


Abstract
Aldehyde oxidase 1 (AOX1) is a cytosolic enzyme highly expressed in liver and plays a key role in metabolizing drugs containing aromatic azaheterocyclic substituents. Rapid metabolism catalyzed by AOX1 can cause a drug to exhibit high clearance, low exposure, and hence decreased efficacy or even increased toxicity (if AOX1 generated metabolites are toxic). There is a need to develop the correlation between AOX1 expression levels and AOX1-substrate clearance. A fast, sensitive, and robust liquid chromatography–tandem mass spectrometry (LC-MS/MS) method was developed to quantify AOX1 in human liver cytosol for the first time. This LC-MS/MS method includes a straightforward ultrafiltration fractionation step and gives great selectivity and wide dynamic range (5.2 pM to 20.7 nM). The AOX1 levels in human liver cytosols of 20 donors were quantified using this method to investigate individual differences in AOX1 expression. No significant individual or gender differences in AOX1 levels were observed, although male donors exhibited a broader distribution than female donors (0.74–2.30 pmol/mg versus 0.74–1.69 pmol/mg, respectively). The AOX1 protein levels measured by LC-MS/MS were consistent with those measured by an enzyme-linked immunosorbent assay. Several donors have a normal AOX1 protein level but low enzyme activity, which might be due to cofactor deficiency, single nucleotide polymorphism, or homodimer dissociation. Cytosols from donors with chronic alcohol consumption had low AOX1-catalyzed carbazeran oxidation activities (<51 µl/min per milligram compared with a median of 455 µl/min per milligram), but preserved similar AOX1 protein expression levels (approximately 15% less than the median value)
Introduction
Aldehyde oxidase 1 (AOX1), a key xenobiotic-metabolizing enzyme, is a member of the molybdo-flavoprotein enzyme family that primarily catalyzes two metabolic pathways: 1) hydroxylation of aromatic N-heterocycles and 2) oxidation of aldehydes to the corresponding carboxylic acids (Pryde et al., 2010; Garattini and Terao, 2011). Aromatic heterocycles are key scaffolds to build pharmacophores in medicinal chemistry and a number of these compounds are putative substrates of AOX1. AOX1 can play an important role in metabolic clearance of drugs containing aromatic azaheterocyclic substituents (e.g., zaleplon, brimonidine). In addition, AOX1 bioactivation of prodrugs is exemplified in famciclovir, which is bioactivated to the potent antiviral agent penciclovir, which suffers from low oral bioavailability due to poor solubility (Rashidi et al., 1997). In addition to its drug metabolism function, AOX1 is also involved in regulation of reactive oxygen species homeostasis (Li et al., 2009). Furthermore, abundant amounts of AOX1 have been observed in adipose tissue and are proposed to play a critical role in adipogenesis and lipid metabolism by modulating peroxisome proliferator-activated receptor-α (Neumeier et al., 2006; Weigert et al., 2008). Accurate quantitation of AOX1 levels in liver cytosol will enhance our understanding of the pharmacological and toxicological effects of drugs and new drug candidates as well as our understanding of the role of this enzyme in physiological functions.
Expression levels and prevalent isoforms of AOX are drastically different between human and preclinical species, which can be a source of cross-species differences in drug metabolic profiles (Dalvie et al., 2010; Diamond et al., 2010). Human tissue expressions of AOX1 were largely estimated by semiquantitative techniques such as a tissue-specific mRNA polymerase chain reaction assay (Garattini and Terao, 2011) and immunohistochemical staining (Moriwaki et al., 2001). According to these studies, liver and adrenal gland are the primary human tissues expressing high levels of AOX1. Liquid chromatography–mass spectrometry (LC-MS)–based quantitation methods have emerged as a promising approach for protein quantitation, owing to their selectivity, robustness, high throughput, and capacity of multiplexing. In a typical LC-MS quantitation workflow, a protein of interest is digested into small proteotypic peptides by proteases with high substrate specificity (e.g., trypsin, Lys-C). Next, a set of suitable proteotypic peptides from the target protein are selected as surrogate measures of protein concentrations. A multiple reaction monitoring (MRM) acquisition method is employed for peptide quantification in conjunction with LC separation (Anderson and Hunter, 2006). In this method, the targeted peptides are first separated by LC. The resolved peptides in LC eluent are ionized into the gas phase, and are introduced into the mass spectrometer for detection. In the MRM mode, targeted peptide ions are preselected through the first mass filter and fragmented by collisional excitation. Fragment ions are then introduced through a second mass filter with preset masses so that only certain sequence-specific fragment ions can be mass analyzed. Compared with the full-scan tandem mass spectrometry (MS/MS) mode, the MRM mode greatly enhances detection selectivity, sensitivity, and duty cycle of targeted peptides. This LC-MS/MS method has enabled the development of a wide array of precise and robust protein quantitation assays for clinical samples (Carr and Anderson, 2008; Fernández Ocaña et al., 2012). There is a great interest in quantifying endogenous human drug metabolizing enzymes and drug transporters to assist better understanding of the absorption, distribution, metabolism, and excretion (ADME) of drugs. LC-MS/MS–based quantitation of the cytochrome P450 enzyme family (Seibert et al., 2009; Sakamoto et al., 2011; Williamson et al., 2011; Heikkinen et al., 2012), transporters (Li et al., 2008; Sakamoto et al., 2011), and UDP-glucuronosyltransferases (Sakamoto et al., 2011) have been reported in the literature to assist the development of predictive clearance models. In this report, we described a fast, sensitive, and robust LC-MS/MS method to quantify absolute AOX1 levels and the method was applied to AOX1 quantification of human liver cytosol. Accurate quantitation of AOX1 levels in human liver cytosol will help develop scaling factors for in vitro–in vivo correlation.
Materials and Methods
Human liver cytosol samples were purchased from Celsis In Vitro, Inc. (Baltimore, MD), including 20 different individual lots (M01–M10 from 10 male donors and F01–F10 from 10 female donors; listed in Supplemental Table 1) as well as a pooled lot (Mix01, pooled from 5 male donors and 5 female donors). Recombinant human AOX1 was ordered from OriGene (Rockville, MD). Rapigest SF was from Waters (Milford, MA). Stable heavy isotope-labeled and light standard peptides were obtained from New England Peptide (Gardner, MA). MS-grade trypsin was acquired from Promega (Madison, WI). Urea, iodoacetamide, dithiothreitol, and other chemicals were from Sigma-Aldrich (St. Louis, MO), unless stated otherwise.
Surrogate Peptide Mapping and Selection.
For peptide mapping, an aliquot of 5 µg recombinant AOX1 was mixed with 4 volumes of 50 mM ammonium bicarbonate buffer (pH 8.0). AOX1 protein was reduced by 5 mM dithiothreitol at 37°C for 30 minutes followed by 10 mM iodoacetamide for 10 minutes in the dark. The final solution was incubated with 0.2 µg trypsin overnight with mild agitation. The tryptic peptide solution was acidified with 10% formic acid and 20 μl of peptide sample was injected onto a fused-core C-18 column (Kinetex 50 × 2.1 mm, 2.6 μm; Phenomenex, Torrance, CA) by the CTC PAL autosampler (Leap Technologies, Carrboro, NC). A 14-min gradient was delivered by a Shimadzu 20D high-performance liquid chromatography (HPLC) system (Shimadzu Scientific Instruments, Columbia, MD) at a flow rate of 350 μl/min using the following gradient: 0 minutes, 5% B; 10 minutes, 40% B; 10.5–12 minutes, 80% B; 12.01–14 minutes, 5% B [mobile phase A (MPA) is 0.1% formic acid in water and mobile phase B (MPB) is 0.1% formic acid in acetonitrile]. A Triple Time-of-Flight (TOF) 5600 mass spectrometer (AB Sciex, Foster City, CA) was used for data acquisition using the information-dependent acquisition mode. Briefly, a full MS1 scan was performed first and the top 20 peptide-like precursor ions (m/z range 400–2000, +2 to +5 charges) in each MS1 scan were selected and fragmented to generate MS/MS spectra. Each precursor ion was excluded for a span of 20 seconds after two acquisitions. Peptide identification was performed on ProteinPilot software (AB Sciex) using the Paragon algorithm. Peptide mapping results were imported into MRMPilot software (AB Sciex) to build MRM transitions in high-resolution mode (targeted MRMHR method).
Aliquots of approximately 1 mg of human liver cytosols (50 μl) were mixed with 150 μl of 8 M urea with a supplement of protease inhibitor cocktail. After brief sonication, protein samples were reduced by 5 mM dithiothreitol at 37°C for 30 minutes followed by incubation in 10 mM iodoacetamide for 10 minutes in the dark. Small molecules and low-molecular-weight protein interferences were removed by ultrafiltration with 50k molecular weight cut off (MWCO) membrane (Millipore, Billerica, MA) according to the manufacturer’s protocol. Retentates were recovered and reconstituted in 0.1% Rapigest and 50 mM ammonium bicarbonate (pH 8.0) digestion buffer and incubated overnight at 37°C with a trypsin/protein ratio of 1 to 50. Dog cytosols were prepared in the same manner and used as matrix solution for standard curve preparation. A cocktail of stable labeled forms of surrogated peptides served as internal standards. The digested cytosolic samples were reinjected and all AOX1 peptides of interest were monitored using the targeted MRMHR method. Three peptides were selected for the final quantitation assay.
LC-PseudoMRM Quantitation.
Peptide mixtures were separated by a 15-minute gradient on a Waters Acquity ultra performance liquid chromatography (UPLC) system at a flow rate of 150 μl/min. MPA consisted of 0.1% formic acid in HPLC-grade water and MPB contained 0.1% formic acid in LC-MS–grade methanol. The LC gradient was initiated with a ramp of 12% B to 48% B in 9 minutes and followed by a steep increase to 95% B in 2 minutes. The column was washed with 95% B for 1 minute and re-equilibrated under initial conditions for 3 minutes. The inlet flow was switched online with the mass spectrometer at 1 minute and diverted to waste at 13 minutes. A dual-pressure linear ion trap (LTQ-Velos) mass spectrometer (Thermo Scientific, Waltham, MA) was operated in the selected ion mode for targeted peptide ions. Typical spray voltage, S-lens voltage, heater temperature, and capillary temperature were set to 3.5 kV, 60%, 200°C, and 250°C, respectively. Precursor ions were fragmented in the collision cell filled with helium gas at the following settings: 10 milliseconds of activation time, 25% normalized collision energy, and 0.25 Q. Peptide quantitation was carried out by integrating the area under the curve of each transition and the whole process was automated in a Xcalibur Quan Browser (Thermo Scientific).
In Vitro AOX1 Activity Assay Using Human Liver Cytosols.
To evaluate the AOX1 activity in various cytosol samples, three probe substrates (carbazeran, zoniporide, and phthalazine) were incubated with liver cytosol samples separately, using an automated procedure on a Sciclone ALH 3000 workstation (Caliper Life Sciences, Hopkinton, MA). Cytosol samples were freshly thawed before incubation and diluted to 1 mg/ml protein concentration in 100 mM potassium phosphate buffer (pH 7.4). The incubation was initiated with the addition of substrate (1 μM, final organic vehicle concentration was 0.01% dimethyl sulfoxide and 0.6% acetonitrile) in a total reaction volume of 200 μl. Reactions were quenched by the addition of cold acetonitrile containing 0.1 μM CP-628347 (Di et al., 2011) at 0, 0.5, 3, 10, 30, 90, and 270 minutes, respectively. Samples were then centrifuged at 3000g (GH3.8A rotor; Beckman Coulter, Brea, CA) for 15 min at room temperature. A 50-μl aliquot of supernatant was combined with equal volume of water and ready for LC-MS analysis. All incubations were performed in quadruplicates.
Bioanalytical Methods for AOX1 Activity Assay.
Aliquots of 20 μl of cytosol incubation samples were introduced onto a Synergi hydro-RP (2 × 10 mm, 2.5 μm; Phenomenex) column with an Aria multiplexing autosampler (Thermo Scientific). An integrated HPLC pumping system (Shimadzu Scientific Instruments) was used for solvent delivery. Samples were then eluted and detected by an API 4000-Qtrap mass spectrometer (AB Sciex) equipped with a Turbo IonSpray source. MPA consisted of 95:2.5:2.5% (2 mM ammonium acetate/methanol/acetonitrile, v/v/v%) and MPB consisted of 90:9.9:0.1% acetonitrile/water/formic acid (v/v/v%). The LC flow rate was 0.6 ml/min. To shorten analysis time, the mobile phase was held at 100% MPA for 10 seconds and then switched to 100% MPB for 15 seconds (for elution) before returning to 100% MPA for re-equilibration. A two-column switch design was used for increased throughput; while one column was in-line with the mass spectrometer for data acquisition, the other column was undergoing sample injection or column re-equilibration. MRM was used to monitor test compounds. The ionization condition and m/z transitions are listed in Table 1 for the three compounds used in AOX1 depletion assays. Peak area ratio of analyte to internal standard was calculated for each injection and was used to determine the substrate depletion rate. In vitro intrinsic clearance values of each cytosol lot were calculated from measured half-lives (t1/2) using Eq. 1:
MRM conditions for the three AOX1 substrates
 (1)
(1)Enzyme-Linked Immunosorbent Assay Methods.
A sandwich enzyme-linked immunosorbent assay (ELISA) method was developed to assess AOX1 protein concentrations in human liver cytosols. Anti-AOX1 antibody (Sigma-Aldrich) was coated onto black Maxisorp plates (Nalge-Nunc, Rochester, NY) by dilution in phosphate-buffered saline and overnight incubation at 4°C. Recombinant human AOX1 protein (OriGene) was serially diluted in an immunoprecipitation lysis buffer (Pierce Chemical, Rockford, IL) as a calibration standard from 5 µg/ml down to 20 ng/ml. Human liver cytosol samples were serially diluted in the same lysis buffer and tested at several dilutions. Samples were incubated on plates, shaking at room temperature for 1 hour. Unbound protein was washed away with a phosphate-buffered saline wash buffer. Bound protein was detected by incubation with biotinylated anti-AOX1 antibody (Sigma-Aldrich) for 1 hour, shaking, at room temperature. The plate was washed again to remove unbound antibody and incubated with horseradish peroxidase–streptavidin (Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 minutes, shaking at room temperature. The plate was washed and signal was developed by the addition of SuperSignal Pico ELISA substrate (Pierce Chemical) and detected on a luminometer (Victor3 plate reader; PerkinElmer, Waltham, MA). Concentrations of AOX1 protein for each sample were determined by comparison of sample signal output to the recombinant human AOX1 calibration curve, fitted to a four-parameter logistic regression. The dynamic range of the assay is approximately 35 ng/ml to 5 µg/ml.
Results
Peptide Mapping of AOX1 Protein and MRM Method Development.
To establish a robust LC-MS/MS quantitation method, a peptide mapping experiment was first conducted on tryptic digests of AOX1 protein standard using a high-resolution hybrid tandem mass spectrometer. The workflow of peptide mapping and targeted MRMHR on a high-resolution TripleTOF 5600 instrument incorporated three preliminary steps: 1) peptide mapping of purified protein and endogenous liver cytosol samples, 2) selection of surrogate peptide candidates, and 3) optimization of MRM transitions without the need of peptide standards in the screening stage. Under optimized digestion conditions, 84.5% sequence coverage was achieved for the recombinant AOX1 protein. A total of 291 distinct peptides were assigned as fragments from AOX1 and 165 of them were above the 95% confidence level. A similar peptide mapping experiment was performed on a crude liver cytosol digestion without fractionation. In the unfractionated cytosol sample, AOX1 protein sequence coverage decreased to 13.3% and only 9 unique peptides were identified with a confidence level above 95% out of 219 distinct peptides detected, indicating a significant ion suppressing effect of coeluents in the unfractionated liver cytosol digests. A list of 33 peptides was preselected using MRMPilot software for further evaluation, in which all 33 peptides were monitored in digested cytosol samples using a targeted MRMHR experiment. Several stringent filter criteria were applied to select suitable surrogate peptides for AOX1 protein quantitation as follows: 1) select peptides that are specific to human AOX1; 2) avoid sequences containing known single nucleotide polymorphisms (SNPs); 3) peptide length should be between 7 and 18 amino acids; 4) avoid labile amino acids including methionine, tryptophan, and cysteine; 5) avoid peptides susceptible to miscleavage; and 6) select peptides with favorable ionization and fragmentation characteristics. After careful screening, three AOX1 peptides (446VFFGEGDGIIR456, 779YIQDIVASTLK789, and 813TGIIAAVTAFAANK826, hereafter abbreviated as VFF, YIQ, and TGI, respectively) with three transitions each were selected as surrogate measures for AOX1 quantity in human liver cytosols.
Figure 1 (top and middle panels) shows the performance of established MRM transitions of three peptides in different matrices using the targeted MRMHR method. The VFF peptide yielded the most intense signal among the three peptides, whereas the TGI peptide attained the lowest signal in all experiments. Although minor chromatographic overlapping was observed for the VFF and YIQ peptides, the specificity of the MRM transitions excluded potential cross-interferences for peak quantitation (as shown by the absence of interfering peak in both chromatograms). In our case, all three transitions for each peptide coeluted tightly together and consistent ratios between individual transition profiles also confirmed the purity of peptide peaks. A comparison of AOX1 in blank buffer (Fig. 1, upper) versus human liver cytosol (Fig. 1, middle) showed that the retention time for all three peptide peaks was delayed slightly in liver cytosol. There was a concomitant loss of signal intensity primarily due to matrix interference.

LC-MS/MS quantitation method development for AOX1 surrogate peptides on 5600 TripleTOF and LTQ Velos platforms. The upper panel shows the selected MRM transitions for the three AOX1 surrogate peptides (446VFFGEGDGIIR456, 779YIQDIVASTLK789, and 813TGIIAAVTAFAANK826) digested from recombinant AOX1 protein. The signal intensity of the peptides is in the order of VFF > YIQ > TGI. For the liver cytosol samples, the same set of AOX1 surrogate peptides was monitored by MRMHR mode. Significant matrix ion suppression was observed, leading to a substantial reduction of MRM signals for the peptides of interest. In the bottom panel, the preliminary MRMHR method was transferred to a refined pseudoMRM method performed on a LTQ Velos instrument with an extended chromatographic time of 15 minutes. The signal-to-noise ratio (S/N) was enhanced by almost 3-fold for all three peptides with 5 times more diluted samples compared with the liver cytosol sample used in the MRMHR mode.
Taking advantage of the highly efficient ion fragmentation and trapping of the LTQ Velos instrument, the targeted MRMHR method was transferred from the TripleTOF 5600 platform to a pseudoMRM method to quantify AOX1 levels in human liver cytosol. Representative chromatograms of the three surrogate AOX1 peptides extracted from liver cytosol are illustrated in Fig. 1 (bottom). There were good agreements of daughter ion selections for the three most intense transitions across the two platforms. Notably, these samples analyzed by the LTQ Velos were 5-fold more diluted than the samples shown in MRMHR by TripleTOF 5600 (middle), whereas the peaks intensities still afforded a 3-fold enhancement on average. For this reason (superior detection sensitivity), the LTQ Velos platform was chosen for the final AOX1 quantitation in human liver cytosols.
AOX1 Quantitation in Human Liver Cytosols.
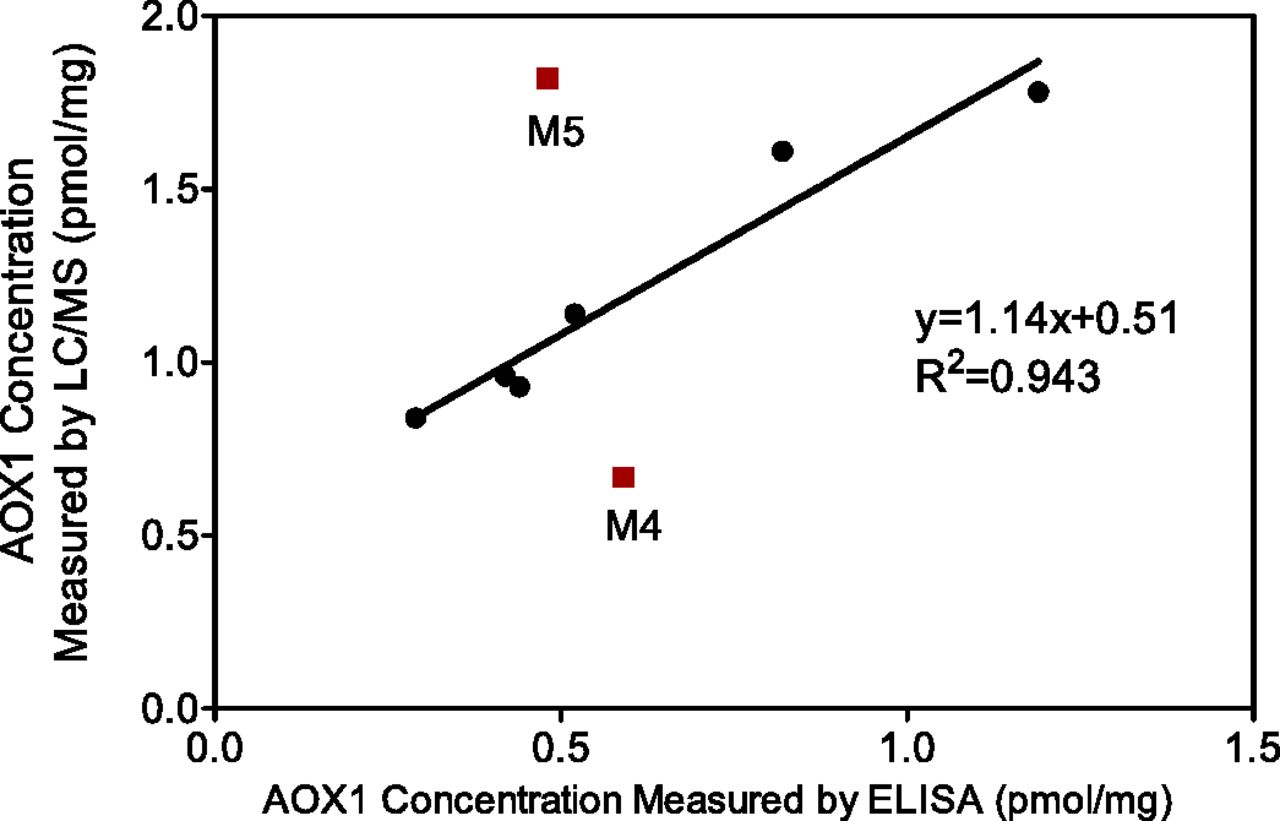
Since liver cytosol is a common matrix to study AOX1-catalyzed metabolism, the LC-MS/MS method was applied to quantitate AOX1 levels in human liver cytosol samples. Figure 2 shows the linear calibration curve for VFF peptide ranging from 6.25 pg/ml to 25 ng/ml. The linear regression gave an equation of y = 1.423x – 0.1313 and an R2 value of 0.9994. In blank dog cytosol matrix, there was no quantifiable human AOX1 peptide signal (Fig. 2), suggesting specificity of the surrogate peptide and absence of interferences from the matrix (AOX deficiency in dogs). The LC-MS/MS method showed robust peptide retention time (7.02±0.02 min) and consistent coelution of the light peptide and stable isotope-labeled internal standard. The dynamic range of the current method covered a 3.5 order of magnitude. Furthermore, the narrow confidence interval value (±5.17%) of internal standard peak intensity across the analysis suggested robust analytical performance of this peptide quantitation method. Two other AOX1 surrogate peptides also gave similar linearity, although the limit of quantitation was higher for these two peptides. Among the 20 liver cytosol samples investigated, cytosols from male donors displayed a broader spread of AOX1 levels (0.74–2.30 pmol/mg) compared with cytosols from female donors (0.74–1.69 pmol/mg). However, there was no significant gender difference (P = 0.26) in terms of the averaged AOX1 levels in each sex (Fig. 3). The AOX1 concentration in the pooled cytosol lot was 1.71 pmol/mg, which is close to the average (1.41 pmol/mg) of the individual lots. AOX1 levels in the liver cytosols of eight individual donors were also measured by an orthogonal ELISA method in parallel. Quantitation results using these two different methods correlated well to each other in general as shown in Fig. 4. The correlation between the two orthogonal quantitative methods provides confidence in the LC-MS/MS–based AOX1 quantitation method in complex biomatrices.

Calibration curve of AOX1 peptide VFFGEGDGIIR prepared in digested dog liver cytosol. The blank matrix (digested dog liver cytosol) did not show traceable interference to the peptide of interest. The LC-MS method showed robust peptide retention time (7.02 ± 0.02 min) and coelution of light peptide and stable isotope-labeled internal standard (SIS) (upper-right corner). The standard curve covered the concentration range of VFF peptide from 6.25 pg/ml to 25 ng/ml with excellent linearity (bottom right corner) (y = 1.4230x – 0.1313; R2 = 0.9994). The insert of calibration plot showed excellent linearity in the low concentration range (6.25 pg/ml to 1.25 ng/ml).

Distribution of AOX1 protein levels in 20 liver cytosols with individual donors. AOX1 levels were measured by LC-MS/MS methods (as described in the Materials and Methods). A narrow range of AOX1 concentration distribution was observed between 0.74 and 2.30 pmol/mg. There were no significant gender differences between the two groups (P = 0.26). However, samples from female donors exhibited slightly tighter AOX1 concentration distribution (0.74–1.69 pmol/mg) compared with samples from male donors (0.74–2.30 pmol/mg).
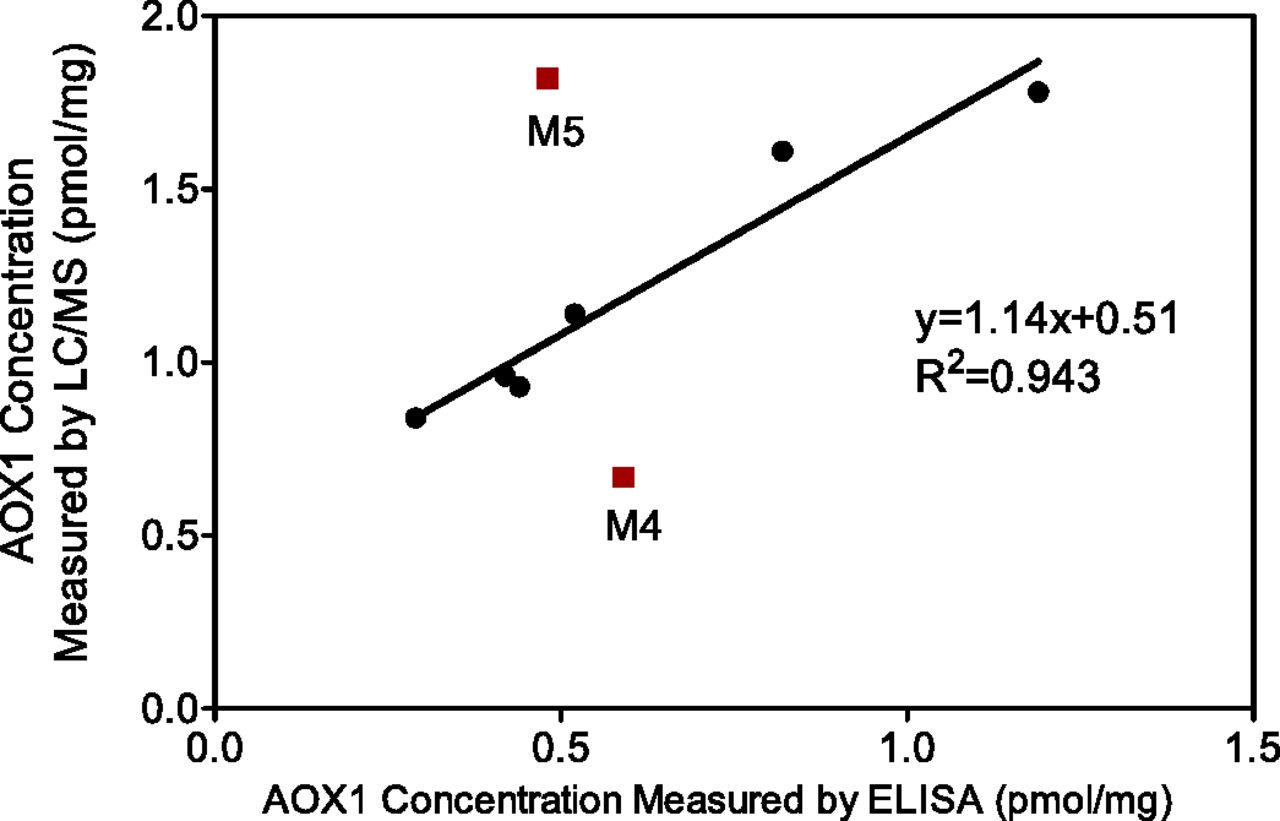
Correlation of AOX1 quantitation results by two orthogonal methods (ELISA and LC-MS/MS) using liver cytosol samples from eight donors. The linear relationship between these two quantitation methods was attained with the exclusion of two outliers (M4 and M5). For the six liver cytosols used for the fitting, the ELISA method gave lower AOX1 expression values compared with the LC-MS/MS method. This may reflect the differences (antigen-antibody binding affinity, purity, and dimer formation) between recombinant AOX1 standard proteins and endogenous AOX1 protein in the corresponding matrices.
In Vitro AOX1 Activity Measurement and Correlation to the AOX1 Expression Levels.
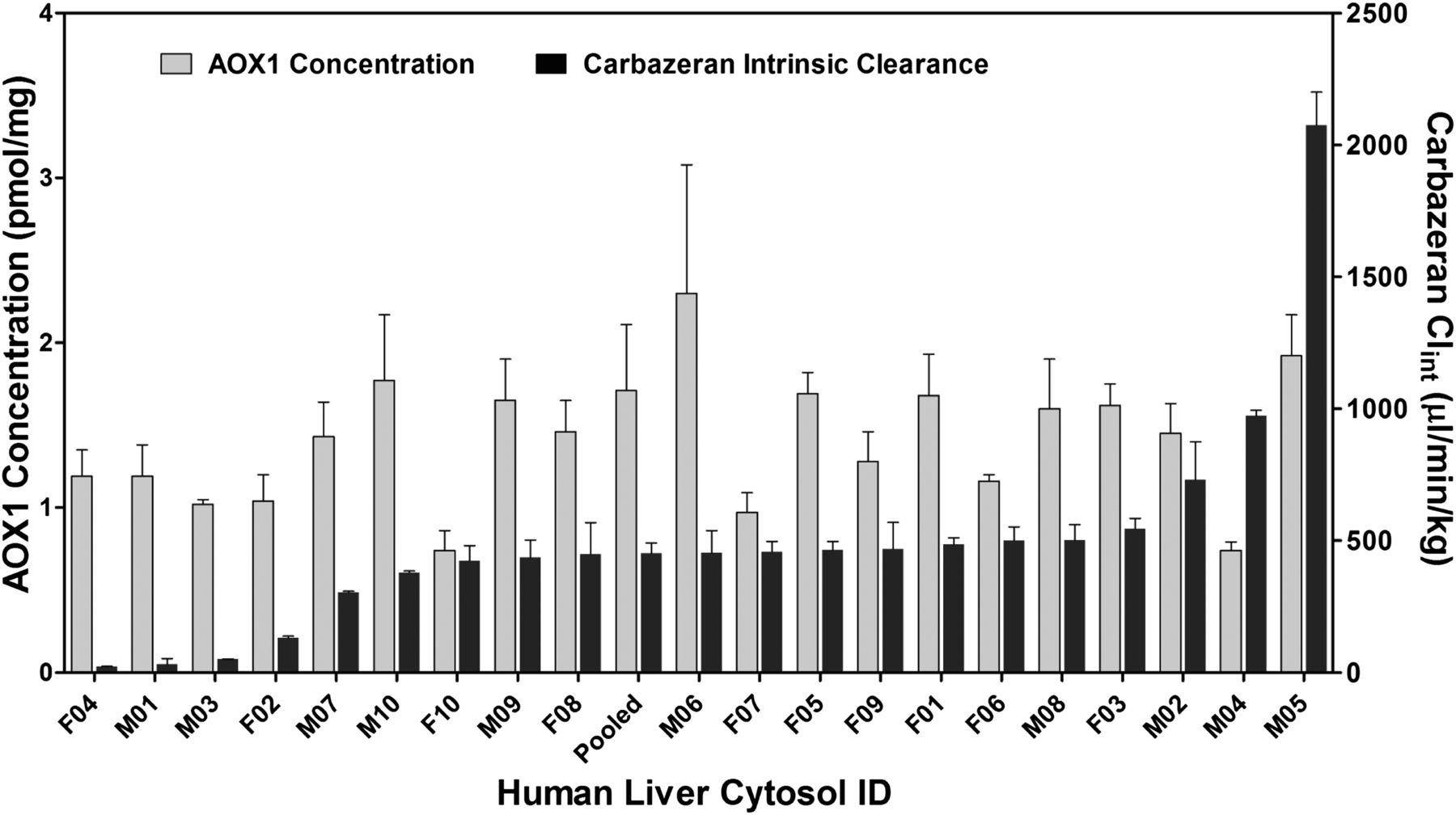
Intrinsic clearance of the 20 different lots of human cytosol were calculated using Eq. 1 and are summarized in Table 2. For all cytosol samples, carbazeran and phthalazine were depleted much faster than zoniporide, with average t1/2 values of 5.1, 3.7, and 42 minutes, respectively. The relative activity ranking of the 20 different cytosol samples was consistent among the three substrates (Fig. 5). The half-lives of phthalazine (Fig. 5A) and zoniporide (Fig. 5B) were plotted against those of carbazeran, respectively. Because of this high concordance of three substrates in reflecting the AOX1 activities, clearances of carbazeran were used to represent the AOX1 activities of different cytosol lots and plotted together with the corresponding AOX1 expression levels measured by LC-MS/MS assay (Fig. 6). The intrinsic clearance of carbazeran spanned >90-fold (23–2075 µl/min per milligram) among the 20 individual cytosol lots, with 13 of 20 lots located within the 30% intervals of the median clearance value (CLint, in vitro approximately 455 µl/min per milligram). The AOX1 activity of the pooled lot also acquired a value (CLint, in vitro approximately 451 µl/min per milligram) close to the median value, which is similar to the observation in the AOX1 expression level measurements. Three cytosol lots (F04, M01, and M03) exhibited substantially lower AOX1 activities (below 51 µl/min per milligram compared with a median value of 455 µl/min per milligram), albeit retained normal AOX1 expression levels. On the other hand, cytosols (M02, M04, and M05) showed abnormally high AOX1 activities (730, 973, and 2075 µl/min per milligram, respectively) and only gave a 20% increase in the AOX1 expression level from the median value.
Intrinsic clearance of the three AOX1 substrates in 21 human liver cytosols

Comparison of in vitro AOX1 activity (expressed as substrate half-lives) of different cytosol lots using different substrates. Strong correlations can be observed between half-lives of phthalazine and those of carbazeran (A), as well as between half-lives of zoniporide and those of carbazeran (B).

Correlation between in vitro AOX1 activity (CLint, in vitro, µl/min per milligram) measured by substrate depletion of carbazeran and AOX1 concentration (pmol/mg) determined by the LC-MS/MS method. The plot is arranged in the order of increasing AOX1 activities from left to right for the 20 individual cytosols and the pooled lot.
Discussion
Individual and Gender Differences in AOX1 Expression and Activity.
Individual variation of drug metabolizing enzymes has been observed in terms of expression level and activity, especially for polymorphic enzymes. For example, up to 6.9-fold differences were observed for UGT1A1 expression level in human liver microsomes (HLM) from 16 donors (Sato et al., 2012). In another HLM study (Kawakami et al., 2011), a >20-fold increase in expression levels was observed for several major cytochrome P450 enzymes (CYP1A2, 2A6, 2C19, and 3A4) in HLMs from 10 donors. Individual variability of enzyme activity tended to be greater than that of protein expression level. In a study of AO activities in liver cytosol of eight different strains of rats, substantial difference was observed (as high as 104-fold between the Sea/SD strain and WKA/Sea strain) (Kitamura et al., 1999). This is consistent with rats having very variable AO. For activity of human AOX1, a >50-fold spread was observed using a probe substrate, benzaldehyde (Sugihara et al., 1997).
In this study, an up to 3.4-fold difference of AOX1expression levels in cytosol samples was observed from 20 different donors by LC-MS/MS assay. Much greater variations were observed for AOX1 activity based on intrinsic clearance values. For example, 19-fold, 43-fold, and 90-fold differences were observed when phthalazine, zoniporide, and carbazeran were used as probe substrates, indicating the different sensitivity of probe substrates in AOX1 (Watanabe et al., 1992). The data suggested that the individual differences are minor in AOX1 expression.
Gender differences have been reported for human xenobiotic-metabolizing enzymes such as cytochrome P450 (Hunt et al., 1992; Lamba et al., 2003; Franconi et al., 2007), sulfotransferases (Kocarek et al., 2008), glutathione transferases (Knight et al., 2007), and UDP-glucuronosyltransferases (Améen et al., 2004; Buckley and Klaassen, 2007). One study with a limited sample size (N = 6) reported that female mice attained higher AO activity than male mice using selected AO substrates (Watanabe et al., 1992). However, such gender differences vanished when a different substrate was used in the same study. AO activity was compared between male and female mammals using the hepatic cytosol system, and a noticeable gender difference was observed for mice (13-fold higher AO activity of male versus female mice) but not for other species, including humans and rats (Klecker et al., 2006). Another study focused on the human hepatic AOX1 activity and interestingly, no difference in AOX1 activity was observed between 10 female and 3 male subjects (Al-Salmy, 2001). In the present study, the sample size was increased to a total of 20 liver cytosols (10 male and 10 female donors), and the data showed no significant gender difference (P = 0.26) in AOX1 expression level by LC-MS/MS (Fig. 3). In terms of activity, male donors appeared to have higher activity than female donors (average carbazeran intrinsic clearance at 394 µl/min per milligram and 593 µl/min per milligram for female and male donors, respectively); however, the gender difference is not statistically significant for AOX1 activity (P = 0.32). There were individuals from both sexes (F04, M01, and M03) that showed abnormally low AOX1 activities, whereas those with abnormally high AOX1 activities (M02, M04, and M05) were all from the male group (Fig. 6).
Correlation between AOX1 Protein Concentration and Enzyme Activity.
For 16 of the 20 different human liver cytosols in this study, the AOX1 protein levels were distributed in a narrow range between 0.97 and 1.77 pmol/mg (median value, 1.45 pmol/mg; 75% percentile, 1.69 pmol/mg), suggesting relatively low variability in individual protein expression. However, the spread of AOX1 activity was substantial for certain lots despite similar protein expression levels. For example, lot M05 had much higher enzyme activity (reason unknown), whereas lots F04, M01, and M03 exhibited substantially lower AOX1 activities. These results suggest an alternative mechanism to manifest AOX1 activity other than AOX1 expression level alone. One possible hypothesis could be that a portion of AOX1 were present in various inactive forms [e.g., SNP (Hartmann et al., 2012), Mo-cofactor deficiency (Schwarz et al., 2009), and homodimer dissociation (Itoh et al., 2007)], which cannot be identified by the three surrogate peptides in the current LC-MS/MS assay. An additional experiment was conducted to test whether the deficiency of the molybdenum cofactor could play a role in AOX1 activity. AOX1-bound molybdenum content in lot F04 and the pooled lot (Mix01) were analyzed by inductively coupled plasma MS, and indeed the Mo concentration in cytosol F04 was 20% lower than that from the pooled lot (data not shown). Mo-cofactor needs to be correctly inserted in order for AOX1 to be active (Garattini et al., 2008). The inductively coupled plasma MS data suggested that active sites of AOX1 in F04 samples may not be fully incorporated with the Mo-cofactor, which would be consistent with the decreased AOX1 activity compared with normal AOX1. However, this still cannot fully account for the drastic differences of AOX1 activities between these two lots as observed in the carbazeran substrate depletion assay. Other possibilities may also contribute to the low AOX1 activity, including protein misfolding, SNP (Hartmann et al., 2012), or chemical/enzymatic modification and disruption of AOX1 homodimer.
Correlations between Alcohol Usage and AOX1 Activity.
The demographic details of the liver cytosol donors revealed that abnormally low AOX1 activities (intrinsic clearance rates for carbazeran at 32 and 23 µl/min per milligram) were observed in two donors with heavy chronic alcohol consumption (M01: 12-pack daily; F04: 3–4 glasses of wine daily for 30 years). To the best of our knowledge, this is the first study to observe that heavy chronic alcohol consumption may also lead to a significant decrease in AOX1 activity. Interestingly, there was no apparent change in the enzyme expression level as measured by LC-MS/MS. Other demographic factors such as age, race, weight, cigarette usage, drug usage, and medication history were not detected to have an impact on AOX1 activity based on this study, albeit the number of samples is too low to make clear conclusions.
In this report, a fast, sensitive, and robust LC-MS/MS method was developed and applied to quantify AOX1 levels in human liver cytosols. The methodology can also be applied for AOX1 quantification in other tissues to aid the development of a scaling factor for prediction of in vivo clearance of AOX1 substrates and to better understand interindividual variability. The LC-MS measured AOX1 concentrations were in good agreement with those measured by an orthogonal ELISA assay. A relatively narrow spread of AOX1 levels was observed in 20 individual donors (<3.5-fold). In vitro AOX1 activities were determined using three representative probe substrates with different sensitivities to human AOX1. The majority of the 20 individual lots showed similar AOX1 activities except for some abnormal lots, in which the measured activities could be up to 450% (maximum value) or 5% (minimum value) of the average. Several lots had very low activity yet retained normal AOX1 protein expression. This suggests that factors other than protein concentration may also affect AOX1 activity; these factors may include protein misfolding, SNP, chemical/enzymatic modification, disruption of AOX1 homodimer, and Mo-cofactor deficiency. Further investigation of these areas would help to deepen the understanding of the protein expression and activity relationship. Pooling of liver cytosols from multiple donors greatly minimized the impact of individual variability, resulting in a mixture with AOX1 level and activity matched to the averaged values. Our results also suggested that heavy chronic alcohol consumption may lead to marked impairment of AOX1 activity but not AOX1 expression levels.
Acknowledgments
The authors thank Tom McDonald for the discussion around AOX1 protein quantification, Tim Strelevitz for providing the substrate for AOX1 activity assay, and Zhaohui Lei for the help in the ICP-MS experiments.
Authorship Contributions
Participated in research design: Fu, Di, Troutman, Obach, Zhang.
Conducted experiments: Fu, Han, Soderstrom, Snyder, Zhang.
Performed data analysis: Fu, Di, Zhang.
Wrote or contributed to the writing of the manuscript: Fu, Di, Soderstrom, Obach, Zhang.
Footnotes
- Received May 31, 2013.
- Accepted July 15, 2013.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AOX1
- aldehyde oxidase 1
- CLint
- intrinsic clearance
- ELISA
- enzyme-linked immunosorbent assay
- HLM
- human liver microsome
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- MPA
- mobile phase A
- MPB
- mobile phase B
- MRM
- multiple reaction monitoring
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- SNP
- single nucleotide polymorphism
- Std
- standard
- TGI
- 813TGIIAAVTAFAANK826
- TOF
- time-of-flight
- VFF
- 446VFFGEGDGIIR456
- YIQ
- 779YIQDIVASTLK789
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}