


Abstract
The purpose of this study was to demonstrate experimentally that alterations of in vivo transporter function at the blood-brain barrier (BBB) in disease and during pharmacotherapy can be reconstructed from in vitro data based on our established pharmacoproteomic concept of reconstructing in vivo function by integrating intrinsic transport activity per transporter molecule and absolute protein expression level at the BBB. Pentylenetetrazole (PTZ)-kindled and spontaneous model of epilepsy (EL) mice were used as models of chemically induced and spontaneous epilepsy, respectively. A mouse model of antiepileptic drug treatment was prepared by consecutive 5-week administration of phenytoin (PHT). Quantitative targeted absolute proteomic analysis of 31 membrane proteins showed that P-glycoprotein (P-gp/mdr1a) protein expression levels were significantly increased in brain capillaries of PTZ (129%), EL (143%), and PHT mice (192%) compared with controls. The brain-to-plasma concentration ratios (Kp brain) of P-gp/mdr1a substrate verapamil were 0.563, 0.394, 0.432, and 0.234 in control, PTZ, EL, and PHT mice, respectively. In vivo P-gp/mdr1a function at the BBB was reconstructed from the measured P-gp/mdr1a protein expression levels and intrinsic transport activity for verapamil per P-gp/mdr1a previously reported by our group. Then, the reconstructed P-gp/mdr1a functional activities were integrated with unbound fractions of verapamil in plasma and brain to reconstruct Kp brain of verapamil. In all mice, reconstructed Kp brain values agreed well with the observed values within a 1.21-fold range. These results demonstrate that altered P-gp functions at the BBB in epilepsy and during pharmacotherapy can be reconstructed from in vitro data by means of our pharmacoproteomic approach.
Introduction
The blood-brain barrier (BBB) is one of the major blood–central nervous system (CNS) interfaces. It expresses multiple functional molecules, including transporters and receptors, and serves to control the traffic of a variety of molecules in and out of the CNS. Consequently, it plays an important role in maintaining brain homeostasis and in restricting the delivery of many therapeutic and diagnostic drugs to the brain. A quantitative understanding of how the molecular transport systems at the human BBB are altered in diseases is essential not only to elucidate the pathophysiological roles of the BBB in human brain, but also to aid development of effective drugs and appropriate pharmacotherapies for CNS diseases.
Imaging technologies such as positron emission tomography have been introduced to evaluate the in vivo activities of BBB transporters in patients with CNS diseases, such as P-glycoprotein (P-gp/MDR1) in epileptic and schizophrenic patients (Arakawa et al., 2010; Feldmann et al., 2013; Syvanen and Eriksson, 2013). However, for most BBB transporters, it is still difficult to synthesize selective probes and inhibitors to enable specific evaluation of the transport activities of the target transporters, because of the effects of other transporters, including functionally unknown transporters, as well as binding to brain tissues.
To solve these problems, we proposed the pharmacoproteomic concept that the in vivo function of a target transporter at the BBB can be reconstructed by integrating the transport activity per target transporter protein measured in in vitro transfected cell lines (transport rate/mole) and the absolute protein expression level of the target transporter in brain capillaries isolated from brain (mole), and we experimentally demonstrated the validity of this approach by using P-gp/mdr1a as a model transporter in normal mice (Uchida et al., 2011a). This result raised the possibility that the in vivo functions of multiple transporters at the human BBB in disease states could be reconstructed from in vitro measured transport activity per target human transporter protein and its absolute expression levels in brain capillaries isolated from diseased brains after surgery or at autopsy, as well as under normal conditions.
Up to 40% of epileptic patients respond poorly to conventional pharmacotherapy, and impaired drug permeation into the brain is considered to be one of the key contributors to therapeutic failure (Loscher and Potschka, 2005). Therefore, it is important to quantitatively understand how the transport system at the BBB changes in epileptic patients and then to accurately predict the drug permeation into the brain.
To apply our established pharmacoproteomic concept to epileptic patients, it is first important to experimentally validate the applicability of the concept for reconstructing the changes of in vivo functions of transporters both in the disease state itself and in the case of treatment with an antiepileptic drug. Pentylenetetrazole (PTZ)-kindled mouse is a well-established chemically induced epilepsy model, but it is difficult to distinguish whether the effects on transporters are caused by the induced seizures or by PTZ administration itself. In contrast, spontaneous model of epilepsy (EL) mouse, which is susceptible to convulsive seizures, has been used as a spontaneous model of epilepsy (King and LaMotte, 1989; Suzuki, 2013) and would allow us to extract the direct effects of epileptic seizures on transporters, unlike the chemically induced models. Thus, the EL mouse should be an appropriate model of the human epileptic disease state for the present purpose. Phenytoin (PHT) is one of the antiepileptic drugs, and its induction properties for transporters and enzymes have been widely studied (Wang et al., 2004; Urquhart et al., 2007; Wen et al., 2008). Therefore, the PHT treatment model should be suitable to investigate whether the alteration of transporter function in response to pharmacotherapy can be reconstructed by means of our approach.
The aim of the present study was to validate experimentally the applicability of our established pharmacoproteomic approach to reconstruct the changes of in vivo BBB transporter function from in vitro data by using PTZ-kindled mice, EL mice, and PHT-treated mice. For this purpose, we employed the quantitative targeted absolute proteomic (QTAP) approach to simultaneously quantify the protein expressions of many transporters (Kamiie et al., 2008; Uchida et al., 2013) in brain capillaries of normal and model mice, to identify candidate transporters potentially contributing to the change of drug permeation across the BBB in epilepsy. Among the 31 membrane proteins examined, P-gp exhibited the largest differences in protein expression levels between normal and model mice. Therefore, we selected P-gp as a model transporter and successfully demonstrated that the increased activity of P-gp at the BBB can be reconstructed in all three mouse models.
Materials and Methods
Chemicals
The 5,5-diphenylhydantoin (PHT) sodium salt, (±)-metoprolol (+)-tartrate salt, and PTZ were purchased from Sigma-Aldrich (St. Louis, MO). D-(−)-Mannitol (mannitol), polyethylene glycol 400 (PEG400), verapamil hydrochloride, and xylitol were purchased from Wako Pure Chemicals (Osaka, Japan). Peptide probes used for quantification of target proteins were synthesized by Thermo Fisher Scientific (Waltham, MA) (>95% purity). All other chemicals were of reagent grade and were obtained commercially.
Mouse Models of Epilepsy and Antiepileptic Drug Treatment
This study was carried out with permission from the Institutional Animal Care and Use Committee at Tohoku University. Male ddY mice and EL mice were purchased from Charles River (Yokohama, Japan) and National Institute of Biomedical Innovation (Osaka, Japan), respectively. Mice were maintained on a 12-hour light/dark cycle in a temperature-controlled environment with free access to food and water. Their plastic cages were cleaned once per week to avoid excessive stimulation.
PTZ-Kindled Epileptic Mouse (PTZ Mouse).
A subconvulsive dose of PTZ (30 mg/kg PTZ dissolved in 30% PEG400) was given intraperitoneally to ddY mice every day for 5 weeks until 15 weeks of age. The convulsive activity was monitored for 30 minutes following each dose of PTZ. The intensity of seizure response was scored according to the following scale (De Sarro et al., 1999): 0, no response; 1, mouth and facial jerks; 2, nodding or myoclonic body jerks; 3, forelimb clonus; 4, rearing, falling down, hind limb clonus, and forelimb tonus; 5, tonic extension of hind limb, status epilepticus, and/or death. Only mice showing at least three consecutive stage 4 or 5 seizures were considered to be kindled mice and were used in this study.
EL Mouse.
The EL mouse is an inbred mutant strain of the ddY mouse that is susceptible to convulsive seizures, and it was selected as a spontaneous model of epilepsy in this study. The EL mice were tossed up to a height of 10–15 cm until seizure occurred (up to a maximum of 50 times) once per week from 6 weeks of age. The intensity of seizure response was scored according to the same scale as that used for PTZ mice. Only mice at 15 weeks of age that had developed stage 4 or 5 seizures for more than 5 consecutive weeks were used in this study.
Antiepileptic Drug PHT Treatment Mouse (PHT Mouse).
A solution of 50 mg/kg PHT in 30% PEG400 was given intraperitoneally to ddY mice every day for 5 weeks until 15 weeks of age. This regimen would have produced a blood concentration of PHT in the therapeutic to slightly higher than therapeutic range, according to the literature (Lothman et al., 1991). Abnormal behavior and loss of weight were not observed during the administration period.
Control Mouse.
Vehicle alone (30% PEG400) was given intraperitoneally to ddY mice (genetic background of EL mice) every day for 5 weeks until 15 weeks of age.
Isolation of Brain Capillaries by Glass Beads Column Method
Mouse brain capillaries were isolated, as described previously (Ohtsuki et al., 2007), with minor modifications. All procedures were carried out at 4°C, except for perfusion. Mice were transcardially perfused with phosphate-buffered saline to remove blood under anesthesia induced with pentobarbital, and the cerebrum was isolated. Ten pooled cerebrums were dissected into 1-mm pieces and homogenized in a Potter-Elvehjem homogenizer with 10 up-and-down nonrotating strokes by hand in 4 volumes of solution B (101 mM NaCl, 4.6 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 15 mM HEPES, pH 7.4) per brain weight. Dextran was added to the homogenate to a final concentration of 16%, and the mixture was centrifuged at 4500g for 10 minutes. The pellet was suspended in solution A (solution B containing 25 mM NaHCO3, 10 mM glucose, 1 mM pyruvate, and 5 g/l bovine serum albumin) and passed through an 85-μm nylon mesh. The filtrate was passed over a column containing 350- to 500-μm glass beads, which was then washed with 50 ml solution A. The capillaries adhering to the beads were detached by gentle agitation in solution A, and the supernatant including the capillaries was collected and centrifuged at 230g for 5 minutes. The pellet was suspended in solution B and centrifuged at 1700g for 5 minutes. The resulting pellet was again suspended in solution B and centrifuged at 1700g for 5 minutes. The pellet was suspended in hypotonic buffer (10 mM Tris-HCl, 10 mM NaCl, 1.5 mM MgCl2, pH 7.4) and sonicated. The capillaries were stored at −80°C after measurement of the protein concentration by the Lowry method using the detergent compatible protein assay reagent (Bio-Rad, Hercules, CA).
QTAP for 31 Membrane Proteins in Isolated Brain Capillaries
Protein expression levels of 31 membrane proteins were simultaneously determined by means of multiplexed selected/multiple reaction monitoring (SRM/MRM) analysis, as described previously (Kamiie et al., 2008; Uchida et al., 2013). Isolated brain capillaries from each mouse model (50 μg protein per sample) were dissolved in denaturing buffer [500 mM Tris-HCl (pH 8.5), 7 M guanidine hydrochloride, 10 mM EDTA], which makes proteins solubilized and denatured, and then the proteins were S-carbamoylmethylated, as previously described (Kamiie et al., 2008). The proteins were extracted by the precipitation with a mixture of methanol, chloroform, and water. The extracted proteins were dissolved in 6 M urea in 100 mM Tris-HCl (pH 8.5), diluted fivefold with 100 mM Tris-HCl (pH 8.5), and treated with tosylphenylalanyl chloromethyl ketone–treated trypsin (Promega, Madison, WI) at an enzyme/substrate ratio of 1:100 at 37°C for 16 hours. The tryptic digests were mixed with internal standard (stable isotope-labeled) peptides and formic acid, and then centrifuged at 4°C and 17,360g for 5 minutes. The supernatants were subjected to high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. Three or four independent measurements were done on three or four separate aliquots of the pooled homogenate from each mouse model, respectively.
High performance LC-MS/MS analysis was performed on an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA) coupled to a triple quadrupole mass spectrometer (API5000 or QTRAP5500; AB SCIEX, Foster City, CA) equipped with a Turbo V ion source (AB SCIEX). Samples equivalent to 33.3 or 3.33 μg protein were injected onto a Waters XBridge BEH130 C18 (1.0 × 100 mm, 3.5 μm) column together with 500 fmol internal standard peptides. Mobile phases A and B consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The peptides were separated and eluted from the column at room temperature using a linear gradient with a 120-minute run time at a flow rate of 50 μl/min. The sequence was as follows: (A:B), 99:1 for 5 minutes after injection, 50:50 at 55 minutes, 0:100 at 56 minutes and up to 58 minutes, and 99:1 at 60 minutes and up to 120 minutes.
The eluted peptides were simultaneously and selectively detected by means of electrospray ionization in a multiplexed SRM/MRM mode, which can quantify many molecules simultaneously by using 300 SRM/MRM transitions (Q1/Q3) at maximum. The dwell time was 8 milliseconds per SRM/MRM transition. Each molecule was monitored with four sets of SRM/MRM transitions (Q1/Q3-1, Q1/Q3-2, Q1/Q3-3, Q1/Q3-4) derived from one set of standard and internal standard peptides. The peptide probes and SRM/MRM transitions reported by Akanuma et al. (2011), Shawahna et al. (2011), Uchida et al. (2011b), and Agarwal et al. (2012) were used. Chromatogram ion counts were determined by using the data acquisition procedures in Analyst software version 1.5 (AB SCIEX). Signal peaks with a peak area count of over 5000 detected at the same retention time as an internal standard peptide were defined as positive. When positive peaks were observed in three or four sets of SRM/MRM transitions, the molecules were considered as expressed in brain capillaries, and their protein expression levels were calculated as the average of three or four quantitative values for each sample. As the LC-MS/MS analysis was performed three or four times for each mouse model, the protein expression levels for target molecules were finally determined as the average values in three or four samples. The limit of quantification was calculated, as described previously (Uchida et al., 2011b).
Determination of Brain-to-Plasma Concentration Ratio of P-gp Substrate Verapamil by intravenous Constant Infusion
Mice were anesthetized with a mixture of ketamine hydrochloride (125 mg/kg body weight) and xylazine hydrochloride (1.22 mg/kg body weight) and cannulated at the right jugular vein with polyethylene tubing (PE-10; Natsume, Tokyo, Japan) for infusion of verapamil dissolved in the infusion buffer (128 mM NaCl, 24 mM NaHCO3, 4.2 mM KCl, 2.4 mM NaH2PO4, 1.5 mM CaCl2, 0.9 mM MgCl2, 9.0 mM D-glucose, pH 7.4). Mice received a 1-minute rapid infusion of verapamil at a rate of 100 μl/min, followed by constant infusion at a rate of 200 μl/h for 45 minutes (Harvard pump 11; Harvard Apparatus, Holliston, MA). Throughout the experiment, mice were kept warm using a hot plate. Blood were collected from the left jugular vein at 45 minutes and immediately centrifuged at 7720g for 5 minutes to obtain plasma. Immediately after the blood sampling, the mice were sacrificed, and the cerebrum of each mouse was collected, weighed, and homogenized in 10 mM ammonium acetate (4 ml: 1 g brain). Then, 180 and 160 μl acetonitrile containing 1% formic acid and metoprolol (internal standard) were added to 20 μl plasma and 40 μl brain homogenate, respectively. The samples were shaken for 20 minutes and centrifuged at 4°C and 17,360g for 5 minutes. Then, 180 μl supernatant was evaporated by centrifugation under vacuum, and the residue was reconstituted in 100 μl 0.1% aqueous formic acid. This solution was centrifuged at 4°C and 17,360g for 5 minutes, and the supernatant was subjected to LC-MS/MS analysis. The brain-to-plasma concentration ratio (Kp brain) was calculated by dividing the brain concentration by the plasma concentration at 45 minutes.
Determination of Unbound Fraction of Verapamil in Plasma Using Equilibrium Dialysis Method
Animals were anesthetized with a mixture of ketamine hydrochloride (125 mg/kg body weight) and xylazine hydrochloride (1.22 mg/kg body weight) on a heating pad to maintain the body temperature. Blood was collected from the jugular vein and immediately centrifuged at 7720g for 5 minutes to obtain plasma. The plasma was adjusted to pH 7.4 with saturated CO2. Five microliters of 50% acetonitrile containing verapamil (50 µM) was added to 495 µl plasma to obtain 0.5 µM final concentration. A total of 125 µl PBS (pH 7.4) and 120 µl plasma containing verapamil was placed in the dialysate and sample sides of a 96-well micro-equilibrium dialysis device (HTD 96b; HTDialysis, Gales Ferry, CT), respectively, with HTD 96a/b Dialysis Membrane Strips (molecular weight cut-off 12–14 K; HTDialysis). The wells were sealed and rotated in a 10% CO2 incubator at 37°C and 80 rpm with a multi-shaker (Tokyo Rikakikai, Tokyo, Japan). After incubation for 6 hours, 5 µl plasma and 50 µl dialysate were collected from the sample and dialysate sides, respectively, and transferred to 96-well format polypropylene plates. A total of 50 µl control PBS and 5 µl control plasma was added to the collected plasma and dialysate samples, respectively. A total of 150 µl acetonitrile containing metoprolol (internal standard) was added (205 µl in total) and vortexed. The samples were centrifuged, filtered at 4°C and 960g for 10 minutes, and subjected to LC-MS/MS analyses. The unbound fraction of verapamil in plasma (fu,plasma) was calculated based on the ratio of the concentrations determined in plasma and dialysate samples.
Determination of Unbound Fraction of Verapamil in Brain Using Brain Slice Uptake Method
Brain slice uptake experiments were performed, as described previously (Kakee et al., 1996), with minor modifications. Mice were anesthetized with a mixture of ketamine hydrochloride (125 mg/kg body weight) and xylazine hydrochloride (1.22 mg/kg body weight), then decapitated, and the brain was immediately removed and immersed in ice-cold oxygenated extracellular fluid (ECF) buffer (122 mM NaCl, 3 mM KCl, 0.4 mM K2HPO4, 25 mM NaHCO3, 1.4 mM CaCl2, 1.2 mM MgSO4, 10 mM D-glucose, 10 mM HEPES, pH 7.4) equilibrated with 95% O2–5% CO2. Cerebral slices, 500 μm thick, were cut using a microslicer (DTK-2000; Dosaka, Kyoto, Japan) and kept in ice-cold oxygenated ECF buffer equilibrated with 95% O2–5% CO2. After preincubation at 37°C for 10 minutes in oxygenated ECF buffer, the brain slices (20–40 mg) were transferred to oxygenated ECF buffer containing 50 nM verapamil at 37°C. After incubation for 6 hours, slices were removed from the solution, dried on filter paper, and weighed. The slices were then homogenized in 9 volumes (w/v) of 10 mM ammonium acetate with an ultrasonic probe (Branson Sonifier 150; Branson Ultrasonics, Danbury, CT). The homogenates were treated according to the same procedure used in the in vivo study and subjected to LC-MS/MS analysis. The unbound fraction in the brain (fu,brain) was calculated using the following equation (eq. 1): where Aslice, Cbuffer, and Vi are the amount of verapamil at 6 hours in the slice, the medium concentration of verapamil at 6 hours measured by LC-MS/MS, and the adherent water volume, respectively.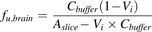 (1)Following incubation of brain slices in ECF buffer containing mannitol for 1, 3, and 5 minutes, Vi was estimated to be 0.0880 ml/g brain from a plot of Aslice/Cbuffer using zero time back-extrapolation.
(1)Following incubation of brain slices in ECF buffer containing mannitol for 1, 3, and 5 minutes, Vi was estimated to be 0.0880 ml/g brain from a plot of Aslice/Cbuffer using zero time back-extrapolation.
LC-MS/MS Analyses of the Compounds
The quantification of verapamil and mannitol was automated by coupling a triple-quadrupole mass spectrometer (API5000; AB SCIEX) to an Agilent 1200 HPLC system (Agilent Technologies). Samples were injected onto an Agilent XDB-C18 column (2.1 × 150 mm, 5 μm) for verapamil or an Inertsil NH2 column (2.1 × 250 mm, 5 μm) for mannitol. The Agilent XDB-C18 and Inertsil NH2 columns were maintained at room temperature and 65°C, respectively. Mobile phases A and B consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Verapamil and mannitol were separated and eluted from the columns at a flow rate of 200 μl/min with a linear gradient (0–2 minutes, 5% B→7 minutes, 100% B→8–20 minutes, 5% B) and under isocratic conditions (73% B for 10 minutes), respectively. The eluted compounds were detected by electrospray ionization (ESI) using SRM/MRM mode; ESI positive 455.3/165.1 for verapamil, ESI positive 268.3/116.1 for metoprolol (internal standard for verapamil), ESI negative 181.0/163.0 for mannitol, and ESI negative 150.9/88.8 for xylitol (internal standard for mannitol).
Reconstruction of In Vivo BBB P-gp/mdr1a Efflux Function and the Brain Distribution of P-gp/mdr1a Substrate Verapamil from In Vitro Experiments
In vivo P-gp/mdr1a efflux function at the BBB is generally defined as the Kp brain ratio (Adachi et al., 2001), which is the ratio of Kp brain in P-gp/mdr1a knockout mice to that in wild-type mice. Using normal mice as a model, we have previously demonstrated that the Kp brain ratio can be reconstructed using the equation presented below (eq. 2) from the in vitro P-gp/mdr1a efflux activity (in vitro P-gp efflux ratio) and protein expression levels of P-gp/mdr1a in mouse P-gp/mdr1a–transfected LLC-PK1 cell monolayer and isolated mouse brain capillaries, according to in vivo reconstruction theory (Uchida et al., 2011a). (2)Furthermore, we have also demonstrated that the Kp brain values of P-gp/mdr1a substrates can be reconstructed from the reconstructed Kp brain ratio by using the following equation (eq. 3) to take account of the unbound fractions of P-gp/mdr1a substrates in plasma (fu,plasma) and brain (fu,brain) (Uchida et al., 2011a).
(2)Furthermore, we have also demonstrated that the Kp brain values of P-gp/mdr1a substrates can be reconstructed from the reconstructed Kp brain ratio by using the following equation (eq. 3) to take account of the unbound fractions of P-gp/mdr1a substrates in plasma (fu,plasma) and brain (fu,brain) (Uchida et al., 2011a).
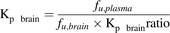 (3)
(3)In the present study, to demonstrate that changes of transporter activity at the BBB in disease states can be reconstructed by applying our established reconstruction theory, we used eq. 2 to reconstruct the in vivo P-gp/mdr1a efflux functions for P-gp/mdr1a substrate verapamil at the BBB (Kp brain ratio) in mouse models of epilepsy and drug treatment from the protein expression levels of P-gp/mdr1a in the isolated brain capillaries from individual mouse models, the reported in vitro P-gp efflux ratio of verapamil, and the reported protein expression levels of P-gp/mdr1a in mouse P-gp/mdr1a–transfected LLC-PK1 cell monolayer (Uchida et al., 2011a). Subsequently, using eq. 3, the Kp brain values of verapamil were calculated from the reconstructed Kp brain ratios and the unbound fractions of verapamil in plasma and brain in the individual mouse models, and the values obtained were compared with the observed Kp brain values. As is clear from eq. 3, the accuracy of Kp brain reconstruction depends on that of Kp brain ratio reconstruction. Therefore, good agreement of the reconstructed and observed Kp brain values indicates accurate reconstruction of the Kp brain ratios. From this point of view, we examined whether the changes of P-gp/mdr1a efflux functions at the BBB in mouse models of epilepsy and treatment could be reconstructed well by means of our pharmacoproteomic approach, based on the reconstructed Kp brain values obtained in this study.
Results
Protein Expression Levels of 31 Membrane Proteins in Brain Capillaries from Mouse Models of Epilepsy (PTZ and EL) and PHT Treatment (PHT).
To find candidate molecules responsible for the change of drug distribution in the brain in epilepsy, the protein expression levels of 31 membrane proteins, including ATP-binding cassette and solute carrier transporters, in brain capillaries of PTZ-kindled, EL, and PHT-treated mice were simultaneously measured by means of LC-MS/MS and compared with those in the vehicle-treated control (Table 1). Drug efflux transporter P-gp/mdr1a showed significantly greater protein expression levels in all three mouse models than in the control: 1.29-, 1.43-, and 1.92-fold greater in PTZ, EL, and PHT mice, respectively. Other drug efflux transporters, mrp4, oat3, and bcrp, also showed significant increases in protein expression levels, but upregulations were observed only in EL mice for mrp4 and oat3, and only in PHT mice for bcrp. These results suggest that the upregulation of P-gp protein expression is the major mechanism of reduced drug permeation into the brain in epilepsy.
Protein expression levels of membrane proteins in brain capillaries isolated from control, pentylenetetrazole, phenytoin, and spontaneous model of epilepsy mice
Brain capillaries were freshly isolated from approximately 3 g cerebrums pooled from 10 mice, using the glass beads column method. Whole-tissue lysates of brain capillaries (50 μg protein) were digested with trypsin to obtain peptides. The protein expression levels of the target proteins were measured by subjecting the peptide samples (33.3 or 3.33 μg protein) to liquid chromatography–tandem mass spectrometry with 500 fmol stable isotope-labeled peptide mixture. Each value represents the mean ± S.E.M. (number of analysis = 3–4).
Distribution of P-gp Substrate Verapamil into the Brain in Mouse Models of Epilepsy and PHT Treatment.
We focused on P-gp, based on the data from quantitative proteomic analysis, and investigated whether the efflux activity of P-gp/mdr1a at the BBB was increased in PTZ-kindled, EL, and PHT-treated mice by determining the brain-to-plasma concentration ratio (Kp brain) of verapamil (Table 2), which is a specific substrate of P-gp and has often been used to evaluate the in vivo function of P-gp at the BBB in positron emission tomography studies. The Kp brain value in PHT mice was significantly lower (2.41-fold) than that in the control mice. The Kp brain values in PTZ and EL mice were 1.43- and 1.30-fold smaller than that in the control mice, respectively, although these differences were not statistically significant. These results suggest that the efflux activity of P-gp/mdr1a at the BBB was increased in PTZ, EL, and PHT mice concomitantly with the increase of P-gp protein expression.
Observed distributions of P-glycoprotein substrate verapamil to brain in control, pentylenetetrazole, phenytoin, and spontaneous model of epilepsy mice
Mice received constant intravenous infusion of verapamil to the jugular vein at a rate of 500 nmol/h per kg following 1-minute rapid infusion (500 nmol/min per kg). The plasma and brain concentrations were measured at 45 minutes, and the brain-to-plasma concentration ratio (Kp brain) values were calculated by dividing the brain concentration by the plasma concentration. Each value represents the mean ± S.E.M. (n = 3–4).
Reconstruction of In Vivo P-gp/mdr1a Function at the BBB in Mouse Models of Epilepsy and PHT Treatment.
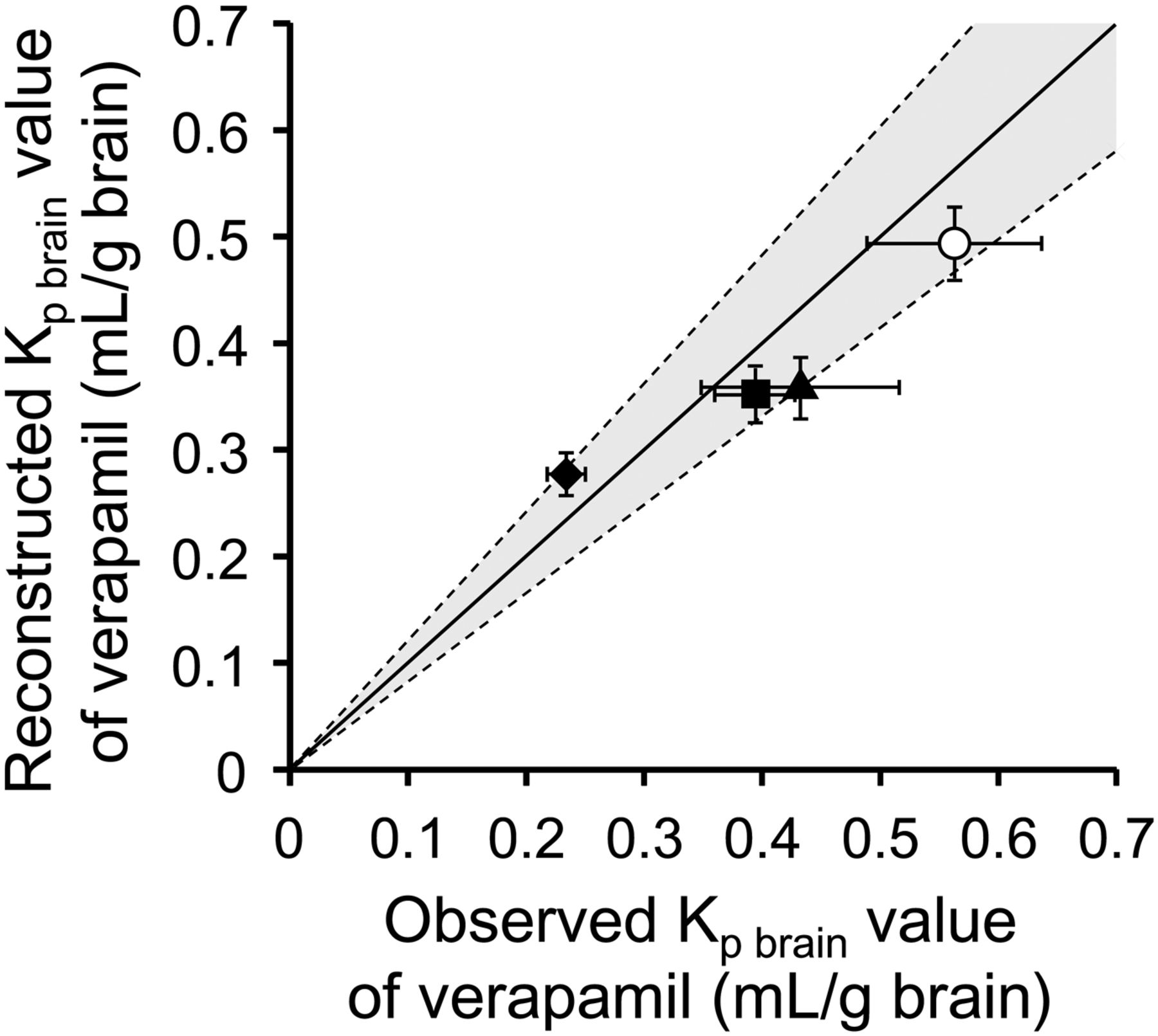
To investigate whether the increased efflux activities of P-gp/mdr1a at the BBB can be reconstructed by means of the pharmacoproteomic-based reconstruction method, we applied this method to PTZ-kindled, EL, and PHT-treated mice. The Kp brain ratio, a parameter of the in vivo P-gp/mdr1a efflux activity at the BBB, was reconstructed according to eq. 2 by integrating the protein expression levels of P-gp/mdr1a in the brain capillaries of PTZ, EL, and PHT mice (Table 1) with the intrinsic efflux activities per P-gp/mdr1a protein for verapamil, taken from Uchida et al. (2011a) (Table 3). The reconstructed Kp brain ratios in the PTZ, EL, and PHT mice were 1.26-, 1.39-, and 1.84-fold higher than that in the control mice, respectively. We then reconstructed the Kp brain values of verapamil by combining these values of the reconstructed Kp brain ratio with the unbound fractions of verapamil in plasma and brain according to eq. 3 (Table 3), and compared the results with the observed Kp brain values (Fig. 1). Good agreement of the reconstructed and observed Kp brain values was observed within a 1.21-fold range in all mice. This indicates that the increased efflux activity of P-gp/mdr1a at the BBB was well reconstructed in all the mouse models.
Reconstructions of Kp brain ratios (P-glycoprotein function at the blood-brain barrier) and Kp brain values of P-glycoprotein substrate verapamil in control, pentylenetetrazole, phenytoin, and spontaneous model of epilepsy mice
Reconstructed brain-to-plasma concentration ratios (Kp brain) were calculated from the reported in vitro P-glycoprotein (P-gp) efflux ratio of verapamil (Uchida et al., 2011a), the reported protein expression level of P-gp/mdr1a in P-gp/mdr1a–expressing LLC-PK1 cell monolayer (Uchida et al., 2011a), and the protein expression level of P-gp/mdr1a in brain capillaries isolated from control, PTZ, EL, and PHT mice, using eq. 2 described in Materials and Methods. Unbound fractions of verapamil in plasma (fu,plasma) and brain (fu,brain) were determined by the equilibrium dialysis method and brain slice uptake method, respectively. Plasma was spiked with 500 nM verapamil and dialyzed against phosphate-buffered saline buffer (pH 7.4) at 37°C for 6 hours (n = 3–4). Brain slices (500 μm) were incubated in extracellular fluid buffer (pH 7.4) containing 50 nM verapamil at 37°C for 6 hours (n = 3–4). The reconstructed Kp brain values were calculated from the reconstructed Kp brain ratios and the fu,plasma and fu,brain values using eq. 3 described in Materials and Methods. Each value represents the mean ± S.E.M. The S.E.M. was calculated according to the law of propagation of error.

Comparison of observed and reconstructed Kp brain values of verapamil in PTZ-kindled, EL, PHT-treated, and control mice. The reconstructed Kp brain values of verapamil were compared with the observed ones. The data were taken from Tables 2 and 3. In all mice, the reconstructed values agreed well with the observed ones within a 1.21-fold range (gray zone), and the largest difference, 1.21-fold, was observed in EL mice. The solid line passing through the origin represents the line of identity, and the broken lines represent 1.21-fold differences. Each symbol represents the mean ± S.E.M. Closed square, PTZ-kindled mouse model of chemically induced epilepsy; closed triangle, EL mutant mouse model of spontaneous epilepsy; closed diamond, mouse model of antiepileptic drug PHT treatment; open circle, vehicle-treated control mice.
Discussion
The present study is the first to clarify the changes in protein expression levels of 31 membrane proteins, mostly transporters, in isolated brain capillaries in mouse models of PTZ-induced epilepsy, spontaneous epilepsy (EL mice), and PHT treatment by means of QTAP analysis. Among the 31 proteins, P-gp showed the greatest change of expression in the model mice compared with the normal controls (Table 1). We then demonstrated that the decreased Kp brain values of P-gp substrate verapamil in epilepsy and during PHT treatment can be well reconstructed (within a 1.21-fold range) from in vitro data by integrating in vitro P-gp transport activity, P-gp protein expression levels, and the unbound fractions in the plasma and brain on the basis of our previously established reconstruction theory (Fig. 1).
In vivo P-gp transport function at the BBB is defined as the Kp brain ratio, which is the ratio of the Kp brain value in P-gp knockout animals to that in wild-type animals (including the model mice), and eq. 3 indicates that the accuracy of reconstruction of the Kp brain values is directly influenced by that of the Kp brain ratio. Therefore, the accurate reconstruction of the Kp brain values in this study has indirectly demonstrated that the Kp brain ratio can also be precisely reconstructed from in vitro data. The reconstructed values of Kp brain ratio (Table 3) indicate that P-gp–mediated efflux is the predominant contributor to the 13.0-, 14.3-, and 18.9-fold smaller brain distributions of verapamil in PTZ, EL, and PHT mice as compared with P-gp knockout mice, respectively. Similarly, it should be possible to reconstruct the BBB P-gp function (Kp brain ratio) in epileptic patients from in vitro human P-gp transport activity, the protein expression levels of human P-gp in the human P-gp–transfected cells, and the protein expression levels of human P-gp in capillaries isolated from diseased brains obtained after surgery or at autopsy. Furthermore, we have previously reported that the reciprocal of Kp brain ratio for P-gp–specific substrate represents the brain-to-plasma unbound concentration gradient, which may be proportional to the levels of pharmacological and adverse effects in the CNS (Uchida et al., 2011a). Therefore, the present results in model mice suggest that our pharmacoproteomics-based reconstruction methodology would be useful for quantitatively predicting changes of in vivo P-gp function at the human BBB in disease states and also could provide useful information to understand the pharmacological and adverse effects of drugs in the diseased CNS.
In the present study, we used not only the classic PTZ-kindled mouse, but also the EL mouse as models of epilepsy. The EL mouse is an inbred mutant strain of ddY mouse that is susceptible to convulsive seizures. It was discovered in 1954, registered internationally in 1964, established as an authentic model of epilepsy by electroencephalography in 1976, and has been widely used as a spontaneous model of epilepsy (Suzuki, 1976, 2013). Despite the similar characteristics of epilepsy in EL mice as compared with human epilepsy, previous BBB studies on epilepsy have generally not used EL mice, but rather have mostly focused on chemically induced model mice (Loscher and Potschka, 2005; Liu et al., 2007). The present study is the first to clarify the quantitative protein expression profile of transporters in the brain capillaries of EL mice and to demonstrate a significant increase of P-gp protein expression level (Table 1). We further established that the increased function of P-gp at the BBB can be reconstructed well on the basis of the increase in the protein expression level in EL mice. It has been reported that approximately 30% of epileptic patients meet the criteria for a diagnosis of autism (Clarke et al., 2005). EL mice also express behavioral abnormalities similar to those seen in persons with autism (Meidenbauer et al., 2011), suggesting that the EL mouse is an appropriate animal model especially for the 30% of epileptic patients with autism. It has been reported that P-gp expression was induced at the BBB in epileptic patients, as seen in the EL mice (Feldmann et al., 2013). For these reasons, we believe the present confirmation of the pharmacoproteomic reconstruction concept in EL mice suggests that this approach would be applicable for predicting in vivo P-gp functions in epileptic patients, especially those with autism.
Pharmacotherapy is standard treatment of patients with epilepsy. Because PHT, among various antiepileptic drugs, has been widely used for studies on the inducibility of transporters and enzymes, we chose the PHT treatment model to investigate whether the change of transporter function in response to pharmacotherapy can be reconstructed by using our pharmacoproteomic concept. As shown in Fig. 1, good agreement was observed between the reconstructed and observed Kp brain values in PHT mice. This result supports the idea that the altered function of P-gp during pharmacotherapy can be reconstructed from in vitro data. To demonstrate clearly that the change of P-gp function was efficiently reconstructed, a slightly higher dose of PHT than the therapeutic dose was used to prepare the model mice in the present study. Although no adverse effects were observed, further studies with therapeutic dosages of PHT and other antiepileptic drugs would be desirable to validate our approach in a more clinically relevant situation.
Only a representative P-gp substrate (verapamil) was used to demonstrate the in vivo reconstruction from in vitro data in the present study, because this reconstruction theory has been experimentally demonstrated using a variety of P-gp substrates both in normal mice and monkeys that have different abundances of P-gp protein expression in the brain capillaries (Uchida et al., 2011a, 2014). Although the principle was demonstrated, the validations using various P-gp substrates with weak or robust efflux activities would further increase the reliability and impact of our pharmacoproteomic approach for the reconstruction in disease conditions and pharmacotherapy. Furthermore, it would be important in the future to investigate whether the brain distributions of antiepileptic drugs in epileptic patients can be predicted from in vitro data by means of our pharmacoproteomic approach.
The Kp brain is determined by four factors, as follows: P-gp protein expression level at the BBB, intrinsic transport activity per P-gp molecule versus the passive diffusion rate, and unbound fractions in plasma and brain. One of the advantages of Kp brain reconstruction is that it enables us to understand the causes of the changes of brain drug distribution in disease states and the contributions of individual factors to these changes. As shown in Table 2, the Kp brain values of verapamil in PTZ, EL, and PHT mice were 1.43-, 1.30-, and 2.41-fold smaller, as compared with the normal control, respectively. No significant difference was observed in plasma or brain unbound fraction between the model and control mice (Table 3). But, in contrast, there were significant increases in the P-gp protein expression levels in all three models compared with the control (Table 1), and the changes of the observed Kp brain values were well reconstructed on the basis of these expression changes (Fig. 1). These results suggest that the main cause of the decreased Kp brain values in the mouse models of epilepsy and PHT treatment is increased protein expression of P-gp.
However, the reconstructed Kp brain values were not identical to the observed ones, and the maximum difference reached 1.21-fold smaller than the observed value in EL mice (Fig. 1). A possible explanation of this discrepancy is that the ratio of the intrinsic transport activity per P-gp molecule to the passive diffusion rate at the BBB in EL mice may be lower than that in the in vitro P-gp–transfected LLC-PK1 cells used for the reconstruction of the Kp brain values in the present study. The intrinsic P-gp transport activity in brain capillaries has been suggested to decrease under inflammatory conditions (e.g., upon TNF-α stimulation) and also under pathologic conditions associated with increased expression of vascular endothelial growth factor in brain (Hawkins et al., 2010; Miller, 2010). In the rodent model of epilepsy, protein levels of vascular endothelial growth factor and inflammatory mediators, including TNF-α, in brain are increased after the induction of seizures (Croll et al., 2004; Vezzani and Granata, 2005; Rigau et al., 2007; Vezzani et al., 2011). Therefore, the intrinsic transport activity per P-gp molecule might be decreased in EL mice. In contrast, the reconstructed Kp brain value was slightly greater than the observed value in PHT mice, suggesting that intrinsic P-gp transport activity at the BBB might be induced by PHT exposure. It would be necessary to clarify the regulatory mechanisms of the intrinsic transport activity per P-gp molecule to achieve more accurate reconstructions of in vivo P-gp function and drug distribution to the brain.
In the present study, several proteins were not detected in the brain capillaries, and the expression levels were determined to be under the limits of quantification (Table 1). However, we cannot completely exclude the possibility that the expressions of some proteins were underestimated so that the expression levels of several proteins were low or under the limits of quantification. This is because some molecules may have been insufficiently solubilized and digested with trypsin. We have previously validated the efficient solubilization and digestion of glut1 in mouse brain capillaries and human P-gp in P-gp–overexpressed cells by comparing quantification values obtained with the present method and with binding assays, including immunoblotting (Kamiie et al., 2008). Furthermore, no bands over 20 kDa were observed by SDS-PAGE after trypsin digestion. The results suggest that the solubilization and trypsin digestion proceed efficiently in the present method. However, this does not necessarily indicate complete solubilization and digestion of all molecules.
In conclusion, our findings indicate that the protein expression level of P-gp showed the largest change among 31 membrane proteins evaluated in brain capillaries of mouse models of epilepsy and PHT treatment, compared with normal controls. We demonstrated that the increased activity of P-gp at the BBB can be reconstructed based on our previously established reconstruction methodology. This validation of the pharmacoproteomic approach in disease models suggests that it would be possible to employ the same methodology to investigate the transport functions at the BBB in patients with other CNS diseases, as well as epilepsy.
Acknowledgments
The authors thank A. Niitomi, N. Handa, Y. Yoshikawa, and K. Tsukiura for secretarial assistance.
Authorship Contributions
Participated in research design: Uchida, Ohtsuki, Terasaki.
Conducted experiments: Uchida.
Contributed new reagents or analytic tools: Uchida.
Performed data analysis: Uchida.
Wrote or contributed to the writing of the manuscript: Uchida, Ohtsuki, Terasaki.
Footnotes
- Received May 13, 2014.
- Accepted July 24, 2014.
This work was supported in part by four grants-in-aid from the Japanese Society for the Promotion of Science (JSPS) for Scientific Research (S) [KAKENHI: 18109002], Scientific Research (A) [KAKENHI: 24249011], Young Scientists (B) [KAKENHI: 23790170], and a JSPS fellowship [KAKENHI: 207291]. This work was also supported in part by two grants for the Development of Creative Technology Seeds Supporting Program for Creating University Ventures and the Revitalization Promotion Program (A-STEP) from the Japan Science and Technology Agency.
T.T. and S.O. are full professors at Tohoku University and Kumamoto University, respectively, and are also directors of Proteomedix Frontiers. This work was not supported by Proteomedix Frontiers, and their positions at Proteomedix Frontiers did not affect the design of the study, the collection of the data, the analysis or interpretation of the data, the decision to submit the manuscript for publication, or the writing of the manuscript, and did not present any financial conflicts. The other author declares no conflicts of interest.
Abbreviations
- BBB
- blood-brain barrier
- CNS
- central nervous system
- ECF
- extracellular fluid
- EL
- spontaneous model of epilepsy
- ESI
- electrospray ionization
- Kp brain
- brain-to-plasma concentration ratio
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- P-gp
- P-glycoprotein
- PEG400
- polyethylene glycol 400
- PHT
- phenytoin
- PTZ
- pentylenetetrazole
- QTAP
- quantitative targeted absolute proteomics
- SRM/MRM
- selected/multiple reaction monitoring
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}