 ± 3.35%.
Metabolism of Icenticaftor
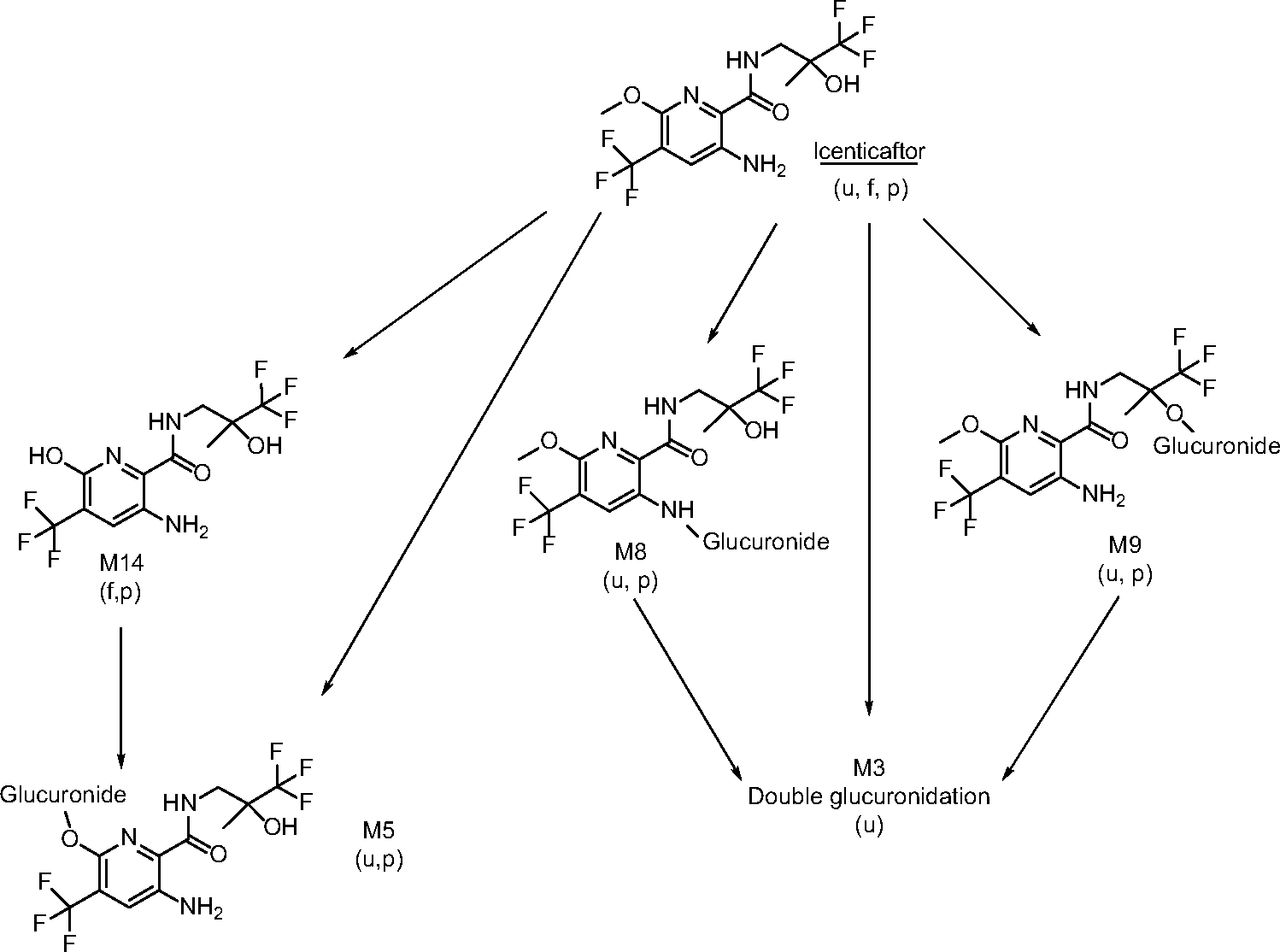
Icenticaftor was extensively metabolized mainly by N-glucuronidation, O-glucuronidation, and/or O-demethylation (Table 2). The concentrations of icenticaftor and metabolites in plasma following a single oral dose of 400 mg [14C]icenticaftor at steady state, and the estimated AUCs are presented in Supplemental Table 1. Unchanged icenticaftor (radioactivity AUC0–48h of 18.5%) and its metabolites, M8 (AUC0–48h of 35.3%) and M9 (AUC0–48h of 14.5%), represented the main circulating entities in plasma (Table 2). M8 and M9 were formed by N-glucuronidation and O-glucuronidation of icenticaftor, respectively. Another metabolite, M5 (formed by O-demethylation and glucuronidation), accounted for 10.7% of the plasma [14C]-AUC0–48h. Other minor metabolites were identified but each accounted for ≤3% of the plasma [14C]-AUC0–48h (Supplemental Table 1). Figure 3 shows a representative metabolite profile in plasma at 4 hours and 48 hours postdose, and Fig. 4 shows the concentration-time course of icenticaftor and its main metabolites in plasma at 0–48 hours postdose.
AUC0–48h, AUCinf, and T1/2 of icenticaftor and its metabolites in plasma based on metabolite pattern analysis

Representative metabolite profile in plasma at 4 and 48 hours postdose. (A) Plasma metabolite profile at 4 hours (Tmax). (B) Plasma metabolite profile at 48 hours. The profiles were obtained using HPLC analysis after direct injection of the plasma extract into the HPLC system and subsequent detection of radioactivity in 96-well plates.

Concentration-time course of icenticaftor and its main metabolites in plasma at 0–48 hours postdose. Metabolite profiles in plasma were determined from plasma pools of six subjects using HPLC analysis, with radioactivity detection at time points 1, 3, 4, 8, 12, 24, and 48 hours.
Metabolites were excreted mainly in urine, with M8, M9, and M5 being the most abundant metabolites (50.1%, 18.4%, and 11.0% of the radioactive dose, respectively; Fig. 5 and Supplemental Table 2). Other metabolites in urine, most of which contributed to ≤1% of the radioactive dose, were mainly formed by oxygenation, N-oxidation, O-demethylation, N-dealkylation and/or glucuronidation(s). Unchanged icenticaftor was the main component in feces (2.68% of the radioactive dose; Fig. 5 and Supplemental Table 2). Minor metabolites detected in feces included M14, M32, and M33, which were formed by O-demethylation, sulfation, and N-acetylation, respectively, of the parent drug. The structures of the metabolites were derived from LC-MS/MS analysis; mass spectral data showing the order of elution and major signals are presented in Supplemental Table 3 and the electrospray mass spectrum of icenticaftor is shown in Fig. 6. Based on all the information gained, the proposed major biotransformation pathway of icenticaftor is depicted in Fig. 7. The proposed minor biotransformation pathway of icenticaftor is shown in Supplemental Fig. 5. Overall, icenticaftor was mainly metabolized by N-glucuronidation, O-glucuronidation, and/or O-demethylation, forming the metabolites M8, M9, and M5; however, N-dealkylation, N-oxidation, oxygenation, C-hydroxylation, formation of a carboxylic acid, sulfation, and N-acetylation also contributed to a minor extent to the biotransformation of icenticaftor.

Representative metabolite profiles in (A) urine and (B) feces. The urine fraction pool (0–96 hours; N = 6), containing 90.4% of the total excreted dose, and the feces fraction pool (0–120 hours; N = 6) were evaluated using HPLC analysis, with subsequent radioactivity detection in 96-well plates.

Electrospray mass spectrum of icenticaftor. (A) Key fragments used for the elucidation of the structure of metabolites. (B) Electrospray ionization, positive ion mode, sample cone voltage of 30 V, trap collision energy ramp of 10–40 eV. The accurate mass measurements were in agreement with the proposed fragmentation. The difference between the calculated and measured masses was ≤0.5 mDa for all indicated fragment ions.

Scheme of the major biotransformation pathway of icenticaftor in humans (Pathway I). The metabolites were detected in plasma, urine, and/or feces. f, feces; p, plasma; u, urine.
In vitro biotransformation data from mouse, rat, monkey, and human hepatocytes indicated that the major metabolites (above 5% of the total radioactivity) were M8 (all four species), M9 (monkey and human), and M16 (rat) (Supplemental Table 4). The main metabolite in mouse, rat, and human hepatocytes was M8 (direct N-glucuronide) and in monkey hepatocytes, M9 (O-glucuronide); however, different profiles for minor metabolites were observed in the different species. A minor metabolic pathway in mouse, rat, and human hepatocytes involved O-demethylation on the pyridine moiety and subsequent glucuronidation (M5). Supplemental Fig. 6 shows the metabolic profiles in human hepatocytes at concentrations of 1–30 μM, with major metabolites M8 and M9 as well as some minor metabolites (e.g., M5 and M25). Supplemental Fig. 7 shows the in vitro metabolic pathways observed across species as described above and in Supplemental Table 4. In vivo metabolic profiles obtained in rats showed that M8 and M9 were the main components in the bile of bile duct–cannulated rats (Supplemental Table 5), while only trace levels were present in feces of intact rats (Supplemental Table 6). Additionally, the larger amount of unchanged drug in the feces of intact rats (9.19% of the dose) indicates hydrolysis of the glucuronides in the intestine of rats by intestinal microbiota. These in vivo data are in line with the in vitro data for metabolism following hepatocyte incubations (Supplemental Table 4).
Excretion
Total radioactivity was excreted mainly in urine (mean: 92.3% of the total dose) and feces (mean: 5.2% of the total dose); more than 95% of the dose was recovered within 96 hours. By 216 hours postdose, excretion of radioactivity was near complete (97.6%; range: 93.4–99.1%), with less than 3% of the dose being excreted after 96 hours. Figure 8 shows a graph for the cumulative excretion of radioactivity in urine and feces.

Cumulative excretion of radioactivity in urine and feces.
Renal excretion of radioactivity was mainly in the form of metabolites (mean ± S.D.: 92.3 ± 4.2% of the dose), with minor amounts of unchanged icenticaftor (3.35% of the total dose within 0–96 hours). In feces, unchanged icenticaftor was the most abundant component (2.68% of the dose), with 1.07% excreted as metabolites. The excretion of radioactivity was complete after 9 days (mean: 97.6%; range: 93.4–99.1%).
Overall, the metabolite investigations in excreta indicated that the majority of the dose (80%) was eliminated by direct glucuronidation (M8, M9), with the remaining dose (18%) eliminated by oxidative pathways partly in combination with glucuronidation, mainly M5, and by other metabolic reactions (2%). For the calculation, it was assumed that 100% of the [14C]icenticaftor dose was recovered and that residual icenticaftor in excreta was formed by hydrolysis of the direct glucuronides in the bladder and intestine.
Systemic clearance (CLss/F) of [14C]icenticaftor was moderate (mean ± S.D.: 56.8 ± 25.6 L/h), and the mean T1/2 of total radioactivity in blood and plasma was 15.4 and 20.6 hours, respectively (Table 1).
In Vitro Phenotyping of Icenticaftor Metabolites
The main metabolic pathways identified in HLMs were mediated by UGT1A9, UGT2B7, CYP1A2, and CYP3A4. Several UGT isoforms (UGT1A3, UGT1A4, UGT1A9, and UGT2B7) were involved in direct glucuronidation of icenticaftor. The formation of the main metabolite M9 (O-glucuronide) was mediated by UGT2B7, with a contribution ratio (fraction of drug systemically available that is converted to metabolites [fm]) of 13% (Table 3). UGT1A9 was predominantly responsible for the formation of the N-glucuronide (M8), which is a major metabolite of icenticaftor in HLMs. UGT1A3 and UGT1A4 were minor pathways (data not shown). Furthermore, UGT1A7 and UGT1A8 (extrahepatic UGT enzyme isoforms) could catalyze M8 formation (data not shown).The formation of M14, M16, M17, M18, and M19 was mediated by CYP1A2 and CYP3A4, with fm 11.6% and 19.4%, respectively (Table 3). UGT1A9 had the lowest unbound Michaelis–Menten constant (Km,u: 0.34 μM).
Summary of in vitro CYP and UGT phenotyping results of icenticaftor
Based on the UGT phenotyping results and the calculated unbound intrinsic clearance values for CYP phenotyping, the contributions of CYPs and UGTs to total icenticaftor metabolism in HLMs (fm) were estimated as 31% and 69%, respectively (Table 3). Further information on the kinetic parameters and identification of human UGT isoenzymes involved in the glucuronidation of icenticaftor is presented in Supplemental Tables 7 and 8 and representative radiochromatograms are presented in Supplemental Figs. 8 and 9.
Safety
Multiple doses of oral icenticaftor 400 mg and a single oral dose of radiolabeled [14C]icenticaftor 400 mg were well tolerated in the healthy male subjects. All six subjects received the dosing according to protocol specifications and completed the study. No deaths, serious AEs, or AEs that led to discontinuation were observed. A total of 24 AEs were reported, which were mild in severity (headache, n = 4; rhinitis, n = 3; nasopharyngitis and somnolence, n = 2 each; abdominal discomfort, catheter-site pain, limb injury, pain in extremity, pollakiuria, n = 1 each). Eleven events of headache reported in four subjects were considered related to the study drug.
Discussion
In a human ADME study, it is assumed that the mean oral absorption can be estimated if the drug and its metabolites are stable against the action of intestinal bacterial enzymes. Initially, the oral absorption of icenticaftor was estimated as 93.4% of the administered dose given that 92.3% of the [14C]icenticaftor dose was excreted in urine and 1.07% was excreted in feces as metabolites. Total excretion was largely a result of the renal excretion of icenticaftor as direct glucuronides. In this study, the main glucuronides, M8 and M9, were not detected in feces, but these may have been hydrolyzed by intestinal microorganisms, as described previously for other drugs (Wilson and Nicholson, 2017). Interestingly, M8 and M9 were the main metabolites identified in the bile (bile duct–cannulated rats), whereas both glucuronides were detected only in trace amounts in the feces of intact rats. This finding suggests that M8 and M9 are not stable against the action of bacterial enzymes in the intestine of rats, a finding that could be extrapolated to humans. Although there are no intravenous data to definitively determine the extent of absorption, given the above findings, absorption could be assumed to be complete for humans (100%), with the major contribution by glucuronidation (80%) largely through renal excretion.
The distribution of icenticaftor within the human body was moderate (Vz/F of 335 L) compared with the volume of water in the body (42 L). Icenticaftor and its metabolites were mainly confined to the plasma compartment, as indicated by the mean blood/plasma AUCinf ratio of 0.579. Therefore, no special affinity of icenticaftor and/or its metabolites for erythrocytes could be concluded.
Peak concentrations of icenticaftor and total radioactivity after oral dosing of 400 mg [14C]icenticaftor to human subjects showed good systemic availability of icenticaftor. In this human ADME study, the steady state AUC (8170 ng*h/ml, Table 1) was identical to the single-dose [14C] AUCinf of icenticaftor (22700 nmol*h/L or 8200 ng*h/ml, Table 2), supporting the validity of the dosing approach. Steady state PK parameters reported for this study were comparable to the results of a previous study of healthy subjects (Kazani et al., 2021). On Day 14 in healthy subjects who received icenticaftor 450 mg twice daily, there was rapid absorption of icenticaftor (Tmax 4 hours), the mean Cmax was 2190 ng/ml, and the AUC0–12h was 12100 ng*h/ml (Kazani et al., 2021).
The PK parameters of radioactivity and icenticaftor in plasma and blood had relatively low to moderate variability for Cmax and AUC, supporting the pooling strategy used for the metabolite pattern investigations. In such cases, time pools across subjects can enable PK calculations for individual metabolites, which may not be detectable in individual subjects, e.g., as for metabolite M5, M14, and M17 (Table 2)
Unchanged icenticaftor and the metabolites M8, M9, and M5 represented the majority of the circulating moieties. Metabolism occurred mainly by N-glucuronidation, O-glucuronidation, and/or O-demethylation and subsequent renal excretion. Approximately 80% of the icenticaftor dose was eliminated by glucuronidation and 18% by glucuronidation and oxidation, with the remaining 2% elimination occurring by other metabolic reactions.
Interestingly, the data from the in vitro studies conducted with rat and human hepatocytes were comparable and revealed M8 and M9 as major metabolites. However, in the systemic circulation of rats, the metabolites M8 and M9 were only of minor importance when compared with those in circulation in humans. Glucuronides were eliminated mainly through the hepatic biliary route in rats compared with the renal route in humans. This indicates a species difference between rats and humans in the elimination of icenticaftor and emphasizes the importance of conducting a human ADME study.
Phenotyping analysis suggested M8 formation through UGT1A9 is likely a major component in icenticaftor metabolism in HLMs (fm = 56%), followed by CYP enzymes (CYP1A2 with partial contribution of CYP3A4; fm = 31%) and UGT2B7 (M9 formation, fm = 13%). The fractional contributions calculated using the in vitro data support the in vivo data, where UGT and CYP fm values of 80–98% and 0.34–18%, respectively, were determined.
In terms of safety, simple O-glucuronides, O-sulfates, and quaternary N-glucuronides can be considered benign from a human safety perspective, but adequate animal exposure may be required for those that undergo chemical rearrangement (e.g., acyl glucuronides) because of reactivity concerns (Luffer-Atlas and Atrakchi, 2017; US Food and Drug Administration, 2020). Notably, the main metabolites of icenticaftor (M8 and M9) were simple O- or N-glucuronides, not acyl glucuronides; therefore, safety concerns were not expected. Additionally, guidelines state that assessment of direct N-or O-glucuronides (M8 and M9, respectively) may not be needed because Phase 2 conjugation reactions generally render a compound more water soluble, thereby eliminating the need for further evaluation (European Medicines Agency, 2012c; US Food and Drug Administration, 2020). Despite this, M8 and M9 metabolites were evaluated in toxicity studies because of the abundance of both metabolites compared with the abundance of the parent drug.
Human ADME studies are typically performed as single-dose studies (Ramamoorthy et al., 2022), which are considered sufficient if there is no dose or time dependency in the first-pass metabolism or elimination of the drug (European Medicines Agency, 2012b). The administration of a radioactive dose of icenticaftor at steady state was deemed necessary to further investigate the rate and route of elimination of icenticaftor following the observation of dose nonlinearity after the administration of a single dose of icenticaftor in a previous investigation. If there is dose- or time-dependent elimination of a drug, then the contribution of different pathways may be different at steady state than at single-dose conditions. Drug accumulation is one of the factors influencing drug dose nonlinearity; however, this may also occur because of other reasons, including saturation of enzymes (Pharmacy180.com). The results would likely have been different if the hADME study had been performed in a single-dose study because different fractions are expected to be metabolized per pathway due to the saturation of at least UGT1A9 (Km,u: 0.34 μM). This is indicated by a more than 2-fold higher AUC and a slightly higher Cmax at steady state than that after a single-dose icenticaftor. The nonlinear PK observed after the administration of both single and multiple doses of icenticaftor is most likely due to saturation of UGT1A9 in the intestine and liver and consequent CL/F decreases with increasing plasma concentrations. This conclusion is supported by the low Km,u value (0.34 μM) of UGT1A9 in the major metabolic pathway.
The present study was tailored to suit the PK characteristics of icenticaftor, in agreement with the guidelines set by applicable health authorities, namely Appendix V of the European Medicines Agency “Guideline on the Investigation of Drug Interactions” and the US Food and Drug Administration draft guidance on the design of human mass balance studies for identifying and quantifying the main elimination pathways in vivo (European Medicines Agency, 2012b; US Food and Drug Administration, 2024). Administering a single dose of radiolabeled icenticaftor when steady-state concentrations have been reached with the nonradiolabeled drug can reveal the metabolic and elimination fate of the radiolabeled drug. While there may be concerns that such studies would not provide information about the saturating behavior of the radiolabeled drug or the metabolic profile of the metabolites at steady state, the European Medicines Agency states that there is no need to estimate AUC0–T (area under the curve [AUC] limited to the end of a dosing interval) because the AUC0–inf of metabolites observed at steady state will reflect the AUC0–T of the metabolite (European Medicines Agency, 2013). Hence, this study supported the development of icenticaftor by providing invaluable insights into its disposition and its low victim drug–drug interaction potential.
Conclusions
Icenticaftor was extensively metabolized, with both the original compound and metabolites confined mainly to the plasma compartment, where the metabolites M8, M9, and M5 represented the main metabolites in circulation. Icenticaftor was eliminated mainly through metabolism and subsequent renal excretion (92.3% excretion through urine [kidneys] and 5.25% through feces [liver/bile]).
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharma AG, Basel, Switzerland. The authors thank Sorcha Mc Ginty; Helen Swainston (Novartis Global Business Solutions, Dublin, Ireland); and Rina Vekaria Passmore (Novartis Global Business Solutions, London, UK) for their medical editorial assistance with this manuscript.
Data Availability
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Material.
Authorship Contributions
Participated in research design: Glaenzel, Huth, Hackling.
Conducted experiments: Glaenzel, Leuthold, Meissner.
Contributed new reagents or analytic tools: Glaenzel, Eggimann; Meissner.
Performed data analysis: Glaenzel, Huth, Hackling, Leuthold, Meissner, Bebrevska.
Wrote or contributed to the writing of the manuscript: Glaenzel, Huth, Eggimann, Hackling, Leuthold, Meissner, Bebrevska.
Footnotes
- Received April 9, 2024.
- Accepted September 9, 2024.
This study was funded by Novartis Pharma AG, Basel, Switzerland.
All authors are employees of Novartis. All authors except LB hold Novartis shares.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- AE
- adverse event
- AUC%extrap
- percent of AUCinf extrapolated
- AUC
- area under the curve
- AUCinf
- area under the concentration-time curve from time zero to infinity
- AUClast
- area under the concentration-time curve calculated from time zero to the last measured time point
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CLint,u
- unbound intrinsic clearance
- CLss/F
- apparent systemic (or total body) clearance from plasma following extravascular administration at steady state
- Cmax
- maximum observed concentration
- COPD
- chronic obstructive pulmonary disease
- CYP
- cytochrome P450
- FEV1
- forced expiratory volume in 1; second
- fm
- fraction of the systemically available drug that is converted to metabolites
- HPLC
- high-performance liquid chromatography
- HPLC-MS
- high-performance liquid chromatography mass spectrometry
- HLM
- human liver microsome
- Km
- Michaelis–Menten constant (substrate concentration producing half-maximal velocity)
- Km,u
- unbound Michaelis–Menten constant
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- PK
- pharmacokinetic
- T1/2
- terminal half-life
- Tlast
- time of last measured concentration
- Tmax
- time to reach peak or maximum concentration
- UGT
- uridine diphosphate glucuronosyltransferase
- Vmax
- maximum velocity (reaction velocity at saturating substrate concentration)
- Vz/F
- apparent volume of distribution during the terminal elimination phase following extravascular administration
- λz
- terminal rate constant
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}