

Hepatic Uptake of Atorvastatin: Influence of Variability in Transporter Expression on Uptake Clearance and Drug-Drug Interactions

Abstract
Differences in the expression and function of the organic anion transporting polypeptide (OATP) transporters contribute to interindividual variability in atorvastatin clearance. However, the importance of the bile acid transporter sodium taurocholate cotransporting polypeptide (NTCP, SLC10A1) in atorvastatin uptake clearance (CLupt) is not yet clarified. To elucidate this issue, we investigated the relative contribution of NTCP, OATP1B1, OATP1B3, and OATP2B1 to atorvastatin CLupt in 12 human liver samples. The impact of inhibition on atorvastatin CLupt was also studied, using inhibitors of different isoform specificities. Expression levels of the four transport proteins were quantified by liquid chromatography tandem mass spectrometry. These data, together with atorvastatin in vitro kinetics, were used to predict the maximal transport activity (MTA) and interindividual differences in CLupt of each transporter in vivo. Subsequently, hepatic uptake impairment on coadministration of five clinically interacting drugs was predicted using in vitro inhibitory potencies. NTCP and OATP protein expression varied 3.7- to 32-fold among the 12 sample donors. The rank order in expression was OATP1B1 > OATP1B3 ≈ NTCP ≈ OATP2B1. NTCP was found to be of minor importance in atorvastatin disposition. Instead, OATP1B1 and OATP1B3 were confirmed as the major atorvastatin uptake transporters. The average contribution to atorvastatin uptake was OATP1B1 > OATP1B3 >> OATP2B1 > NTCP, although this rank order varied among individuals. The interindividual differences in transporter expression and CLupt resulted in marked differences in drug-drug interactions due to isoform-specific inhibition. We conclude that this variation should be considered in in vitro to in vivo extrapolations.
Introduction
Transporter-mediated hepatic uptake of atorvastatin is the rate-determining step in the elimination of the drug in vitro (Watanabe et al., 2010) and in vivo (Maeda et al., 2011). At least four members of the solute carrier (SLC) superfamily transport atorvastatin in various in vitro systems. These are the three organic anion transporting polypeptide transporters, OATP1B1 (SLCO1B1), OATP1B3 (SLCO1B3), and OATP2B1 (SLCO2B1) (Choi et al., 2011; König, 2011; Karlgren et al., 2012b), and the sodium taurocholate cotransporting polypeptide (NTCP, SLC10A1) (Choi et al., 2011). All four proteins are expressed in the basolateral membrane of human hepatocytes, where they mediate uptake of substrates into the hepatocytes from the blood (Cui et al., 2003; Keitel et al., 2005).
The importance of OATP1B1 in hepatic uptake has been emphasized because OATP1B1 genetic variants with reduced function and OATP inhibition have been associated with greater systemic exposure of atorvastatin in clinical studies (Lau et al., 2007; Pasanen et al., 2007). However, in a recent study, NTCP was shown to contribute significantly to the hepatic uptake of three different statins (pitavastatin, fluvastatin, and rosuvastatin) with 24%–45% of overall active uptake (Bi et al., 2013). These results indicate that NTCP may play a more important role in statin uptake than previously assumed. The importance of NTCP in atorvastatin uptake has not yet been clarified and needs to be addressed.
Our study investigated the contribution of NTCP to hepatic atorvastatin uptake using a protein expression–based prediction model (Karlgren et al., 2012b) and in vitro hepatocyte experiments. We combined in vitro uptake kinetics with protein quantification to assess the contribution of each transporter to atorvastatin uptake clearance (CLupt) in livers from 12 individuals with varying expression of the four uptake transporters. We also investigated the influence of interindividual transporter expression and isoform-specific inhibition on atorvastatin clearance at clinically relevant concentrations.
Materials and Methods
Compounds
Atorvastatin was kindly provided by AstraZeneca (Mölndal, Sweden). Atazanavir was acquired from Toronto Research Chemicals Inc. (Toronto, ON, Canada). Cyclosporine, gemfibrozil, and rifampicin were purchased from Sigma-Aldrich (St. Louis, MO), and lopinavir from Abbott Laboratories (Chicago, IL). All other chemicals were of analytic grade and purchased from commercial sources.
Cloning and Establishment of Stable NTCP-HEK293 Cells
Total human liver RNA was obtained from Clontech (Mountain View, CA), and cDNA was generated by reverse transcription using the SuperScript III first-strand synthesis supermix (Invitrogen, Carlsbad, CA). A NTCP-pcDNA5/FRT vector was constructed in two steps. First, the NTCP open reading frame (ORF) was amplified from the human liver cDNA using Platinum Pfx DNA polymerase (Invitrogen) and the gene-specific primer pair 5′-CTAGAAGCTTATGGAGGCCCACAACGCGTC-3′/5′-CTAGGGTACCGGGGCTGTGCAAGGGGAGCAGT-3′. The restriction sites introduced by the primers are underlined. The polymerase chain reaction (PCR) product was cloned into the HindIII/KpnI site of the vector p3xFLAG-CMV14 (Sigma-Aldrich). The inserted NTCP ORF was verified by DNA sequencing analysis and found to be identical to the NCBI SLC10A1 reference sequence (NM_003049).
Next, the NTCP-p3xFLAG-CMV14 was used as template in a second amplification step with Platinum Pfx DNA polymerase (Invitrogen) and the primer pair 5′-CTAGAAGCTTATGGAGGCCCACAACGCGTC-3′/5′-CTAGCTCGAGCTACTTGTCATCGT CATCCT-3′. HindIII and XhoI restriction sites introduced by the primers are underlined. The PCR product, consisting of the NTCP ORF with a FLAG-tag, was cloned into the HindIII/XhoI site of the expression vector pcDNA5/FRT (Invitrogen). The inserted sequence was verified by DNA sequencing analysis. Human embryonic kidney (HEK) Flp-In-293 cells (Invitrogen) were transfected with the constructed NTCP-pcDNA5/FRT expression vector and further selected using hygromycin B (Invitrogen), as previously described elsewhere (Karlgren et al., 2012a).
Cell Cultivation
Mock-transfected HEK Flp-In-293 cells and cells stably expressing NTCP or either of the three OATP transporters [established and characterized by Karlgren et al. (2012a,b)] were cultivated as described elsewhere (Karlgren et al., 2012a). Passages between 10 and 30 were used throughout the study.
Transport Experiments in HEK293 Cells
Two days before the transport experiments, OATP2B1-expressing cells were seeded in 96-well CellBind plates (Corning, Amsterdam, Netherlands) at a density of 100,000 cells per well. Cells expressing NTCP, OATP1B1, or OATP1B3 were seeded in 24-well plates at a density of 600,000 cells per well for 2 days (OATP1B1, NTCP) or 3 days (OATP1B3) before the experiments. For all cultures in 96- or 24-well plates, Flp-In medium without phenol red and hygromycin B was used. Cell density was optimized by a computer-assisted experimental design (MODDE 7.0, Umetrics, Umeå, Sweden) (Karlgren et al., 2012a).
The following experimental procedure was used in both the kinetic and inhibition experiments. At start of the experiment, cells were washed twice with prewarmed Hank’s balanced salt solution (HBSS), pH 7.4, followed by incubation with prewarmed substrate or substrate/inhibitor solutions for 2 minutes at 37°C. The incubation was terminated by adding ice-cold Dulbecco’s phosphate-buffered saline (DPBS), followed by two to three washes with ice-cold DPBS. The cells were dried, and the intracellular drug accumulation was quantified by ultra-high performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS). All transport experiments in HEK293 cells were run in duplicate (24-well format) or triplicate (96-well format) on at least two independent separate occasions.
Kinetic Characterization of NTCP- and OATP-Mediated Atorvastatin Uptake.
The uptake of atorvastatin in HEK293 cells stably expressing NTCP was linear up to 6 minutes in the concentration range of 0.1–800 µM (data not shown). To assess the kinetics of the NTCP-mediated uptake of atorvastatin, we incubated NTCP-HEK293 cells for 2 minutes with increasing concentrations of atorvastatin (0.1–500 µM). The initial uptake rate was plotted against the substrate concentration. The resulting uptake curve was fitted to the Michaelis-Menten equation with the addition of a nonsaturable passive diffusion rate component (eq. 1) using GraphPad Prism version 5.04 (GraphPad Software, La Jolla, CA):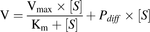 (1)where V is the uptake rate, Vmax is the maximal uptake rate (at saturating substrate concentration), [S] is the substrate concentration, Km is the substrate concentration at which the uptake rate is half of Vmax and Pdiff is the passive diffusion.
(1)where V is the uptake rate, Vmax is the maximal uptake rate (at saturating substrate concentration), [S] is the substrate concentration, Km is the substrate concentration at which the uptake rate is half of Vmax and Pdiff is the passive diffusion.
The Michaelis Menten kinetics of OATP1B1-, OATP1B3-, and OATP2B1-mediated uptake of atorvastatin were previously determined elsewhere (Karlgren et al., 2012a,b). Km and Vmax data were used from these studies.
Concentration-Dependent Inhibition of NTCP- and OATP-Mediated Atorvastatin Uptake.
The half-maximal inhibitory concentrations (IC50 values) of atazanavir, cyclosporine, gemfibrozil, lopinavir, and rifampicin for NTCP-, OATP1B1-, OATP1B3-, and OATP2B1-mediated atorvastatin uptake were determined in vitro using stably transfected HEK293 cells overexpressing each of the transporters. The inhibitors were selected to cover three well-known clinically interacting drugs causing in vivo atorvastatin area under the plasma concentration-time curve (AUC) changes to varying extents (cyclosporine, gemfibrozil, and rifampicin). In addition, two drugs (atazanavir and lopinavir) that interact clinically with other statins were included.
The substrate concentration was set to 1 µM, and uptake was measured at 7 to 12 inhibitor concentrations: 0.01–40 µM (atazanavir), 0.01–25 µM (cyclosporine), 0.01–1000 µM (gemfibrozil), 0.01–10 µM (lopinavir), and 0.01–630 µM (rifampicin). Cells incubated with 1 µM atorvastatin were used as a reference in the calculations of the remaining active uptake in the presence of the compound of interest. In all experiments, uptake in mock-transfected cells was subtracted from the total uptake to correct for the passive permeability. The resulting inhibition data were fitted to eq. 2 using GraphPad Prism version 5.04 (GraphPad Software) to estimate an IC50 value: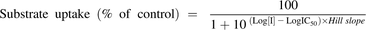 (2)where [I] is the inhibitor concentration and the Hill slope describes the steepness of the curve. The equation is equal to the four-parameter equation when the top plateau of the curve is constrained to 100% and the bottom plateau is fixed to 0% in the data fitting.
(2)where [I] is the inhibitor concentration and the Hill slope describes the steepness of the curve. The equation is equal to the four-parameter equation when the top plateau of the curve is constrained to 100% and the bottom plateau is fixed to 0% in the data fitting.
As previously defined, a compound was considered to be a specific inhibitor of a transporter if the IC50 value was at least 10-fold lower than the IC50 values of the other three transporters (Karlgren et al., 2012b). On the basis of the IC50 values, the corresponding inhibition constants (Ki) were calculated assuming competitive inhibition (eq. 3): (3)
(3)
UPLC-MS/MS Analysis.
Intracellular atorvastatin was extracted with 200 µl acetonitrile/water (60:40) spiked with 50 nM warfarin as internal standard, followed by centrifugation at 2465g at 4°C for 20 minutes. Atorvastatin concentration in the supernatant was determined using UPLC-MS/MS with the following analytic system: Acquity UPLC with a reversed phase BEH C18 column (2.1 × 50 mm, particle size 1.7 µm) (Waters, Milford, MA) and a mobile gradient consisting of acetonitrile, formic acid, and water, coupled to a Waters Xevo triple quadrupole with electrospray ionization interface.
Protein Concentration.
In all uptake kinetic or inhibition experiments, total protein content was measured in representative wells using the BCA Protein Assay Reagent Kit (Pierce Biotechnology, Rockford, IL).
Human Liver Tissue
Normal excess human liver tissue was obtained from liver resections performed at the Department of Surgery, Uppsala University Hospital (Uppsala, Sweden), as approved by the Uppsala Regional ethics review board (ethical approval no. 2009/028). All donors gave their informed consent. Twelve snap-frozen liver tissue samples were used for analysis of hepatic protein expression. Another five human liver tissue specimens were used for hepatocyte isolation and subsequent uptake experiments. A summary of donor characteristics can be found in Supplemental Table 1. All donors were of Caucasian origin. The donors had no history of human immunodeficiency virus (HIV) or hepatitis infection.
Transport Experiments in Human Hepatocytes
Primary hepatocytes were isolated using a two-step collagenase perfusion technique described elsewhere (Lecluyse and Alexandre, 2010). The cells were suspended in DMEM supplemented with 5% fetal bovine serum, penicillin-streptomycin (100 U ml−1 and 100 µg ml−1, respectively), 4 µg ml−1 insulin, and 1 µM dexamethasone, and plated on collagen I-coated 24-well plates (BD Biosciences, Franklin Lakes, NJ) at a density of 375,000 cells per well. The cells were allowed to attach for 3 hours at 37°C and 5% CO2 atmosphere. After attachment, the medium was replaced with hepatocyte maintenance medium (Lonza, Basel, Switzerland) supplemented with penicillin-streptomycin, insulin-transferrin-selenium (10 µg ml−1, 5.5 µg ml−1, and 5 ng ml−1, respectively), and 0.1 µM dexamethasone.
NTCP-Mediated Uptake of Atorvastatin.
Twenty-four hours after seeding, the cells were washed twice with either prewarmed modified Krebs-Henseleit bicarbonate (KHB) buffer (1.2 mM MgSO4, 0.96 mM KH2PO4, 4.83 mM KCl, 118 mM NaCl, 1.53 mM CaCl2, 23.8 mM NaHCO3, 12.5 mM HEPES, 5 mM glucose, pH 7.4) or sodium-free KHB (NaCl and NaCHO3 replaced with choline chloride and KHCO3, respectively, pH 7.4). This was followed by a preincubation with the sodium-containing/sodium-free KHB for 10 minutes at 37°C. The preincubation medium was removed, and drug transport was initiated by adding either atorvastatin (1 µM) or taurocholate (1 µM) in KHB or sodium-free KHB. The substrate concentration was selected to be in the vicinity of transporter Km (linear range) without violating the limit of detection in the mass spectrometric analysis. Uptake was terminated after 2 minutes by adding ice-cold DPBS. Cells were washed three times with DPBS and then dried. Intracellular accumulation of atorvastatin and taurocholate was determined with UPLC-MS/MS as described earlier. The experiment was run in quadruplicate on two separate occasions using hepatocytes isolated from two different donors.
Inhibition of Atorvastatin Uptake.
At start of the experiment, cells were washed twice with prewarmed HBSS, pH 7.4, followed by incubation with prewarmed substrate or substrate/inhibitor solutions for 0.5, 1.0, 1.5, or 2.0 minutes at 37°C. Atorvastatin concentration was set to 1 µM for reasons explained earlier. Because the hepatocyte experiments required a higher substrate concentration than that reached in vivo (due to sensitivity limitations in the mass spectrometric analysis), the inhibitor concentration was scaled up to 3 times the predicted unbound inlet concentration to the liver to give a similar extent of uptake inhibition as in our in vivo predictions. Uptake was terminated at designated time points by adding ice-cold DPBS followed by three washes. Acetonitrile was added to stop atorvastatin metabolism and was then let evaporate. Intracellular drug accumulation was quantified by UPLC-MS/MS as described earlier. Atorvastatin uptake was plotted against incubation time, and the initial uptake rate in absence and presence of inhibitor was determined from the slope of the curves. The experiment was run in triplicate on three separate occasions using hepatocytes isolated from two different donors and one in-house batch of cryopreserved hepatocytes from a third donor.
Quantitative Protein Expression Analysis
Targeted Protein Quantification of NTCP in Human Liver and in NTCP-HEK293 Cells.
HEK293 cells stably transfected with NTCP were harvested and frozen down as previously described for HEK-OATP cells (Karlgren et al., 2012b). Membrane fractions from the pellet of HEK293 cells were extracted and digested with trypsin using the protocol by Qui and colleagues (Qiu et al., 2013). The abundance of NTCP in HEK293 cells stably expressing the transporter relative to that in a representative human liver sample (previously prepared and analyzed for OATP1B1, OATP1B3 and OATP2B1 protein expression (Karlgren et al., 2012b)) was determined by peptide-based liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) measurements. An isotope-labeled peptide, unique for NTCP, was used as an internal standard to quantify the corresponding surrogate peptide of NTCP protein in both the cell line and human liver sample. Each sample was analyzed in duplicate (technical repeats).
Quantitative Proteomic Analysis of Interindividual Differences in Protein Expression in the Human Liver Samples.
The protein expression levels of NTCP, OATP1B1, OATP1B3, and OATP2B1 in 12 human liver samples were determined from previously obtained in-depth label-free mass spectrometry data (Karlgren et al., 2012b) using the total protein approach as described by Wisniewski et al. (2012). The plasma membrane constituted approximately 10% of the crude membrane fraction analyzed.
In Vitro to In Vivo Extrapolations
Prediction of Hepatic Intrinsic Uptake Clearance from Protein Expression Levels.
Hepatic intrinsic uptake clearance (CLint, uptake) of atorvastatin was predicted from the maximal transport activity (MTA), calculated according to eq. 4 (Karlgren et al., 2012b). Briefly, the ratio of the protein expression in a representative human liver sample to that in the overexpressing cell line (obtained using targeted peptide-based protein quantification) was used as a scaling factor to convert the maximal transport rate observed in vitro to a theoretical maximal transport activity in vivo (see eq. 5).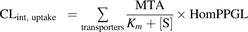 (4)where [S] is the maximal unbound plasma concentration of atorvastatin and HomPPGL is milligrams of homogenate protein per gram of liver tissue:
(4)where [S] is the maximal unbound plasma concentration of atorvastatin and HomPPGL is milligrams of homogenate protein per gram of liver tissue: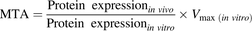 (5)Using previously determined MTA values for atorvastatin transport by OATP1B1, OATP1B3, and OATP2B1 (Karlgren et al., 2012b), and the MTA value obtained herein for NTCP, we predicted the intrinsic uptake clearance in the reference liver tissue sample. Uptake clearance for each of the 12 individuals was then predicted from the relative protein expression between different individuals, using the representative liver sample as reference.
(5)Using previously determined MTA values for atorvastatin transport by OATP1B1, OATP1B3, and OATP2B1 (Karlgren et al., 2012b), and the MTA value obtained herein for NTCP, we predicted the intrinsic uptake clearance in the reference liver tissue sample. Uptake clearance for each of the 12 individuals was then predicted from the relative protein expression between different individuals, using the representative liver sample as reference.
Prediction of Drug-Drug Interactions.
In vitro inhibition data (Ki) were used to predict the impact of inhibition on atorvastatin intrinsic uptake clearance according to eq. 6 (Karlgren et al., 2012b): (6)The inhibitor concentration [I] in the predictions was the estimated maximal unbound concentration of the inhibitor at the inlet to the liver (Iu, max, in), as reported by Yoshida et al., 2012.
(6)The inhibitor concentration [I] in the predictions was the estimated maximal unbound concentration of the inhibitor at the inlet to the liver (Iu, max, in), as reported by Yoshida et al., 2012.
Data Presentation and Statistical Analysis
Data are expressed as mean ± standard deviation unless otherwise stated. Differences in hepatic uptake in the presence and absence of sodium were assessed using Student’s t test. P < 0.05 was considered statistically significant.
Results
Atorvastatin Uptake in NTCP- and OATP-expressing HEK293 cells.
Atorvastatin uptake was 8-fold higher in cells expressing NTCP compared with mock-transfected cells (Fig. 1A). The uptake followed Michaelis-Menten kinetics (Fig. 1B). The Km was determined to 185 ± 108 µM and Vmax to 2260 ± 1184 pmol min−1 mg protein−1, respectively (Table 1). OATP1B1-, OATP1B3-, and OATP2B1-mediated atorvastatin uptake kinetics were determined elsewhere (Karlgren et al., 2012b) and are given in Table 1.

Atorvastatin uptake in NTCP-HEK293 cells. (A) Uptake of atorvastatin in cells expressing NTCP compared with passive uptake in mock-transfected cells. Data represent the mean and standard deviation of 12 independent experiments, each run in triplicate. (B) Kinetic profile of NTCP-mediated uptake of atorvastatin and passive diffusion in mock-transfected cells. Atorvastatin uptake in HEK293 cells either stably transfected with NTCP or mock-transfected was measured over a range of 10 concentrations between 0.1 and 800 µM. Data represent the uptake rate measured in duplicate in one representative experiment.
Kinetic parameters of NTCP-, OATP1B1-, OATP1B3-, and OATP2B1-mediated atorvastatin uptake in stably transfected HEK293 cells
Interindividual Variability in Protein Expression of NTCP, OATP1B1, OATP1B3, and OATP2B1 in Human Liver Samples.
Twelve human liver tissue samples were analyzed for expression of NTCP, OATP1B1, OATP1B3, and OATP2B1 using label-free mass spectrometry. The four transport proteins were detected in all 12 samples. The rank order in expression was OATP1B1 > OATP1B3 ≈ NTCP ≈ OATP2B1 (Fig. 2, A–D; Table 2). All of the uptake transporters displayed considerable variability in protein expression among the 12 individuals, ranging from 3.7-fold (OATP1B1) to 32-fold (OATP1B3) (Table 2). However, the summed expression of the four transporters in the 12 livers only varied 2.3-fold.

Label-free protein quantification of uptake transporters in human liver tissue. Protein expression levels of OATP1B1 (A), OATP1B3 (B), OATP2B1 (C), and NTCP (D) in crude membrane fractions from 12 human livers. Protein abundance were calculated from the ratio of the summed signal intensities of the peptides identifying each protein to the signal intensities of all peptides identified for the human liver proteome (>4000 proteins). The last bar represents the arithmetic mean expression for the 12 samples with standard deviation.
Protein expression levels of uptake transporters in human liver membrane fractions
Predictions of the Intrinsic Uptake Clearance and Contribution of NTCP, OATP1B1, OATP1B3, and OATP2B1 to Atorvastatin Uptake In Vivo.
The contribution of NTCP, OATP1B1, OATP1B3, and OATP2B1 to atorvastatin uptake in vivo (Fig. 3) was predicted from atorvastatin uptake kinetics (Table 3) by correcting for the difference in membrane protein expression between the cell lines and the human liver samples. NTCP was predicted to play a minor role in the uptake, with 1.5% to 10% of total transporter-mediated uptake. Instead, OATP1B1 and OATP1B3 were confirmed to be the major uptake transporters with relative contributions of 26% to 89% and 1.8% to 60% to the total active uptake, respectively. In 3 of the 12 individuals (subjects 5, 6, and 9), OATP1B3 was the primary transporter responsible for atorvastatin uptake into the liver. In contrast, the third hepatic OATP transporter, OATP2B1, generally played a less important role in the uptake, with 3.2–30% of total active uptake (Fig. 3). The rank order in average contribution was OATP1B1 > OATP1B3 >> OATP2B1 > NTCP.

Atorvastatin hepatic uptake. Prediction of in vivo intrinsic hepatic uptake clearance (CLint,uptake) of atorvastatin based on protein expression data in human liver and in vitro cell models. The dotted line represents the mean intrinsic uptake CL for the 12 livers. The hepatic clearance via passive diffusion (CLpassive) was predicted to be the same as that observed in HEK293 cells (120 μL min−1 g liver−1). OATP1B1 and OATP1B3 were predicted to contribute to the majority of the hepatic atorvastatin uptake.
Half-maximal inhibitory concentrations, IC50, and corresponding inhibition constants, Ki, calculated assuming competitive inhibition
For the 12 livers studied, a mean atorvastatin intrinsic uptake clearance of 2030 ml min−1 was predicted (95% CI, 1440–2620 ml min−1). Assuming the same passive diffusion of atorvastatin across the cell membrane of hepatocytes and HEK293 cells, transporter-mediated active uptake was predicted to dominate, with 90% ± 2% (85%–93%) of overall atorvastatin uptake.
NTCP-Mediated Uptake of Atorvastatin in Human Hepatocytes.
NTCP plays a more significant role in the uptake of pitavastatin, fluvastatin, and rosuvastatin than what we have predicted for atorvastatin. We thus further investigated the validity of our predictions by studies in human hepatocytes. In this model, the sodium-independent OATP transporters are active in parallel with the sodium-dependent NTCP. Hence, removal of sodium ions would incapacitate the NTCP transporter but not the OATP transporters.
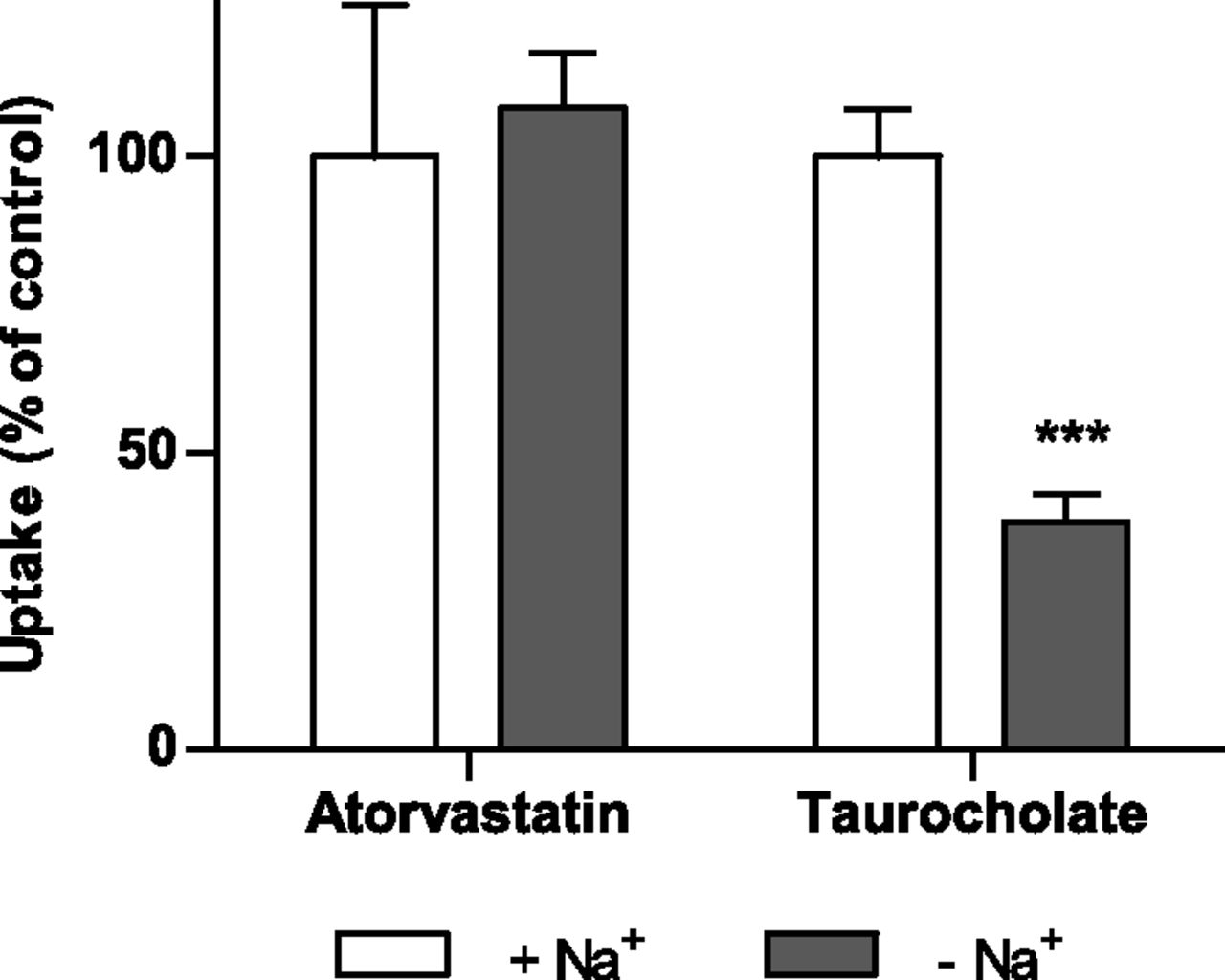
No statistically significant difference in atorvastatin uptake in sodium-containing as compared with sodium-free buffer was observed, whereas the uptake of the prototypical NTCP substrate taurocholate was reduced by more than 50% after removal of sodium (P < 0.001) (Fig. 4). This result is consistent with our predictions of a low contribution of NTCP to hepatic atorvastatin accumulation.

Atorvastatin and taurocholate uptake in human hepatocytes in sodium-containing (control) and sodium-free buffer after 2 minutes of incubation with a concentration of 1 µM. Data are shown as mean ± S.D. (n = 4) from a representative experiment. ***P < 0.001 compared with control.
Inhibition of NTCP- and OATP-Mediated Atorvastatin Uptake In Vitro.
The dose-dependent inhibition of NTCP-, OATP1B1-, OATP1B3-, and OATP2B1-mediated atorvastatin uptake in HEK293 cells with atazanavir, cyclosporine, gemfibrozil, lopinavir, and rifampicin is presented in Fig. 5, A–E. These drugs inhibit plasma clearance of atorvastatin or other statins in clinical drug-drug interaction (DDI) studies. The corresponding IC50 values are summarized in Table 3. We defined an inhibitor as selective if the IC50 value was at least 10 times lower than those of the other three transporters. On the basis of this definition, atazanavir, lopinavir, and rifampicin were defined as selective OATP1B1 inhibitors for atorvastatin uptake (Fig. 5, A, D, and E). On the other hand, OATP2B1- and NTCP-mediated atorvastatin uptake was stimulated by rifampicin in a dose-dependent manner (Fig. 5E). All the uptake transporters except OATP2B1 were strongly inhibited by cyclosporine (Fig. 5B). In contrast, gemfibrozil only interacted weakly with OATP1B1-, OATP2B1-, and NTCP-mediated transport of atorvastatin (IC50 > 50 µM), Fig. 5C.

Inhibition of OATP1B1-, OATP1B3-, OATP2B1-, and NTCP-mediated uptake of atorvastatin by atazanavir (A), cyclosporine (B), gemfibrozil (C), lopinavir (D), and rifampicin (E) in stably transfected HEK293 cells. The cells were incubated with 1 µM atorvastatin and increasing concentrations of the potential inhibitors for 2 minutes at 37°C. Data represent mean ± S.E.M.
Predictions of the Influence of DDIs on Atorvastatin Uptake Clearance.
The influence of coadministration of atazanavir, cyclosporine, gemfibrozil, lopinavir, or rifampicin on hepatic atorvastatin uptake clearance at clinically relevant concentrations was predicted for each human liver sample (Fig. 6). In these predictions, the maximal unbound inlet concentration to the liver was used as the inhibitor concentration, whereas the maximal unbound plasma concentration was used for atorvastatin. Due to the variability in protein expression and isoform-specific inhibition (see previous discussion), marked differences in inhibition patterns were observed. For instance, coadministration of cyclosporine (an inhibitor of NTCP, OATP1B1, and OATP1B3) resulted in a low variability in interindividual inhibition (40%–57% remaining active uptake clearance). In contrast, the OATP1B1-specific inhibitors atazanavir, lopinavir, and rifampicin showed greater variability between individuals, with 20%–57%, 33%–76%, and 36%–81% remaining active uptake CL, respectively.

Impact of drug-drug interactions on predicted atorvastatin uptake clearance. Remaining intrinsic uptake clearance upon coadministration of five different inhibitors is shown for the 12 livers. Coadministration of gemfibrozil was predicted to have a minor impact on atorvastatin uptake CL, whereas coadministration of atazanavir, cyclosporine, lopinavir, and rifampicin was predicted to give a more pronounced reduction in atorvastatin uptake.
This can be explained by the isoform specificity of these compounds. Cyclosporine inhibits both OATP1B1 and OATP1B3 with similar potency, but atazanavir, lopinavir, and rifampicin inhibit OATP1B1 with much higher potencies than OATP1B3. The summed contribution of OATP1B1 and OATP1B3 to atorvastatin uptake CL showed lower interindividual variability than the OATP1B1 contribution alone for these 12 livers. Hence, as illustrated here by atazanavir, lopinavir, and rifampicin, large differences in isoform-specific inhibitor potencies translate to high interindividual variability in the inhibition of the hepatic atorvastatin uptake (Fig. 6).
Overall, coadministration with atazanavir, an inhibitor with a relatively high estimated concentration at the inlet to the liver, was predicted to give the largest effect on atorvastatin uptake. Gemfibrozil, on the other hand, was predicted to have a minimal effect on atorvastatin uptake clearance due to its weak or nonexisting inhibition of OATP1B1, OATP1B3, OATP2B1, and NTCP.
Inhibition of Atorvastatin Uptake in Human Hepatocytes.
To verify our DDI predictions, we investigated the inhibition of the initial atorvastatin uptake in human hepatocytes from three donors (Fig. 7). Two of the hepatocyte batches were used directly after isolation, and one was a cryopreserved plateable batch from our collection of human hepatocytes. The uptake of atorvastatin was linear up to 2 minutes. The measured atorvastatin uptake clearance without inhibition was 5–12 µl min−1 mg protein−1, which is similar to that measured independently in sandwich-cultured hepatocytes (unpublished data kindly provided by Dr. El-Kattan, Pfizer). In line with our predictions, cyclosporine was found to inhibit the uptake of atorvastatin by 44%–74% whereas gemfibrozil did not inhibit the uptake at all. Atazanavir inhibited the uptake by 26%–45%, which is in the lower range of our predictions. The other two inhibitors showed less inhibition than predicted from the liver tissue samples (lopinavir 0–30%, and rifampicin 0–21%).

Inhibition of atorvastatin uptake by atazanavir, cyclosporine, gemfibrozil, lopinavir, and rifampicin in human hepatocytes. Atorvastatin (1 µM) was incubated in the presence or absence of the potential inhibitors. Uptake clearance was determined from the slope of the curves. Data are shown as mean ± S.D. (n = 3) from a representative experiment.
Discussion
In a previous study, we introduced an expression-based model to assess the contribution of each OATP to atorvastatin uptake clearance in vivo (Karlgren et al., 2012b). Herein, we applied this model to study interindividual differences in atorvastatin uptake clearance using liver tissue samples from 12 individuals. We also determined the contribution of NTCP to hepatic atorvastatin uptake for the first time.
NTCP and OATP protein levels were determined by label-free mass spectrometry. Our measured transporter abundances were comparable to previously reported hepatic expression data, albeit we found higher levels of OATP1B1 (Kimoto et al., 2012; Ohtsuki et al., 2012; Bi et al., 2013; Prasad et al., 2014). We speculate that this could be a result of the sample preparation used in our study (Wisniewski et al., 2009), including the membrane fractionation, solubilization, and tryptic digestion. The higher expression of OATP1B1 relative to NTCP, OATP1B3, and OATP2B1 confirms its importance in hepatic drug disposition.
Consistent with other reports (Nies et al., 2013), we noted considerable interindividual variability in transporter expression. The observed variability could be a result of differences in gene regulation, polymorphisms, and/or epigenetic profiles (Ivanov et al., 2012; Nies et al., 2013), but it was outside the scope of this study to investigate this further.
Transporter abundances were used to determine the contribution of each transporter to atorvastatin uptake. Our method of predicting the transporter contribution to uptake clearance builds on the assumptions that all protein quantified in the isolated membrane fraction is available to transport (Karlgren et al., 2012b). Intracellular pools or posttranslationally inactivated protein is not accounted for. Hence, the term maximal transport activity (MTA), which represents an upper limit in transport capacity. Although OATP transporters are reported not to be stored in intracellular vesicular compartments, NTCP has been shown to be subject to recycling from endosomal compartments (Roma et al., 2008). Thus, it cannot be excluded that the abundance of NTCP in the cell membrane was overestimated. Nevertheless, this does not influence the interpretation of the results, as the contribution of NTCP to atorvastatin uptake was minor.
Interestingly, the contribution of NTCP to atorvastatin uptake was much lower than previously reported for pitavastatin, fluvastatin, and rosuvastatin (Bi et al., 2013). Instead, we confirmed that OATP1B1 and OATP1B3 were the primary transporters involved in atorvastatin uptake in vivo. The importance of OATP1B1 in hepatic atorvastatin uptake has been emphasized in other studies (Pasanen et al., 2007; Amundsen et al., 2010), but our data suggest that OATP1B3 is almost as important. The relatively high contribution of OATP1B3 to the uptake of atorvastatin differs from other statins, such as simvastatin acid and pitavastatin (Hirano et al., 2004; Elsby et al., 2012). This observation is supported by a study on the clinical pharmacokinetics of atorvastatin, in which OATP1B1 was predicted to account for 47% of the total atorvastatin hepatic uptake (Shitara et al., 2013). We attribute the remainder of the active uptake mainly to OATP1B3.
In contrast to OATP1B1 and OATP1B3, the third OATP transporter, OATP2B1, did not contribute substantially to hepatic uptake of atorvastatin. Instead, OATP2B1 may be important in the uptake of statins into skeletal muscle cells. OATP2B1 expression has been localized to the sarcolemmal membrane of human skeletal muscle fibers, suggesting that this protein has a key role in statin-related adverse effects in this tissue (Knauer et al., 2010). OATP2B1 is also expressed in the human intestine and has been found to alter the AUC of drugs such as fexofenadine (Imanaga et al., 2011). It may thus have an important role in intestinal absorption of statins.
Transporter-mediated DDIs were predicted for a set of clinical inhibitors with different isoform specificity using in vitro inhibitory data. Because OATP inhibition is substrate-dependent (e.g., Noe et al., 2007; Soars et al., 2012), the in vitro inhibitory capacity was determined with atorvastatin as the victim drug. There were clear differences in OATP inhibition pattern when using atorvastatin as substrate instead of prototypical model substrates (Table 4). For instance, atazanavir, cyclosporine, and rifampicin showed a strong-to-moderate inhibition of OATP2B1-mediated uptake of estrone-3-sulfate, but no inhibition of atorvastatin transport. The substrate-dependent inhibition observed here and by others could be a result of multiple binding sites/domains on the OATP transporters (Noe et al., 2007; Miyagawa et al., 2009). We therefore recommend using the substrates of concern, rather than model substrates, in in vitro studies for predicting in vivo interactions.
Comparison of half-maximal inhibitory concentrations, IC50, determined in vitro using either atorvastatin or an endogenous substrate
Atorvastatin uptake increased in the presence of rifampicin in a concentration-dependent manner in OATP2B1- and NTCP-expressing HEK293 cells. Because the contribution of these transporters to overall atorvastatin uptake was limited, this stimulation was not accounted for in our DDI predictions. Stimulation of OATP uptake has been reported previously in in vitro studies (Grube et al., 2006). To our knowledge, though, it has never been observed in vivo. There is evidence of in vivo stimulation of efflux transport by the multidrug resistance associated protein 2 (Mrp2) in rats (Heredi-Szabo et al., 2009), but the degree of potentiation is much less than that observed in vitro in the same study.
Marked differences in the predicted extent of DDIs were observed between the individuals as a result of the variability in transporter contribution to atorvastatin uptake (Fig. 6). This illustrates the importance of investigating the relative contribution of all transporters involved in the uptake of drug substrates to correctly assess the impact of DDIs of various isoform-specific inhibitors. Our MTA-based approach can easily be applied for this purpose and gives accurate predictions of transporter contribution without the need of either in vivo studies of reduced-function genetic variants or human hepatocyte experiments with transporter-specific substrates (relative activity factor) (Hirano et al., 2004). It should be noted that the expression-based scaling factor is in vitro system-specific and thus needs to be determined for each cell line used. Once it has been established, it can be applied to any substrate.
In vitro experiments in human hepatocytes confirmed our DDI predictions. Although the hepatocytes were isolated from different tissue samples than those used for our predictions, the results were in good agreement. Lopinavir and rifampicin, however, gave less inhibition than predicted, and the inhibition by atazanavir was in the lower range of our predictions. This could indicate that the hepatic OATP1B1 expression/availability was lower in these hepatocyte batches than in the human liver tissue samples (Kimoto et al., 2012; Lundquist et al., 2014).
The DDI predictions were included to illustrate the possible consequence of large interindividual variability in transporter expression on the extent of impaired hepatic uptake, potentially translating to variability in systemic exposure. Our predictions agreed qualitatively with clinical observations. Gemfibrozil was predicted to have a low impact on atorvastatin uptake CL in vivo. The reported AUC changes upon concomitant administration of this drug with atorvastatin are also relatively small (1.2- to 1.4-fold) (Backman et al., 2005; Whitfield et al., 2011). In contrast, cyclosporine and rifampicin were predicted to reduce the hepatic uptake by approximately 50%, and these compounds increase atorvastatin AUC to a large extent in vivo (>7-fold AUC change; Asberg et al., 2001; Hermann et al., 2004; Lemahieu et al., 2005; Lau et al., 2007; He et al., 2009). Although hepatic uptake plays a major role in clinically observed DDIs with atorvastatin as the victim drug, inhibition of cytochrome P450 3A4 (CYP3A4)-mediated metabolism and efflux in both the intestine and liver is likely to contribute to the AUC changes seen in vivo. Gemfibrozil and rifampicin are CYP3A4 noninhibitors (Wen et al., 2001; Maeda et al., 2011), but cyclosporine is a potent inhibitor of CYP3A4, and its pronounced effect on atorvastatin AUC may reflect this complexity.
In conclusion, our study has shown that quantification of drug transport protein expression can advance our understanding of interindividual differences in hepatic uptake and DDIs. We found substantial differences in the expression of NTCP, OATP1B1, OATP1B3, and OATP2B1 in 12 human liver samples. As a consequence, the contribution of these transporters to hepatic uptake influenced the potential for DDI with coadministered drugs at the individual level. We confirm a dominating role of OATP1B1 and OATP1B3 in the uptake clearance of atorvastatin whereas NTCP and OATP2B1 play minor roles. Our study provides proof-of-concept that differences in transporter expression must be taken into account in predictions of variability in drug clearance and clinical DDIs for drugs that are substrates of several transporters.
Acknowledgments
The authors thank Drs. Ulf Haglund, Frans Duraj, and Jozef Urdzik at Uppsala University Hospital for their skillful contribution in clinical sampling, Drs. Xi Qiu and Emi Kimoto at Pfizer for their contribution to the protein quantification, Dr. Ewa Ellis at Karolinska Institutet for kindly providing one of the hepatocyte batches used in the DDI experiments, and Cherendeep Dhanda for valuable contributions in the transport experiments.
Authorship Contributions
Participated in research design: Vildhede, Karlgren, Artursson.
Conducted experiments: Vildhede, Karlgren, Svedberg, Wisniewski, Lai, Norén.
Contributed new reagents or analytic tools: Wisniewski, Lai.
Performed data analysis: Vildhede.
Wrote or contributed to the writing of the manuscript: Vildhede, Karlgren, Svedberg, Wisniewski, Lai, Norén, Artursson.
Footnotes
- Received November 29, 2013.
- Accepted May 5, 2014.
↵1 Current affiliation: Pharmaceutical Candidate Optimization, Bristol-Myers Squibb, Princeton, New Jersey.
This work was supported by the Swedish Research Council [grant approval no. 2822]; the Swedish Fund for Research without Animal Experiments; the Lars Hierta Memorial Foundation; and O.E. and Edla Johansson’s Scientific Foundation.
Part of this work was presented as follows:
Karlgren M, Vildhede A, Wisniewski J, and Artursson P (2012) Variability in OATP protein expression and influence on atorvastatin uptake and drug-drug interactions. Abstract P289. 19th International Symposium on Microsome and Drug Oxidations (MDO)/12th European Regional ISSX Meeting; 2012 June 17–21; Noordwijk aan Zee, the Netherlands. International Society for the Study of Xenobiotics, Washington, DC.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the plasma concentration-time curve
- CLupt
- uptake clearance
- CYP
- cytochrome P450
- DDI
- drug-drug interaction
- DMEM
- Dulbecco’s modified eagle medium
- DPBS
- Dulbecco’s phosphate-buffered saline
- HEK
- human embryonic kidney
- IC50
- half-maximal inhibitory concentration
- KHB
- Krebs-Henseleit bicarbonate
- Km
- Michaelis-Menten constant
- MTA
- maximal transport activity
- NTCP
- sodium taurocholate cotransporting polypeptide
- OATP
- organic anion transporting polypeptide
- ORF
- open reading frame
- SLC
- solute carrier
- UPLC-MS/MS
- ultra-high performance liquid chromatography coupled with tandem mass spectrometry
- Vmax
- maximal uptake rate
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}