


Abstract
Cytochrome P450 (CYP) superfamily represents the major drug-metabolizing enzymes responsible for metabolizing over 65% of therapeutic drugs, including those for pediatric use. CYP-ontogeny based physiologically based pharmacokinetic (PBPK) modeling has emerged as useful approach to mechanistically extrapolate adult pharmacokinetic data to children. However, these models integrate physiological differences in the pediatric population including age-dependent differences in the abundances of CYP enzymes. Conventionally, developmental changes in CYP enzymes have been reported using protein abundance and activity data from subcellular fractions such as microsomes, which are prone to high technical variability. Similarly, the available pediatric pharmacokinetic data suffer from the lack of specific CYP substrates, especially in younger children. In the present study, we used viable hepatocytes from 50 pediatric (age, 1 day–18 years) and 8 adult human donors and carried out global proteomics-based quantification of all major hepatic CYP enzymes, including orphan enzymes that have not been studied previously. While CYPs 2B6, 3A5, 4A11, 4F3, and 4V2 did not show a significant association with age, all other quantified isoforms either increased or decreased with age. CYPs 1A2, 2C8, 2C18, and 2C19 were absent or barely detected in the neonatal group, while CYP3A7 was the highest in this group. The >1 to 2 years age group showed the highest total abundance of all CYP enzymes. The age-dependent differences in CYP enzymes reported in this study can be used to develop ontogeny-based PBPK models, which in turn can help improve pediatric dose prediction based on adult dosing, leading to safer drug pharmacology in children.
SIGNIFICANCE STATEMENT We quantified the age-dependent differences in the abundances of hepatic CYP enzymes using a large set of viable pediatric and adult hepatocytes using quantitative global proteomics. We report for the first time the ontogeny in the abundance of CYP enzymes in human hepatocytes, especially, orphan CYPs 20A1, 27A1, 51A1, 7B1, and 8B1 and CYP4 subfamily of enzymes. Our study provides important data about CYP ontogeny that can be used for the better prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling.
Introduction
Pharmacokinetics (PK) and pharmacodynamics of drugs may undergo alterations due to interindividual variability caused by factors such as genetics, age, sex, and health conditions, thereby impacting sensitivity to drug effects, encompassing adverse reactions or insufficient efficacy (Hines et al., 2009). Changes in physiological processes affecting drug PK in the pediatric population are nonmonotonic in nature, meaning they do not correlate with body size or surface area (Emoto and Johnson, 2022). Therefore, the use of simple allometric scaling for dose estimations in children based on adult dosing leads to inaccurate predictions, which could lead to toxicity or lack of efficacy, particularly in neonates and infants (Johnson, 2008; Johnson and Ke, 2021). This underlines the significance of understanding the developmental changes in physiology including the abundance of drug-metabolizing enzymes and transporters to improve the accuracy of prediction of drug clearance in children.
Cytochrome P450 (CYP) enzymes play a pivotal role in drug metabolism, responsible for metabolizing over 65% of drugs, including essential pediatric medications such as codeine, midazolam, acetaminophen, ibuprofen, and morphine (O’Donnell and Rosen, 2014). The ontogeny of CYP enzymes has been extensively studied over the years and classified on the basis of developmental patterns of expression (Hines, 2007, 2013). However, these ontogeny profiles have predominantly been generated using mRNA expression, Western blotting-based protein abundance, microsomal activity data using probe substrates, or available in vivo data using suboptimal substrates (Van Groen et al., 2021). Targeted quantitative proteomics is now increasingly used to quantify differences in the protein content of drug-metabolizing enzymes and transporters from children to adults (Streekstra et al., 2021). However, proteomic analysis of enriched subcellular fractions has revealed variable enrichment of CYPs in human liver microsomes when compared with homogenates of human liver and human hepatocytes (Wegler et al., 2021). This technical variability can be either due to contamination of microsomes with other organelles or the loss of proteins during early fractionation (Wiśniewski et al., 2016). To reduce this variability, scaling factors like microsomal protein per gram of liver (MPPGL) are used with the assumption that all proteins in the fraction are equally scalable, despite variability in protein enrichment (Wiśniewski et al., 2016). However, these scalars are also prone to variability, as shown by a study by Zhang et al. that reported a 19-fold variability in MPPGL values determined from 128 livers (Zhang et al., 2015). Further, MPPGL also shows age-dependent variability of around 5- to 10-fold at a given developmental stage (Leeder et al., 2022). These factors lead to challenges in accurately determining the ontogeny of CYP enzymes using microsomal data.
To address these challenges, we leveraged viable human hepatocytes for the first time and characterized the ontogeny of CYP enzymes including orphan hepatic CYPs by a quantitative proteomics approach. We performed proteomics analysis of 50 pediatric and 8 adult hepatocytes to provide a comprehensive characterization of the developmental changes in CYP enzymes during early human life. These data not only contribute to a deeper understanding of hepatic drug metabolism but also holds significant implications for refining pediatric physiologically based pharmacokinetic (PBPK) models, thus improving the accuracy of pediatric drug dosing and reducing the risk of adverse events.
Experimentals
Materials
Methanol, dimethyl sulfoxide, mass spectrometry (MS)-grade acetonitrile, potassium di-hydrogen phosphate, dipotassium hydrogen phosphate, and formic acid were procured from Fisher Scientific (Fair Lawn, NJ). Acetone was purchased from Sigma-Aldrich (St. Louis, MO). A bicinchoninic acid kit for total protein quantification was purchased from Pierce Biotechnology (Rockford, IL). Ammonium bicarbonate, dithiothreitol, iodoacetamide, and MS-grade trypsin were procured from Thermo Fisher Scientific (Rockford, IL). Human serum albumin and bovine serum albumin were purchased from Calbiochem (Billerica, MA) and Thermo Fisher Scientific, respectively. INVITROGRO HT medium (thawing media) and INVITROGRO KHB medium were received from BioIVT (Baltimore, MD). Diclofenac was purchased from Sigma-Aldrich.
Procurement and Preparation of Human Hepatocyte Samples
Cryopreserved adult (n = 8) and pediatric (n = 50) human hepatocytes were provided by BioIVT (Westbury, NY). The samples were categorized into different groups based on age: 0 to 12 days (n = 7); 26 days to 1 year (n = 10); >1 to 2 years (n = 4); >2 to 6 years (n = 18); >6 to 12 years (n = 6); >12 to 18 years (n = 5); and > 18 years (n = 8). The demographic information of the samples is provided in Supplemental Table 1. The hepatocytes were thawed and resuspended according to a previously described protocol (Ahire et al., 2023). The vial was immersed in a 37°C water bath for ∼120 seconds, poured into prewarmed INVITROGRO HT medium (10 mL), mixed, and centrifuged at 50 xg and room temperature for 5 min. The supernatant was discarded, and the pellet was resuspended with 2 mL of INVITROGRO KHB medium. The cell viability and count were determined using the trypan blue exclusion method with Auto T4 cellometer (Nexcelom Bioscience, Lawrence, MA). The cells were then used for proteomics and diclofenac activity assay.
Protein Quantification in Hepatocyte Samples
The total protein concentration was quantified using a bicinchoninic acid assay kit following the vendor protocol, and the hepatocytes were digested using protocol described previously (Subash et al., 2023). One million hepatocytes were mixed with 300 μL of Solubilization Buffer of the Mem-PER Plus Membrane Protein Extraction Kit and incubated for 60 min at 300 rpm (4°C). Briefly, 100 mM ammonium bicarbonate, 2 μg/mL bovine serum albumin, and 50 mM dithiothreitol were added to 80 μL of hepatocyte sample (containing 1 million hepatocytes) and mixed, followed by gentle shaking at 300 rpm for 10 min at 95°C for denaturation and reduction. The sample was cooled down to room temperature for 10 min and then alkylated with 10 μL of 500 mM iodoacetamide for 30 min in dark. It was then protein precipitated with 1 mL ice-cold acetone, incubated at –80°C, and then centrifuged at 16,000 × g and 4°C for 15 min. The pellet was washed with ice-cold methanol and then centrifuged at 16,000 × g and 4°C for 15 min. The obtained pellet was dried at room temperature and resuspended in 60 μL of 50 mM ammonium bicarbonate. Trypsin digestion was carried out by adding 20 μL of trypsin and incubating for 16 h at 37°C with gentle shaking (300 rpm). The digested sample was quenched with 5 μL of 0.5% formic acid, vortex-mixed, and centrifuged at 16,000 × g and 4°C for 10 min, and the supernatant was transferred into a liquid chromatography-mass spectrometer vial for analysis.
Global Proteomics Analysis of Digested Hepatocytes Using the Total Protein Approach
Global proteomics data acquisition of the digested samples was carried out using EASY-nLC 1200 series system coupled with Q-Exactive-HF MS instrument (Thermo Scientific, San Jose, CA) in the data-independent acquisition (DIA) and positive ionization modes. The digested sample was injected, and the peptides were separated using PepMap RSLC C18 column (2 μm, 250 × 0.075 mm, Thermo Scientific). The column was set at 40°C, with a flow rate of 300 nL/min, and 0.1% formic acid in water and 0.1% formic acid in 80% acetonitrile as mobile phase A and B, respectively. The gradient program (%B) was: 0 to 5 minutes (2% to 6%), 5 to 60 minutes (6% to 30%), 60 to 65 minutes (30% to 100%), and 65 to 80 minutes (100%). The MS ionization was performed using an electrospray ionization source at a spray voltage of 1.7 kV. The MS was operated in full scan mode with a resolution of 120,000 for MS1 and 30,000 for MS2 and automatic gain control target of 3 × 106 for MS1 and 1 × 106 for MS2, with a maximum injection time of 55 milliseconds, capillary temperature of 275°C, and 50 S-lens radiofrequency (RF) level. The m/z scan range was 350 to 1100.
The MS data (.RAW files) were analyzed using DIA-NN software (v1.8.1) with Homo sapiens proteome library for human hepatocyte samples. The maximum number of missed trypsin cleavages was set as two with the maximum number of variable modifications as five and false discovery rate of 1%. The following modifications were included: carbamidomethylation of cysteine residues, acetylation of protein N-termini as fixed modifications, and oxidation of methionine as a variable modification. All other parameters were default DIA-NN settings.
The protein abundance was determined using total protein approach (Sharma et al., 2023). The protein concentration (pmol/mg protein) was determined using the spectral peptide intensities (raw intensities) and the protein molecular weight (eq. 1).
 where, MS intensityi and Total MS intensity refer to sum of MS spectral intensities of all peptides of protein i and sum of MS spectral intensities of all the peptides in the sample, respectively, and MWi is the molecular weight of protein i.
where, MS intensityi and Total MS intensity refer to sum of MS spectral intensities of all peptides of protein i and sum of MS spectral intensities of all the peptides in the sample, respectively, and MWi is the molecular weight of protein i.
Determination of the CYP Ontogeny Profile
The ontogeny profile and the age at which the CYP content values reached 50% of the adult values (age50) was determined by fitting the data to an ontogeny equation using GraphPad Prism (ver. 8.4.3)
 where Y is the fraction of CYP content (of adult) at age X and Y0 is the fraction of CYP content (of adult) at birth, Plateau is the maximum average relative CYP content or activity, (i.e., 1), X is age in years, and K is the Hill coefficient (Bhatt et al., 2019).
where Y is the fraction of CYP content (of adult) at age X and Y0 is the fraction of CYP content (of adult) at birth, Plateau is the maximum average relative CYP content or activity, (i.e., 1), X is age in years, and K is the Hill coefficient (Bhatt et al., 2019).
4-Hydroxydiclofenac Formation Assay in Human Hepatocyte Samples
Diclofenac 4-hydroxylation activity assay was performed in 24-well plate format using 0.1 × 106 hepatocytes per well. The reaction was performed in triplicate and initiated by spiking diclofenac (final diclofenac concentration: 200 μM, <1% dimethyl sulfoxide) in 295 μL of hepatocyte suspension followed by incubating the well plates at 37°C and 5% CO2. At the incubation endpoint (60 min), 100 μL of the reaction mixture was quenched into 200 μL of ice-cold acetonitrile. The quenched reaction mixture was vortexed and centrifuged at 10,000 × g and 4°C for 5 min, and the the supernatant was transferred into a vial for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of 4-hydroxydiclofenac.
The samples from the activity assays were analyzed using the same nanoLC-high-resolution mass spectrometry used for proteomics analysis. nanoLC conditions were set at 300 nL/min flow rate and 1 μL injection volume using mobile phase A: 0.1% formic acid in water, and mobile phase B: 0.1% formic acid in 80% acetonitrile, in a gradient mode. The spray voltage was kept at 1.7 kV, and capillary temperature was 300°C. Detailed nanoLC conditions and MS method are provided in Supplemental Tables 2 and 3. The data were analyzed in data-independent acquisition mode.
Statistical Analysis
Statistical analysis was done using GraphPad Prism (v8.4.3; San Diego, CA) and Microsoft Excel (v2209; Redmond, WA). All the graphs were developed using GraphPad Prism. To study the association of age with protein abundance, the age groups were analyzed using the Kruskal–Wallis test followed by Dunn’s multiple comparison test. The continuous age-dependent abundance data of proteins were analyzed using a nonlinear allosteric sigmoidal model.
Results
Age-Dependent Abundance and Variability of Hepatic CYP Enzymes
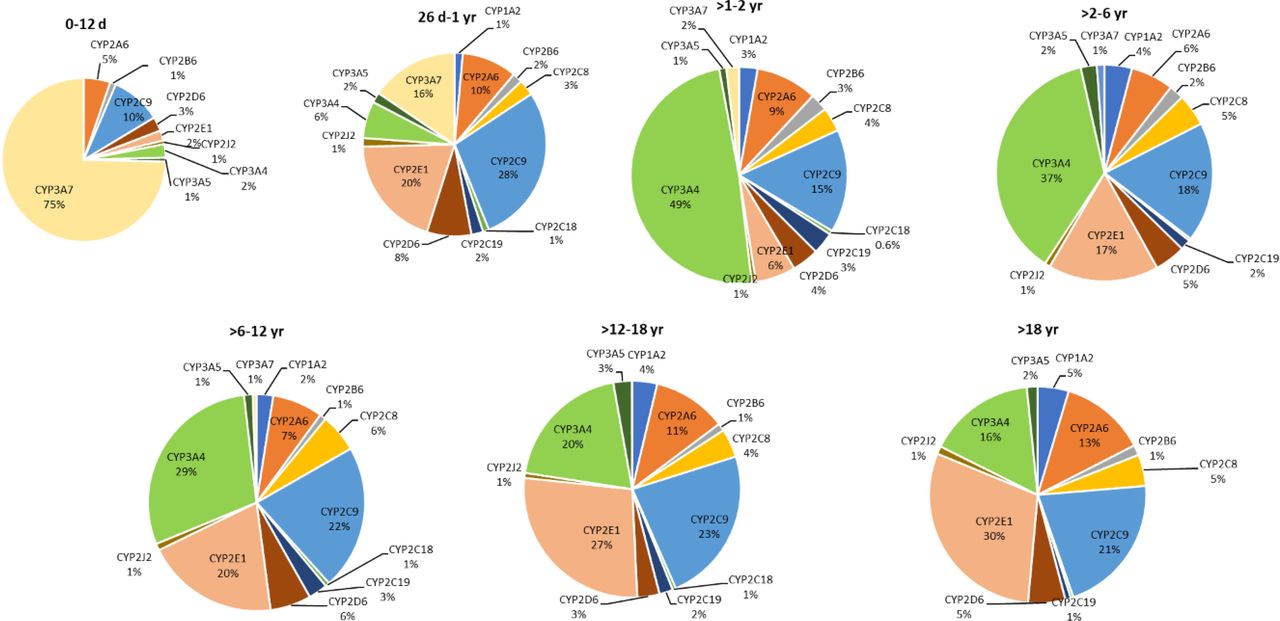
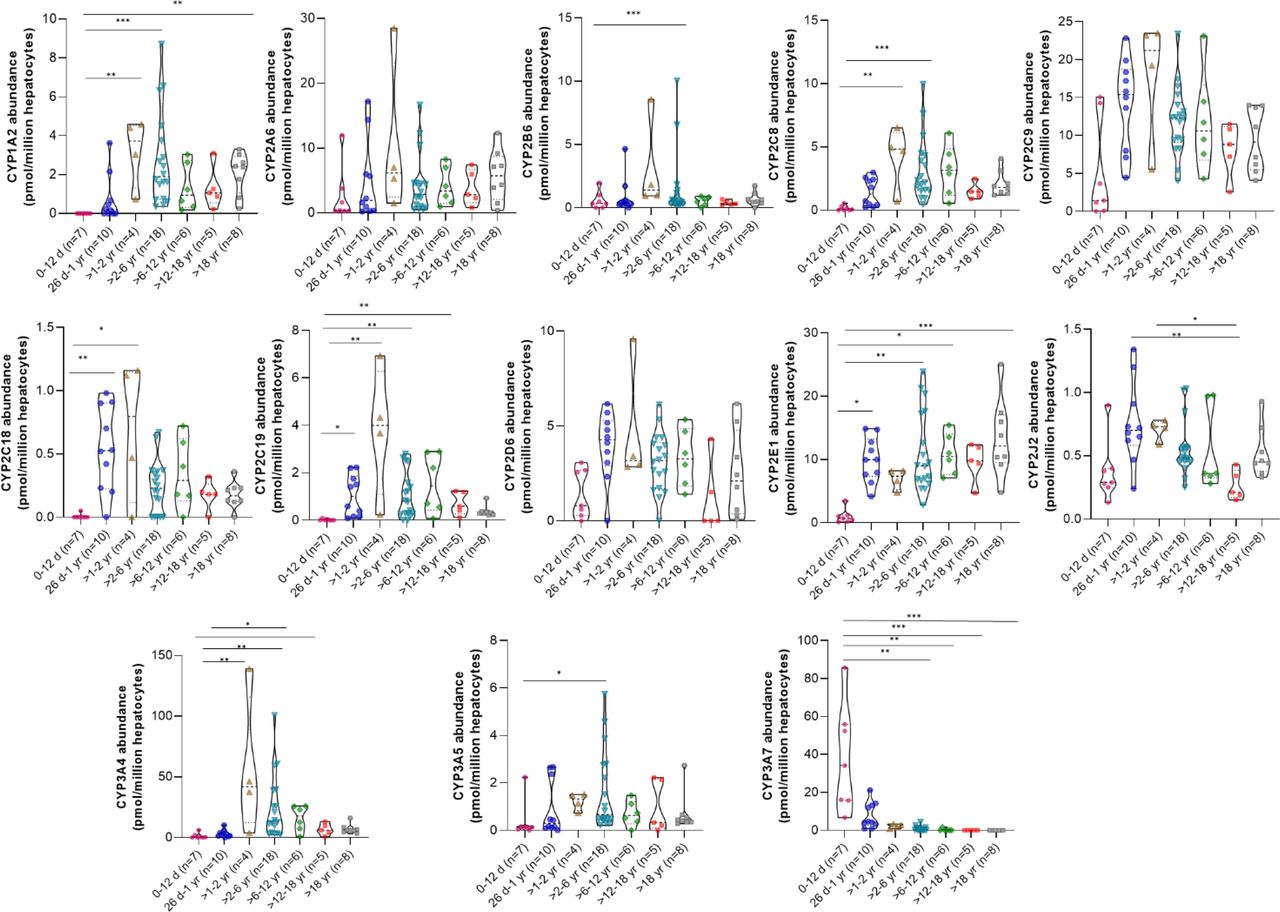
Although the overall distribution of the major drug-metabolizing CYP enzymes (CYP1A-3s and CYP4s) was variable across different hepatocyte samples (Fig. 1, A and B), the average data of different age groups showed age dependence (Supplemental Table 4). CYP3A7 was the most abundant enzyme in the 0- to 12-day age group (75%), which then reduced to 2% in >1 to 2 years and was not expressed in later age groups (Fig. 2). CYP3A4, on the other hand, was not detected in the neonatal samples but was the most abundant isoform in >1 to 2 years (49%) with a slight decline in later age groups and was around 16% in adult samples. In the 0- to 12-day age group, CYPs 1A2, 2C8, 2C18, and 2C19 were absent or significantly lower as compared with the levels detected in the later groups (Fig. 3). The proportion of these isoforms in higher age groups was 1% to 4% (Fig. 2). CYPs 2D6, and 2J2 showed lower levels in the 0- to 12-day age group, followed by a 2- to 3-fold increase in 26 days to 1 year, with no significant change in the later age groups after the initial increase (Fig. 3). CYPs 2B6 and 3A5 showed consistent levels across all age groups until adulthood. CYP2E1 increased up to ninefold in the 26-day to 1-year age group as compared with the 0 to 12 days to account for 20% of the total, following which it decreased to 6% in > 1 to 2 years with a further increase in older age groups to reach 30% in adults (Fig. 2).

Relative protein abundances of cytochrome P450 enzymes (A: CYP1-3 isoforms and B: CYP4 isoforms) in 58 cryopreserved human hepatocytes (age, 1 day to 63 years).

Pie charts representing the fractional abundance of hepatic CYPs1A2, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, and 3A7 across different age-groups (n = 58, age ranging from 1 day to 63 years). The size of each pie chart corresponds to the sum of all protein amounts in the respective age group.

Categorical age-dependent data of drug-metabolizing CYP450 enzymes in 58 cryopreserved human hepatocytes (age, 1 day to 63 years). The truncated violin plot indicates the range, median, and 25th and 75th percentiles. Statistical comparisons between different age groups were conducted using the Kruskal–Wallis test followed by Dunn’s multiple comparison test, with significance levels indicated as follows: P value * < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001.
CYPs 4A11, 4F3, and 4V2 showed comparable levels across age groups. CYP4F12 was most abundant in the 26-day to 1-year age group (Fig. 4). Conversely, CYP4F11 showed an opposite trend with a fourfold increase from 0 to 12 days to >1 to 2 years with abundances remaining constant until adulthood. CYP4F2 was lowest in the 0- to 12-day age group with an increase in levels in later age groups until adulthood. The relative abundances and age-wise categorization of other CYP isoforms (CYPs 20A1, 27A1, 51A1, 7B1, and 8B1) are provided in Supplemental Figs. 1, A and B.

Categorical data for CYPs 4A11, 4F2, 4F3, 4F11, 4F12, and 4V2 in 58 cryopreserved human hepatocytes (age 1 day to 63 years). The truncated violin plot indicates the range, median, and 25th and 75th percentiles. Statistical comparisons between different age groups were conducted using the Kruskal–Wallis test followed by Dunn’s multiple comparison test, with significance levels indicated as follows: P value * < 0.05; ** < 0.01; *** < 0.001; **** < 0.0001.
Protein abundances of CYPs 2B6, 2C19, 2D6, 3A4, and 3A5 in a few samples in the higher age groups was zero (undetected), likely due to genetic polymorphisms associated with these isoforms. These isoforms also exhibited high interindividual variability across age groups (Fig. 3). As technical variability was ruled out through analysis of membrane marker protein calnexin (Supplemental Fig. 2), the observed variability in these isoforms can be reasonably attributed to biological factors.
The ontogeny equation was used to model the protein abundance levels for each of the CYP isoforms (Supplemental Fig. 3) and to compute the age50 values (Supplemental Table 5). The age50 with 95% confidence intervals could be determined only for CYPs 1A2, 2E1, 3A7, 4F2, and 4F11 with the values of 71, 17, 26, 22, and 43 days, respectively.
Classification of CYPs Based on Ontogeny Profiles
Based on the developmental trajectories, the detected CYPs were classified into three categories as described previously (Hines, 2013). The class 1 proteins showed the highest expression after birth and were reduced to low levels by 2 years of age. CYPs 3A7 and 4F12 belonged to class 1. The abundance of the class 2 proteins remained relatively constant throughout the age range and included CYPs 2B6, 3A5, 4A11, 4F3, and 4V2. Class 3 included proteins that were expressed at negligible/low levels at birth and significantly increased after birth to reach adult levels. These included CYPs 1A2, 2A6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 4F2, and 4F11.
A summary of ontogenic patterns of the various CYP isoforms with their potential clinical implications is provided in Table 1.
Summary of ontogenic patterns of CYP isoforms and their potential clinical impact
Protein–Protein Correlations between CYP Enzymes
The protein–protein correlations of the normalized protein abundance values of the detected CYP isoforms were determined (Fig. 5). CYP3A4 correlated with CYPs, 2A6, 2B6, 2C8, and 2C9 (r > 0.6 and P value < 0.05). Although CYP3A7 sharply declines within the first year of age, in the samples greater than 1 year, a positive correlation was observed in its abundance with CYP2B6, 2C8, 2C9, and 3A4. CYP4V2 also showed a positive correlation with other CYP4 classification of enzymes signifying similar regulatory mechanisms of these enzymes. CYP2D6 and CYP3A5 did not show significant correlation with other drug-metabolizing enzymes, likely due to high genetic polymorphism in these enzymes.

Significant protein-protein correlations among hepatic CYP450 enzymes in cryopreserved human hepatocytes (>1 year of age samples). Correlations are deemed significant (red highlight) if the Pearson correlation coefficient is equal to or greater than 0.5, with a corresponding P value of less than 0.05.
Association of Sex With the Abundance of Hepatic CYP450 Enzymes
The Mann–Whitney test indicated the abundance of CYP20A1 was significantly associated with sex, with the abundances in males being higher than that in females (Fig. 6). There was no significant association of sex with the abundance of the remaining CYP isoforms (Supplemental Fig. 4).

Sex-dependent differences in the abundance of CYP20A1 in cryopreserved human hepatocytes (>1 year of age samples). The truncated violin plot depicts the range, median, and 25th and 75th percentiles of protein abundance values. Sex-dependent variations were assessed using the Mann–Whitney test; * P value < 0.05.
Ontogeny of 4-Hydroxydiclofenac Formation in Human Hepatocytes
The formation of 4-hydroxydiclofenac showed an increasing trend with age (Supplemental Fig. 5A) with a positive correlation between CYP2C9 content (Supplemental Fig. 5B). The correlation of CYP2C9 abundance and activity validates the observed ontogeny with the abundance values. This also reflects that the hepatocytes are functionally viable and proteomics data are reflecting the activity.
Discussion
As part of the Best Pharmaceuticals for Children Act, the Food and Drug Administration provides a 6-month market exclusivity to investigational drugs when clinical trials include pediatric patients. (https://www.fda.gov/drugs/development-resources/best-pharmaceuticals-children-act-bpca). However, there are several challenges in conducting clinical trials in pediatric populations due to logistical issues with recruitment, ethical considerations, and the risk of exposing this vulnerable population to potential toxic effects of drugs (Lagler et al., 2021). To circumvent these challenges, the advancement of PBPK modeling has enabled the integration of information on the ontogeny of organ development and pathways involved in drug disposition to guide safe and efficacious dose projections in children (Johnson et al., 2022), for example, the incorporation of hepatic CYP3A ontogeny to develop a PBPK model that provides mechanistic insight into factors contributing to variability in tacrolimus pharmacokinetics in adult and pediatric renal transplant patients (Emoto et al., 2019). Although LC-MS/MS-based quantitative proteomics has generated protein abundance data of drug-metabolizing enzymes and transporters across different age groups to support PBPK modelling (Streekstra et al., 2021; Chu et al., 2022), currently available ontogeny data have limitations such as small sample sizes and reliance on microsomes of S9 fraction data. To improve the quality of pediatric PBPK models, it is important that any available sample is leveraged and analyzed to enrich ontogeny data. Utilizing precious human hepatocytes samples, we investigated the ontogenic profiles of CYP enzymes without the potential technical variability associated with subcellular fractionation. The quality of hepatocyte quality was also confirmed by the consistent levels of the markers of endoplasmic reticulum (calnexin), plasma membrane (NaKATPase), and cytosol (GAPDH) across age groups. Moreover, previous work (Kumar et al., 2019) has demonstrated that cryopreserved hepatocytes from the same vendor (BioIVT) exhibit similar proteomic levels of transmembrane proteins compared with liver tissue. However, the same study showed that the levels of drug transporters depend on the subsequent culture of cryopreserved hepatocytes. Since we did not culture the cryopreserved hepatocytes before proteomics analysis, based on the previous study, we can assume that there is no or minimal effect on CYP abundance. More importantly, since microsomal preparation involves significant loss of the endoplasmic reticulum due to centrifugation (Harwood et al., 2014), hepatocytes provide a much better model than microsomes for estimating CYP ontogeny. The correlation of diclofenac oxidation with CYP2C9 also confirms the functional viability of the hepatocytes used.
More than 50% of clinically used drugs are primarily metabolized by approximately 20 members in CYP1, CYP2, CYP3, and CYP4 subfamilies (Zanger and Schwab, 2013; Song et al., 2021). The abundance of these enzymes can directly impact clinical outcomes of substrate drugs, and the gene expression, protein abundance, and activity of CYP enzymes changes with age (Jin and Zhong, 2023). Given the significant role of CYPs in drug metabolism, mechanistic predictions of these changes across different age groups are crucial for ontogeny-informed PBPK modeling and for age-appropriate drug dosing in the clinic.
In this study, we report for the first time the ontogenic changes in the hepatic abundance of CYPs 20A1, 27A1, 51A1, 7B1, and 8B1 and the CYP4 subfamily of enzymes, which have not been studied previously. Most of these isoforms are involved in the metabolism and regulation of endogenous compounds such as cholesterol, steroids, and bile acid synthesis (Nebert et al., 2013). Nevertheless, an understanding of age-dependent changes in these proteins provides important information about their potential physiological roles during human development. Our results also identified and validated key cytochrome P450 isoforms that are reported to exhibit significant developmental changes during early life using subcellular fractions such as microsomes and cytosols. For example, CYP3A7 is known to catalyze the 16 alpha-hydroxylation of dehydroepiandrosterone, a physiologically important reaction for the formation of estriol in pregnancy, and is also involved in metabolism of potential environmental pollutants like aflatoxin B1 (Hashimoto et al., 1995; Lacroix et al., 1997; Ohmori et al., 1998). CYP3A7 was indeed the most abundant (75%) of all the major drug-metabolizing isoforms in the 0- to 12-day age group and decreased to 2% by 1 year, with a switch to CYP3A4. This replacement of CYP3A7 by CYP3A4 is highlighted by a negative correlation in the samples of less than 1 year age group. The >1 to 2 years age group showed the highest total abundance of all CYP enzymes, with CYP3A4 representing ∼50% of the total abundance, indicating developmental maturation of CYP enzymes in this age group. The majority of the isoforms show an increase in expression beyond the 26-day to 1-year age group and reach the adult levels by early childhood (>2–6 years). This observation corroborates with the reported mRNA, protein levels, and catalytic activity data (Van Groen et al., 2021). For example, CYP1A2 protein expression was absent in the 0- to 12-day age group in agreement with a reported study (Sonnier and Cresteil, 1998) showing a similar observation in microsomes prepared from neonatal and fetal livers. Similarly, the in vivo metabolic clearance of caffeine, a prototype substrate of CYP1A2, has been shown to be significantly lower in infants as compared with adults (Aranda et al., 1979). Interestingly, even though CYP1A1 or CYP1A2 protein expression is not detected in fetal livers, caffeine N-demethylation activity has been reported in this age group (Cazeneuve et al., 1994), which is likely attributed to be associated with CYP3A7. Age-dependent changes in fm of CYPs, sulfotransferases, and uridine diphosphate-glucuronosyltransferases toward acetaminophen metabolism and fmAO versus fmUGT in metabolism of carbazeran also underscore the need for understanding enzyme composition across different age groups (Ladumor et al., 2019; Subash et al., 2024).
Our data do not agree with other reports in a few instances. For instance, CYP2E1 protein levels and in vitro hydroxylation of chloroxazone (CYP2E1 probe) were shown to increase gradually and attain adult levels only by 1 to 10 years of age (Vieira et al., 1996). This is not in agreement with the results of our study, which show a sharp increase in the levels of CYP2E1 from the 0- to 12-day age group and reaching adult values in the 26-day to 1-year group itself. This discrepancy could be due to several reasons including a limited number of pediatric samples used in the previous study, the use of liver microsomes as a matrix, and quantification using semiquantitative immunoquantification methods (Vieira et al., 1996). Another example is that of CYP2C19, which has been reported to be the most dominant CYP2C enzyme during fetal development using Western blotting and microsomal activity assays and is classified as a class 2 enzyme (Hines, 2007, 2013). In the present study, however, CYP2C19 was barely detected in the 0- to 12-day group, with its levels increasing at 26 days to 1 year, and behaves like a class 3 enzyme. Therefore, we believe that the use of viable hepatocytes and LC-MS/MS proteomics places higher confidence in our results as compared with in microsomes and cytosols using conventional electrophoretic or activity-based methods.
The CYP4F family of enzymes is involved in the metabolism of fatty acids, xenobiotics, therapeutic drugs, and signaling molecules (Jarrar and Lee, 2019). Our results show that CYP4F12 showed highest abundance in the 26 days to 1 year age group, whereas CYP4F2 and 4F11 showed an increase from neonates to adults. CYP4F2 and CYP4F11 are involved in the metabolism of vitamin K and arachidonic acid, with CYP4F2 playing the major role in the regulation of synthesis of clotting factors (McDonald et al., 2009; Hirai et al., 2013; Michaels and Wang, 2014). In addition to endogenous substrates, CYP4F11 is involved in the metabolism of erythromycin and chlorprom azine, CYP4F12 metabolizes the antihistaminic drug ebastine, and CYP4F2 and CYP4F3 metabolize the multiple sclerosis drug fingolimod, all of which are also administered to pediatric patients (Ginsburg and Eichenwald, 1976; Estelle et al., 1993; Ahmed et al., 2021; Piri Cinar et al., 2023). Thus, it is important to consider the ontogeny of these isoforms prior to dosing drugs that are substrates for these various CYP4 isoforms.
While our study provides comprehensive insights into CYP ontogeny in pediatric hepatocytes, the exact mechanisms of enzyme regulation during development are still unknown. Transcription factors like hepatocyte nuclear factor 4 alpha, CCAAT/enhancer binding proteins, the GATA family of zinc-finger transcription factors, and epigenetic and regulatory factors such as DNA methylation and histone modifications have been shown to play roles in the age-related expression of CYP enzymes (Czekaj and Skowronek, 2012; Park et al., 2015). For example, hepatocyte nuclear factor 4 alpha is a general regulator supporting the expression of major drug-metabolizing CYPs (Jover et al., 2001). It has been reported to modulate the activity of the basal CYP3A4 reporter gene and a PXR/CAR mediated reporter gene, which in turn mediates fetal liver CYP3A7 and pediatric liver CYP3A4 expression levels (Gerbal-Chaloin et al., 2002; Tirona et al., 2003; Stevens, 2006). DNA methylation can suppress gene expression; further, a higher DNA methylation of CYP3A4 regulatory sites in fetal compared with adult has been suggested to partially explain the absence of CYP3A4 before birth (Kacevska et al., 2012). Further investigations, however, are needed to better understand the factors contributing to age-related variations in enzyme expression and activity.
In conclusion, our study contributes to the growing body of knowledge on the ontogeny of CYP enzymes in pediatric hepatocytes, highlighting the importance of considering developmental changes in drug metabolism pathways. The comprehensive ontogeny data from our study can be used to build proteomics-informed PBPK models that can help optimize pediatric pharmacotherapy. Thus, a better understanding of CYP ontogeny will facilitate the development of personalized medicine approaches tailored to the unique metabolic profiles of pediatric patients, improving drug efficacy and safety in this vulnerable population.
Acknowledgments
The authors would like to sincerely thank Scott Heyward, BioIVT, for helping in the procurement of the pediatric and adult hepatocytes used in this study. The authors would also like to acknowledge the assistance provided by Siavosh Naji-Talakar, Aarzoo Thakur, Deepak Ahire, Guihua Yue, and Dilip Kumar Singh from Washington State University.
Data Availability
The authors declare that all the data supporting the findings of this study are contained within the paper or provided in the Supplemental Material.
Authorship Contributions
Participated in research design: Subash, Prasad.
Conducted experiments: Subash
Performed data analysis: Subash, Prasad.
Wrote or contributed to the writing of the manuscript: Subash, Prasad.
Footnotes
- Received June 8, 2024.
- Accepted September 11, 2024.
The work was supported primarily by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH) [Grant R01 HD081299].
B.P. is cofounder of Precision Quantomics Inc. and a recipient of research funding from Bristol Myers Squibb, Genentech, Gilead, Merck, Novartis, Takeda, and Generation Bio. S.S. declares no competing interests for this work.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CYP
- cytochrome P450
- DIA
- data-independent acquisition
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MPPGL
- microsomal protein per gram of liver
- MS
- mass spectrometry
- PBPK
- physiologically based pharmacokinetic
- PK
- pharmacokinetics
- Copyright © 2024 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}