


Abstract
In drug development, considerable efforts are made to extrapolate from in vitro and preclinical findings to predict human drug disposition by using in vitro-in vivo extrapolation (IVIVE) approaches. Use of IVIVE strategies linked with physiologically based pharmacokinetic (PBPK) modeling is widespread, and regulatory agencies are accepting and occasionally requesting model analysis to support licensing submissions. Recently, there has been a drive to improve PBPK models by characterizing the absolute abundance of enzymes, transporters, and receptors within mammalian tissues and in vitro experimental systems using quantitative targeted absolute proteomics (QTAP). The absolute abundance of proteins relevant to processes governing drug disposition provided by QTAP will enable IVIVE-PBPK to incorporate terms for the abundance of enzymes and transporters in target populations. However, most studies that report absolute abundances of enzymes and transporter proteins do so in enriched membrane fractions so as to increase the abundance per sample, and thus the assay’s sensitivity, rather than measuring the expected lower abundance in the more biologically meaningful whole cells or tissues. This communication discusses the balance between protein enrichment and potential loss during the preparation of membrane fractions from whole cells or tissues. Accounting for losses with recovery factors throughout the fractionation procedure provides a means to correct for procedural losses, thereby enabling the scaling of protein abundance from subcellular fractions to whole-cell or organ abundances. PBPK models based on corrected abundances will more closely resemble biological systems and facilitate development of more meaningful IVIVE scaling factors, producing more accurate quantitative predictions of drug disposition.
Introduction
Transporter proteins are expressed in numerous organs throughout the body and can contribute significantly to drug disposition. Therefore, the impact of transporter proteins on parameters influencing drug disposition, including drug-drug interactions (DDIs), requires careful consideration. Incorporating the abundance of pharmacokinetically relevant proteins, including transporters, into the requisite organs of physiologically based pharmacokinetic (PBPK) models will improve the prediction of temporal profiles of drug disposition throughout the body. Such models will be able to integrate transporter kinetics determined by in vitro assays to predict drug disposition and DDI through in vitro–in vivo extrapolation (IVIVE) (Varma et al., 2012). Thus, knowledge of the expression of transporter proteins in both the in vitro and in vivo systems is required to generate scaling factors to bridge differences in transporter expression between both systems (Neuhoff et al., 2013).
Reports quantifying the abundance of transporter proteins have emerged that use liquid chromatography linked to tandem mass spectrometry (LC-MS/MS) and quantitative immunoblotting techniques in both mammalian and in vitro experimental systems (Li et al., 2009; Ohtsuki et al., 2012; Tucker et al., 2012; Prasad et al., 2013). The quantification of absolute transporter protein abundances in tissues and in vitro systems is routinely undertaken in enriched membrane fractions procured after subcellular fractionation. The preparation of enriched samples and extraction of the most relevant matrix (e.g., plasma cell membrane) are not uniform, and many aspects are subject to interoperator or laboratory-to-laboratory variability. However, estimates of procedural losses of target proteins incurred throughout membrane fractionation have not been reported. Our group showed that the recovery factor (RF) for enzymes might be dependent on the procedure and the operator (Barter et al., 2008). However, on application of the relevant RF, estimation of the abundance in the tissue becomes less sensitive to operator differences, and variability between laboratories should be reduced. To our knowledge, there have been no reports on RF values pertinent to drug transporters. Lack of comparability in reported abundance values could be due partly to potential differences in recovery, which also hinder the application of protein abundance data to biologically meaningful applications such as PBPK models. Such models require parameters that define the functional transporter complement of plasma membranes in living tissues. Currently, this is only approximated by transporter abundance in processed subcellular fractions without considering the confounding effect of the variability in the recovery inherent to the different techniques and operator handling used to enrich the matrix with respect to target proteins. This communication postulates that by measuring suitable membrane protein markers or target transporter protein abundances in the original, intermediate, and endpoint membrane fractions, protein loss during subcellular fractionation can be calculated. RFs determined from procedural losses will enable the determination of the native absolute abundance in cells and organs required for incorporation into PBPK models.
Subcellular Fractionation in Membrane Proteomics
For more than 50 years, a reduction in the complexity of cellular material has been advocated to study organelle function (Neville, 1960). Organelle enrichment is generally achieved by differential centrifugation techniques that use multiple steps to obtain the desired fraction for functional studies (Fleischer and Kervina, 1974). When attempting to reduce sample complexity, an assessment of the purity of the targeted organelle fraction(s) is routinely performed by undertaking activity assays in which an enzyme(s) residing specifically within an organelle is used as a marker of purity. Assessment of the marker enzyme’s activity in fractions derived throughout the enrichment procedure and comparison with activity in the whole-cell fraction enable evaluation of enrichment or contamination. Furthermore, recovery and/or losses of target proteins within these organelle fractions may be assessed with enzyme activity balance sheets (Blitzer and Donovan, 1984).
Quantification of transporter proteins with a quantitative targeted absolute proteomics (QTAP) strategy is thought to require subcellular fractionation to reduce sample complexity to prevent the muffling of targeted, low abundance integral membrane proteins, thus enhancing detection sensitivity in LC-MS/MS (Huber et al., 2003). QTAP comprises 1) the selection of standard isotope–labeled (SIL) proteotypic peptides used as surrogates of the whole protein for quantification of protein abundances in the sample; 2) purification of the subcellular fraction(s) that contain the proteins to be quantified; 3) enzymatic digestion of the proteins into proteotypic peptides; and 4) simultaneous quantitation of the SIL and proteotypic peptides in the target biologic matrix by LC-MS/MS (Ohtsuki et al., 2011). Various approaches to obtain total or plasma membrane subcellular fractions are used to quantify the abundances of transporter proteins, where a differing number of homogenization, centrifugation, and incubations steps are applied (Li et al., 2009; Miliotis et al., 2011; Ohtsuki et al., 2012). However, when using procedures involving several stages to obtain a membrane fraction, it is inevitable that protein losses from the target organelle fraction will occur, leading to an underestimation of protein abundances. An additional, and no less important, consideration to protein losses is contamination of the target organelle with other organelle fractions (i.e., endoplasmic reticular and mitochondrial fractions). Such contamination is likely to vary with the differing fractionation protocols. It has been postulated recently that the plasma membrane accounts for 10% of the crude membrane fraction procured from a “native” membrane protein-extraction kit (Vildhede et al., 2014). The small proportion of plasma membrane constituting this fraction, in which the functionally relevant protein abundance can only be assessed, may result in an underestimation of the target protein abundances, comparable to a “dilution effect,” leading to inaccuracies in assessing the relationship between transporter-protein abundance and function. Moreover, since protein expression is usually quoted as moles per mass of protein in the subcellular fraction, differing complements of organelles and protein in the fractions being quantified limit comparison of results between studies. Within an IVIVE strategy, functional transporter abundances are advocated for the generation of mechanistic scaling factors and are incorporated into organ models within PBPK models (Harwood et al., 2013). Any underestimation in transporter abundance and function within the PBPK model could lead to a predicted reduction in the impact of a transporter protein(s) function on drug disposition after simulation, resulting in pharmacokinetic outcomes that are not observed in the clinic. Therefore, when translating abundance data into PBPK models, an accurate characterization of protein abundance in the living tissue is critical.
Protein Losses in Centrifugation: The Utility of Recovery Factors
The well-established scaling factor, microsomal protein per gram of liver (MPPGL) is required to estimate metabolic whole-organ intrinsic clearances from microsomal fractions within an IVIVE framework. Microsomal protein (MSP) fractions are generated from liver preparations by using differential centrifugation procedures after liver tissue homogenization. During the preparation of microsomal fractions from intact liver tissue, considerable losses of MSP were demonstrated (47%), which can hamper the extrapolation of metabolic clearance from subcellular fractions to intact tissue (Wilson et al., 2003). In a consensus study in which collated MPPGL data from the literature were presented, MPPGL values were corrected for losses incurred during preparation in all 10 studies (Barter et al., 2007). Generating RFs is achieved by calculating the fractional loss of MSP by measuring the cytochrome P450 (P450) content in the initial liver homogenate fraction and microsomal fraction (eq. 1). The resulting RF (1 − fractional loss of MSP) can be used to calculate a corrected MPPGL (eq. 2) (Barter et al., 2008).
From a physiologic perspective, obtaining protein abundances within an entire organ is a valuable parameter for estimating the impact of that protein’s function on drug disposition. An approach to calculating whole-organ abundance has been proposed using CYP3A and CYP2E1 abundances to determine the cytochrome content within intact livers (Lipscomb et al., 2003). The calculation combines concurrent quantification of CYP3A and CYP2E1 in whole liver homogenates and the MSP fraction with the scaling factors MPPGL and liver weight to obtain whole-organ abundance. Obtaining data on enzyme abundances in both initial homogenate and MSP fractions with accompanying protein content data allows evaluation of the potential CYP3A and CYP2E1 loss and the generation of sample specific RFs:
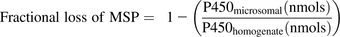 (1)
(1)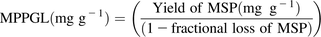 (2)
(2)Accounting for Protein Losses in QTAP Abundance Analysis
Currently, accurate QTAP quantification of transporter abundance requires that the following assumptions are met or that suitable correction factors be found to account for discrepancies: 1) qualification of the surrogate peptide(s) used as standards; 2) membrane extraction is complete, with negligible losses of target transporter proteins from the starting material through to the final enriched membrane fraction; 3) the enriched membrane fraction is completely solubilized in the requisite buffer before digestion; and 4) enzymatic digestion is complete (Ji et al., 2012; Prasad and Unadkat, 2014).
Few studies quantifying transporter abundances have assessed whether the quantitative assays that are deployed meet these assumptions. Two studies have applied isotope-labeled proteins as internal standards embedded in cells and tissues enabling target proteins to be traced through the entire workflow determining both losses and enrichment. Such standards are produced using the stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2003) and stable isotope labeling by amino acids in mammals (SILAM) methods (Kruger et al., 2008). The SILAC method labels the proteins of cultured cells with SIL amino acids (commonly, lysine and arginine) spiked into the growth media. Similarly, SILAM labels the proteins of mammals via a diet enriched with SIL amino acids. Such methods have been applied to assess peptide recovery during digestion in one case and to validate the utility of a SIL approach through a complete QTAP assay’s sample preparation and analysis in a second case. In the first case, the manner in which SILAC controls incomplete tryptic digestion, but not membrane recovery, was reported for an assay quantifying the organic anion-transporting polypeptides (OATPs) OATP1B1, OATP1B3, and OATP2B1 in liver samples (Balogh et al., 2012). In the second case, quantification of sodium taurocholate cotransporting polypeptide (NTCP) in liver samples was used to compare a SILAM protocol; the internal standard was added before membrane purification, with a SIL peptide protocol, in which the SIL peptides were added to the sample after tryptic digestion. Comparable abundances of NTCP were quantified between the SIL and SILAM approaches (i.e., a comparable signal intensity at the mass spectrometry detection system) (Qiu et al., 2013). The SILAM assay provides a quality control for the complete workflow of the SIL peptide assay, yet an assessment of the membrane recovery and target protein loss has not been undertaken. Similar signal intensities measured in the standard and native peptide are not indicators of the potential for protein losses during fractionation or otherwise. A loss of protein during the fractionation procedure would lead to lower signal intensities of the selected transitions at the detector system of the mass spectrometer and lower protein abundance quantifications in the sample for both standard and the native protein.
An alternative, less direct, method to estimate loss during fractionation is to attempt quantification of target proteins in discarded, cytosolic, and nonplasma membrane–enriched fractions. This approach has been applied to another OATP1B1, OATP1B3, and OATP2B1 quantifying assay (Ji et al., 2012). However, abundances were not quantified in the starting lysate fraction; thus, a full proteomic balance sheet for the target protein cannot be evaluated. The sensitivity of the assay applied to discarded fractions must also be questionable since a substantial proportion of target protein might be lost to a discarded fraction or substantially diluted by cytosolic and other proteins that it was rendered undetectable by the assay, a “muffling” effect. In addition, the stability of target proteins in these fractions may affect the resulting abundances. Many, but not all, QTAP-based studies report assessment of the efficiency of tryptic digestion of proteins (Zhang et al., 2011; Ji et al., 2012; Groer et al., 2013). Such assessments provide important quality assurance since they demonstrate minimal bias introduced from incomplete peptide release; however, such assessments do not address protein recovery from whole tissues.
Ideally, an approach that estimates target protein loss between whole tissue and the purified membrane fraction, that enables the scaling of abundances in subcellular fractions to accurately reflect whole cells or organs abundances, is required for PBPK models. The estimate is constructed from the total protein content, determined by a BCA or similar assay and peptide abundance determined by LC-MS/MS, in both whole-cell lysate (total protein) and purified membrane fractions. Near complete release of peptides during digest would require confirmation via another quality-control procedure to ensure that peptide abundance remained an accurate surrogate measure for each protein’s abundance, analogous to enzyme mass balance sheets (Huber et al., 2003).
Although not explicitly mentioned in the report, to our knowledge, Ohtsuki et al. (2013) have provided the only data sets that enable the calculation of RF. They reported transporter protein abundances for both whole-cell lysate and the plasma membrane fractions of a blood-brain barrier cell model (Fig. 1) (Ohtsuki et al., 2013). Differential enrichment of the proteins was achieved, ranging from 1.3 for 4F2 cell-surface heavy chain antigen to 11 for P-glycoprotein. An assessment of protein yield when generating enriched membrane fractions in Caco-2 cells grown on Transwell filters relative to the starting total protein was recently undertaken in our laboratory. These data suggest that relative to the starting whole-cell protein fraction, the plasma membrane fraction constitutes only 1 to 2% of the total protein using a differential centrifugation fractionation procedure. Based on these data, the expected enrichment in transporter protein abundance in the starting total protein fraction versus the plasma membrane fraction would be 50- to 100-fold (eq. 3). If the plasma membrane yield in the blood-brain barrier cell model used in Ohtsuki et al. (2013) were similar to our Caco-2 yield, the fold enrichments in protein abundances from the whole-cell fraction to the plasma membrane are lower than expected from the Caco-2 protein yield data. However, it should be noted that the protein yields in each enriched fraction will be dependent on the biological system under study as well as the procedures used. An unexpected result of differential enrichment was reported in a previous study by the same authors that found a higher mean abundance of the sinusoidal plasma membrane marker, Na/K-ATPase, in 17 human livers in the total membrane versus the plasma membrane fraction (Fig. 2) (Ohtsuki et al., 2012). The different enrichments for each of the proteins quantified poses a problem for the strategy of estimating sample preparation loss for all quantified proteins from the loss of a single high-abundance protein (i.e., Na/K-ATPase) between whole-cell lysate and plasma membrane fraction. Our own experience, based on Caco-2 cell lines, also indicates some discrepancy in enrichment between mucosal and serosal proteins (unpublished data). This discrepancy could be due to 1) losses of protein when obtaining the plasma membrane from the preceding total membrane fraction; 2) differential recovery of peptide during digestion, perhaps as a result of differing biological matrix; and/or 3) differential protein sequestration to intracellular storage vesicles before cell lysis. Therefore, an RF for each peptide may be required.
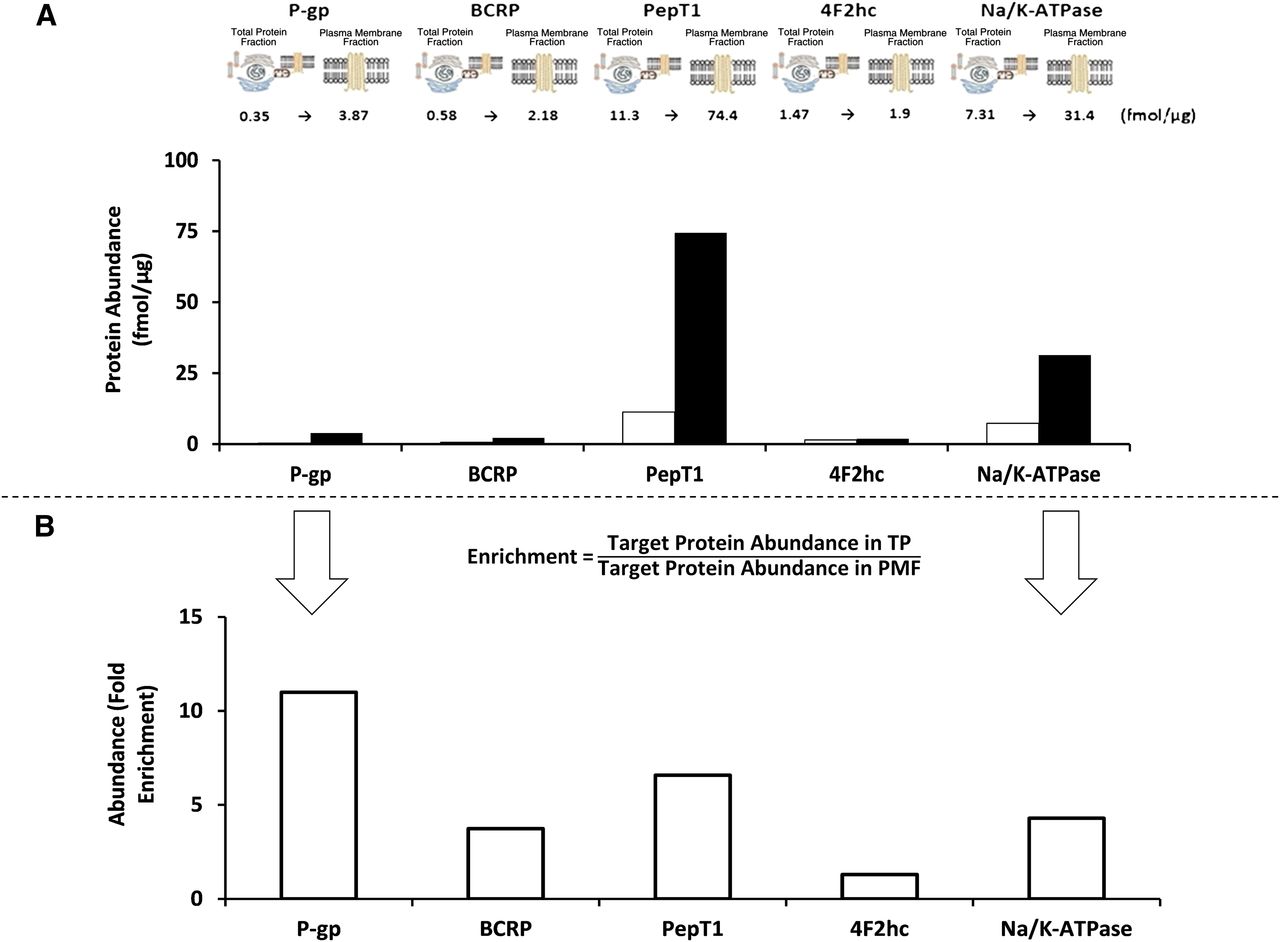
(A) The fold enrichment (“actual enrichment”) in selected peptide abundances measured in the whole cell and plasma membrane fraction of matched samples of the blood-brain barrier cell model (hCMEC/D3) is provided from data reported in Ohtsuki et al. (2013). The text above the bars refers to abundance in femtomoles per microgram (fmol/µg) of protein, with the abundances in the whole-cell fraction given as the first number and white bars and the abundances after the arrow and black bars as the abundance in the plasma membrane fraction. (B) The whole cell and plasma membrane fraction abundance ratio (“the actual enrichment”) has been calculated from eq. 4 in this communication and is provided in the histogram. BCRP, breast cancer resistant protein; 4F2hc, 4F2 cell-surface heavy chain antigen antigen; PepT1, peptide transporter 1; P-gp, P-glycoprotein.

The Na/K-ATPase abundance in total membrane (white bar) and plasma membrane fractions (black bar) from human livers (n = 17) has been extracted and graphically plotted from data published in Ohtsuki et al. (2012). The text above the bars refers to abundance in femtomoles per microgram (fmol/µg) of membrane protein.
Characterizing loss is vital for the incorporation of protein abundance into PBPK models without distortions from peptide-dependent biases inherent to the QTAP assays. Approaches to characterize such biases require formal studies to quantify the balance between protein enrichment and losses; completeness of protein digestion in the biological matrices of whole-cell lysate and plasma membrane fractions; and subcellular distribution of target proteins by GFP tagging and confocal microscopy. Such characterization should guarantee quality and reproducibility to ensure confidence in QTAP assays for transport proteins and high-quality data suitable for IVIVE-PBPK models.
Correcting for Protein Losses in Centrifugation
Accurate determination of the functional abundance of transporter protein(s) in the whole organs of individuals is essential for predicting temporal drug profiles in whole body PBPK models. Whole-organ transporter abundances have not yet been reported in the literature; therefore, scaling factors such as the plasma membrane protein per gram of liver and liver weight are required to scale the abundance levels reported in the membrane fraction to the whole organ (Fig. 3). It is also noteworthy that the plasma membrane protein per gram of liver in itself may require application of an RF to correct for losses in centrifugation in a similar manner to that reported for MPPGL (Barter et al., 2008).
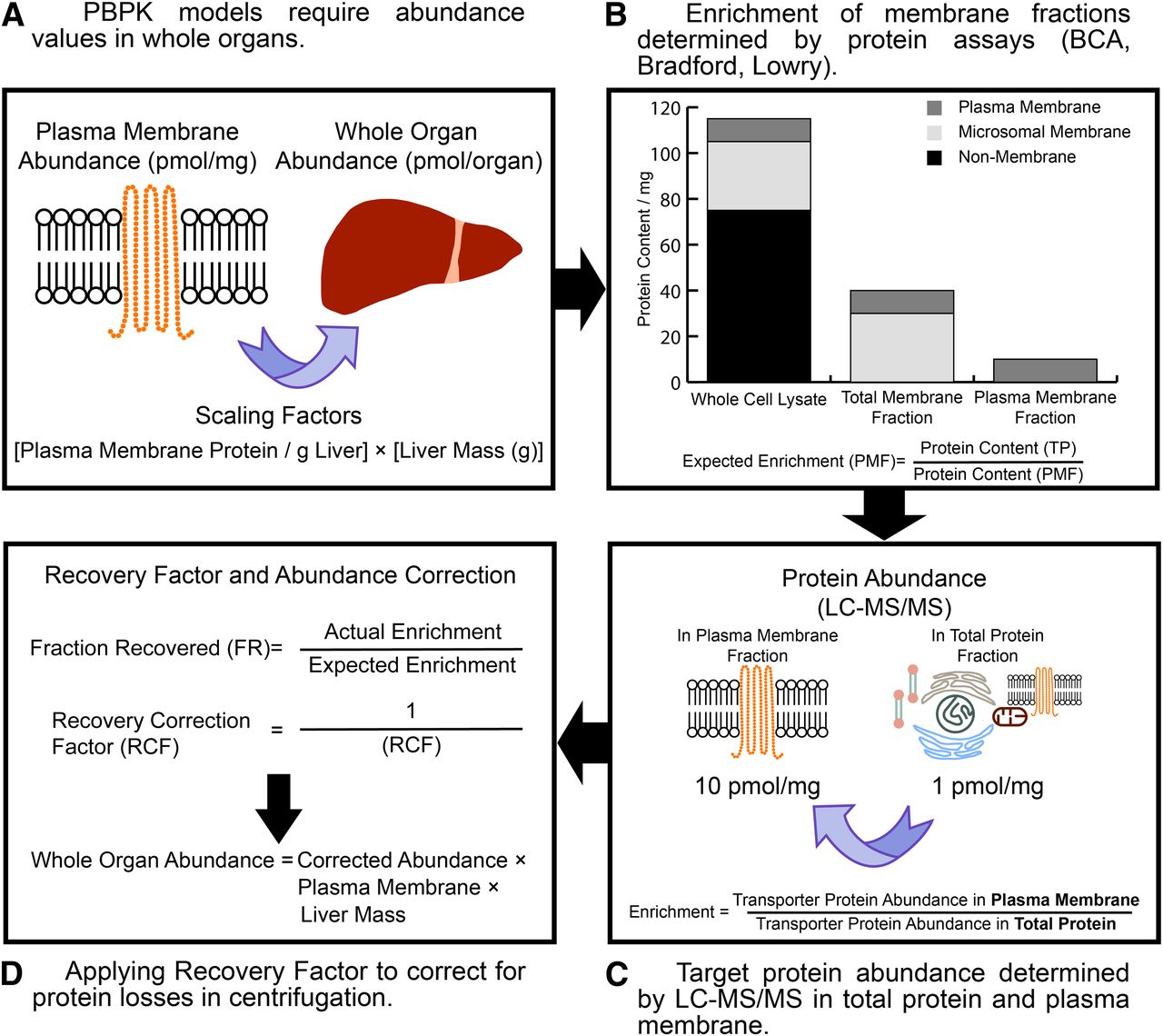
A schematic describing the generation of recovery factors (RFs) to correct transporter abundances in membrane fractions for protein losses encountered during centrifugation. Whole-organ protein abundances require generating from membrane fractions using the appropriate physiologic scaling factors (i.e., plasma membrane protein per gram of liver, PMPPGL) (A). By measuring protein content in the starting total protein (TP) and the endpoint membrane fraction (i.e., the plasma membrane fraction, PMF), the expected enrichment of the target protein can be obtained (B). Quantifying the target protein abundances in the starting total protein and endpoint membrane fractions provides the actual target protein enrichment (C). If the expected and actual enrichments are matched, the RF is by definition 1. If there is a disparity between the expected and actual enrichment, an RF can be generated and applied to the endpoint membrane abundance and is scaled to obtain whole organ abundances (D).
A value for the expected enrichment of the membrane fraction (eq. 3) is required before assessing the final transporter abundances. This value is obtained by performing a protein assay to calculate the yield of protein in the starting total protein fraction, which could be a whole-cell lysate or tissue homogenate, and the crude or plasma membrane fraction for which transporter abundance determinations have routinely been performed (Fig. 3):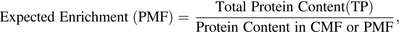 (3)where CMF is the crude membrane fraction and PMF is the plasma membrane fraction.
(3)where CMF is the crude membrane fraction and PMF is the plasma membrane fraction.
The actual enrichment of the target protein is calculated from the abundance(s) of target protein(s) quantified in the total protein fraction and the membrane fraction in QTAP assays (eq. 4):
 (4)
(4)To correct for losses of protein throughout centrifugation the fraction recovered (FR) is determined as the ratio of the actual and expected enrichments for a transporter isoform (eq. 5) and together with eq. 6 can be used to generate an RF that is used to correct the abundances generated in the membrane fraction (eq. 7):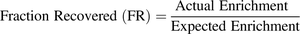 (5)
(5)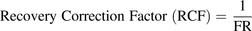 (6)
(6)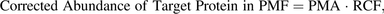 (7)where PMA is the measured abundance of target protein in the plasma membrane sample.
(7)where PMA is the measured abundance of target protein in the plasma membrane sample.
The corrected protein abundance is scaled to the whole-organ abundance using the requisite scaling factors for the evaluated organ. The limitation to this strategy is the ability to quantify lower abundance peptides in the total protein fraction. In this instance, a transporter from the same membrane (apical or basal) and protein subfamily that can be quantified could be used as the basis for a recovery factor. Using the total protein abundance is unlikely to be applicable for incorporation into IVIVE-PBPK models as there are no assurances that the selected peptide that is measured is derived from a fully formed protein (i.e., protein translation is incomplete) resident in the plasma membrane. Furthermore, assessing peptide losses downstream of subcellular fractionation stages should not be neglected and therefore requires incorporation into QTAP workflows (i.e., membrane solubilization, peptide digestion efficiency, chromatographic stability, and transition through the mass spectrometer).
On incorporation of transporter protein abundances into an IVIVE-PBPK workflow, any underestimation of transporter abundance could lead to inaccuracies in 1) the generation of scaling factors that are used to scale between the in vitro and in vivo systems and 2) scaling the transporter activity within the in vivo system to predict transporter isoform clearance in the whole organ. Thus, any differences in procedural losses of target proteins between the in vitro and in vivo system (i.e., a matrix-specific effect) could lead to a bias in the generation expression based scaling factors used in an IVIVE approach (Harwood et al., 2013; Vildhede et al., 2014). Furthermore, a transporter isoforms maximal flux capacity is typically related to its protein abundance (Li et al., 2010). Therefore, an underestimation in whole organ transporter abundances will impact on the prediction of drug disposition and the magnitude of DDI exhibited in that individual.
A recent study incorporated absolute transporter protein abundance data into an IVIVE strategy. This study (Vildhede et al., 2014) quantified the absolute transporter abundances of hepatic uptake transporters OATP1B1, OATP1B3, OATP2B1, and NTCP and the canalicular efflux transporter P-glycoprotein in human livers, isolated hepatocytes, and human embryonic kidney cell line HEK293 single transfected cells (Vildhede et al., 2014). A scaling strategy incorporating the abundances of the uptake transporters was developed, and a relative expression factor approach was used to account for the differences in transporter expression between the in vitro and in vivo systems. An in vitro Vmax was used to assign a fractional contribution of each isoform to an overall uptake clearance, and the variability in the abundance of the transporters in 12 livers enabled an estimate of the expected variability in the magnitude of DDI between individuals. Two key assumptions within this strategy are 1) that activity (Vmax) is proportional to expression, which may not be the case for overexpression systems (Tang et al., 2002), and 2) the recovery of each transporter isoform throughout the procedure is uniform and not biased by the matrix in which it resides. Any differences in the magnitude of losses of transporter proteins should be accounted for, which could affect the scaling factors used to predict transporter-mediated clearance and DDI, thus hindering the accuracy of the IVIVE strategy. Indeed, incorporation of the abundance and activity into a PBPK model should enable the success of this strategy to be ascertained in regard to recovery of pharmacokinetic profiles in human populations. Numerous studies have attempted to predict the impact of hepatic uptake transporters on drug disposition in IVIVE-PBPK, all of whom concluded that there was a considerable underprediction in liver uptake when using a bottom-up approach to IVIVE scaling (Jones et al., 2012; Varma et al., 2012, 2013; Yeo et al., 2013; Jamei et al., 2014). In these studies, to recover the observed pharmacokinetic profile, it was necessary to fit the input parameters by statistical means or correcting transporter-based scaling factors. A comparison of the approach taken by Vildhede et al. (2014) with these studies will permit an assessment of the predictive success of this strategy with reference to human pharmacokinetics and to those studies previously performed.
In a further study by Prasad et al. (2014) that used QTAP methodologies for transporter abundance quantification, the impact of genotype-related increases in hepatic transporter expression on the OATP1B1 probe substrates repaglinide and rosuvastatin were predicted using a PBPK model [Simcyp V12.0, Simcyp (now part of Certara), Sheffield, UK] (Prasad et al., 2014). In these simulations in which repaglinide and rosuvastatin had already been verified with regard to their recovery of human pharmacokinetic profiles (Yeo et al., 2013; Varma et al., 2013; Jamei et al., 2014), the impact of a haplotype-dependent increase in expression agreed with clinical pharmacokinetic observations, where clinical data were available. It is anticipated that further avenues using strategies to combine abundance and activity data generated from human systems, with similar studies employing in vitro systems engineered to possess important genetic polymorphism, could be applied in early drug development, where IVIVE-PBPK is used to predict drug disposition without clinical data. However, convincing evidence for the prospective predictive success in recovering pharmacokinetic profiles, particularly for SLC transporters, is still lacking, and translation of the in vitro system phenotype may not always occur to in vivo (Seithel et al., 2008).
It is a challenge to establish a robust IVIVE strategy for drugs interacting with hepatic uptake transporters during drug development, where clinical pharmacokinetic profiles of a compound are not available for model optimization. As models progress toward incorporating a fully mechanistic transporter IVIVE-PBPK approach that incorporates absolute transporter abundances, the necessary scaling methods required to relate empirical measurements in accessible samples to biological systems are being developed (Harwood et al., 2013; Prasad et al., 2014; Vildhede et al., 2014). Prediction of transporter-mediated drug disposition by populating such models with both accurate in vivo transporter abundances and transporter activity data extrapolated from in vitro assays require further development. Characterizing procedural losses of transporter proteins during sample preparation for QTAP methods, and so establishing recovery factors, will improve the accuracy of in vivo transporter abundance estimates and so facilitate the progress toward prediction of transporter-mediated drug disposition by IVIVE-PBPK. Furthermore, in light of variations observed in absolute abundances between laboratories, for example, hepatic OATP1B1 abundance determination (Prasad et al., 2014; Vildhede et al., 2014), a multicenter study evaluating the consistency and comparability of the preparation steps and analytic outcome are warranted. We are not aware of such comparisons being made on the same set of samples.
Concluding Remarks
A lack of methodologic standardization can hinder the implementation of biological data into physiological models. Therefore, the aim of this communication is to address correcting for protein losses throughout centrifugation procedures, together with reporting protein yields from the membrane fraction under study (Tucker et al., 2012; Prasad et al., 2013), to enable scaling of functional membrane protein abundances to the whole organ. Establishing procedural protein losses that are incurred in subcellular fractionation and using the appropriate RFs to each peptide quantified will facilitate the development of more meaningful IVIVE-PBPK models that more closely resemble the biological system.
Acknowledgments
The authors thank the reviewers for comments that helped with expanding the discussions in this commentary and Eleanor Savill for assistance with manuscript preparation.
Authorship Contributions
Participated in research design: Harwood, Russell, Neuhoff, Rostami-Hodjegan.
Performed data analysis: Harwood, Russell, Neuhoff.
Wrote or contributed to the writing of the manuscript: Harwood, Russell, Neuhoff, Warhurst, Rostami-Hodjegan.
Footnotes
- Received April 4, 2014.
- Accepted July 24, 2014.
This work was supported by a grant from The Royal Commission for the Exhibition of 1851.
This work was contributed to Orbito IMI project (http://www.imi.europa.eu/content/orbito) as a sideground.
Abbreviations
- DDI
- drug-drug interaction
- IVIVE
- in vitro-in vivo extrapolation
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MPPGL
- microsomal protein per gram of liver
- NTCP
- sodium taurocholate cotransporting polypeptide
- OATP
- organic anion–transporting polypeptide
- PBPK
- physiologically based pharmacokinetic modeling
- QTAP
- quantitative targeted absolute proteomics
- RF
- recovery factor
- SIL
- stable isotope labeled
- SILAC
- stable isotope labeling by amino acids in cell culture
- SILAM
- stable isotope labeling by amino acids in mammals
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Subcellular Fractionation in Membrane Proteomics
- Protein Losses in Centrifugation: The Utility of Recovery Factors
- Accounting for Protein Losses in QTAP Abundance Analysis
- Correcting for Protein Losses in Centrifugation
- Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters