


Visual Overview

Abstract
This study focused on the mechanistic interpretation of ex vivo oxidation of a candidate drug in blood plasma samples. An unexpected lipid peroxide–mediated epoxidation followed by a dramatic rearrangement led to production of a five-membered oxazole derivative from the original six-membered pyrazinone-carboxamide core of a human neutrophil elastase inhibitor, 6-(1-(4-cyanophenyl)-1H-pyrazol-5-yl)-N-ethyl-5-methyl-3-oxo-4-(3-(trifluoromethyl)phenyl)-3,4-dihydropyrazine-2-carboxamide (AZD9819). The rearranged oxidation product 2-(1-(4-cyanophenyl)-1H-pyrazol-5-yl)-5-(N-ethylacetamido)-N-(3-(trifluoromethyl)phenyl)oxazole-4-carboxamide was characterized by accurate-mass tandem mass spectrometry fragmentations, by two-dimensional NMR and X-ray crystallography of an authentic standard, and by incorporation of an 18O atom from molecular 18O2 to the location predicted by our proposed mechanism. The lipid peroxide–mediated oxidation was demonstrated by using human low-density lipoprotein (LDL) in pH 7.4 phosphate buffer and by inhibiting the oxidation with ascorbic acid or l-glutathione, two antioxidants effective in both plasma and the LDL incubation. A nucleophilic mechanism for the epoxidation of AZD9819 by lipid hydroperoxides explains the prevention of its ex vivo oxidation by acidification of the plasma samples. The discovery of the lipid peroxide–dependent oxidation of an analyte and the means of prevention could provide valuable information for biotransformation and bioanalysis.
Introduction
Accurate measurement of drug molecule concentrations in blood plasma samples is essential for pharmacokinetic and toxicokinetic studies in drug discovery and development (Li et al., 2011; Rozet et al., 2011). Correct identification of metabolites in plasma samples is critical for safety assessment of a drug candidate (Ma and Chowdhury, 2011; Penner et al., 2012; Gao et al., 2013). The occurrence of ex vivo instability complicates the analysis by making metabolite identities insecure and measured drug concentrations uncertain. Although the instability of some molecules could be predicted by the presence of certain chemically or biologically labile moieties (Erve, 2008; Li et al., 2011; Mitchell, 2014), in other cases, ex vivo instability can be unexpected.
One such unexpected instability was discovered during the preclinical development of an inhaled drug candidate, 6-(1-(4-cyanophenyl)-1H-pyrazol-5-yl)-N-ethyl-5-methyl-3-oxo-4-(3-(trifluoromethyl)phenyl)-3,4-dihydropyrazine-2-carboxamide (AZD9819) (Cutting et al., 2011), an inhibitor of human neutrophil elastase for potential treatment of chronic obstructive pulmonary disease (Sjö, 2012; Lucas et al., 2013). A mono-oxidation product of AZD9819 was detected in plasma ex vivo and in vitro. The occurrence of this oxidation was empirically prevented by acidification following plasma sample collection (e.g., with 2 M phosphoric acid added at approximately 1.5% of the plasma volume) in preclinical and clinical development studies. Two intriguing findings emerged from our investigation of this otherwise seemingly simple mono-oxidation. First, the mono-oxidation product was in fact a drastically rearranged oxidation product (ROP). Second, we pinpointed lipid peroxides as the cause of the initial epoxidation step in our proposed mechanism of ROP formation. This cause of oxidation of drug molecules in blood plasma has not been reported previously.
The ROP formation is distinct in terms of both the cause of oxidation and subsequent rearrangements from two previous reports of rearranged oxidative metabolites of pyrazinone-containing thrombin inhibitors developed at Merck (Singh et al., 2003; Subramanian et al., 2003). Oxidative rearrangements in the Merck cases occurred via cytochrome P450 (P450)–mediated epoxidation at the C=C bond in a pyrazinone ring (of 3-amino-6-methyl-pyrazinone and 3-amino-6-chloro-pyrazinone derivatives) (Singh et al., 2003; Subramanian et al., 2003). This is no surprise because P450 enzymes are known to be capable of epoxidation (Parkinson, 2001; Guengerich, 2007). However, ROP was not produced by cytochrome P450 in liver in our case with AZD9819. Our finding of the role of lipid peroxides in the mechanism of ROP formation described in this article could help in troubleshooting and preventing the instability of some other xenobiotic or endogenous organic molecules in plasma samples.
Materials and Methods
AZD9819 was synthesized by a reported procedure (Cutting et al., 2011), with spectral properties as follows; mass spectrometry (MS): protonated molecular ion [M+H]+ mass-to-charge ratio (m/z) 493.1592 ± 0.0011 (S.D., n = 14; Calculated m/z 493.1594); 1H NMR (600 MHz, CD3SOCD3): δ 1.02 (t, J = 7.2 Hz, 3H), 1.88 (s, 3H), 3.19 (qd, J = 7.0, 6.5 Hz, 2H), 6.77 (d, J = 1.8 Hz, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.0 Hz, 1H), 7.88 (t, J = 8.0 Hz 1H), 7.89 (d, J = 8.7 Hz 2H), 7.93 (d, J = 1.8 Hz 1H), 7.95 (s, 1H), 7.96 (d, J = 8.0 Hz 1H), 8.71 (t, J = 8.0 Hz 1H); 13C NMR (150 MHz, CD3SOCD3): 14.5, 18.7, 33.6, 109.3, 111.2, 118.5, 122.5, 123.7 (q, J = 272.5 Hz), 123.9, 124.8 (q, J = 3.5 Hz), 126.6 (q, J = 3.6 Hz), 130.5 (q, J = 32.6 Hz), 131.3, 132.0, 133.4, 137.4, 138.4, 141.2, 142.4, 143.0, 143.3, 154.4, 161.3 (complete assignment available in Supplemental Figs. 1 and 2)
The Synthetic Standard of 2-(1-(4-cyanophenyl)-1H-pyrazol-5-yl)-5-(N-ethylacetamido)-N-(3-(trifluoromethyl)phenyl)oxazole-4-carboxamide (ROP).
AZD9819 (1 g, 0.002 mol) was slurried in acetonitrile (50 ml) and water (50 ml) at 25°C. Sodium perborate tetrahydrate (0.38 g, 0.0024 mol) was dissolved in water (20 ml) and added slowly to the reaction mixture over approximately 3 hours. During the addition, the pH of the reaction mixture was maintained at 8 by the addition of 0.1 M hydrochloric acid solution. After addition of the perborate, the reaction was stirred for 20 hours, diluted with water (100 ml), and extracted with dichloromethane (2 × 50 ml). The combined dichloromethane extracts were washed with water (2 × 50 ml), dried over anhydrous sodium sulfate, and evaporated under reduced pressure to leave the product ROP as a white powder [yield = 0.85 g, 83%; MS: [M+H]+ m/z 509.1544 ± 0.0011 (S.D., n = 8; Calculated m/z 509.1543); 1H NMR (600 MHz, CD3SOCD3): δ 0.90 (t, J = 6.9 Hz, 3H), 1.93 (br s, 3H), 3.55 (q, J = 6.9 Hz, 2H), 7.32 (d, J = 1.9 Hz, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.83 (d, J = 8.6 Hz, 2H), 8.03 (d, J = 8.6 Hz, 2H), 8.05 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 8.26 (s, 1H), 10.53 (s, 1H); 13C NMR (151 MHz, CD3SOCD3): δ 13.1, 21.8, 42.6, 111.2, 112.2, 116.8 (q, J = 3.8 Hz), 118.2, 120.6 (q, J = 3.5 Hz), 124.1 (q, J = 272.2 Hz), 124.3, 125.9, 126.5, 129.4 (q, J = 31.6 Hz), 129.8, 129.9, 133.5, 138.8, 141.9, 142.7, 148.5, 149.8, 158.2, 169.3 (complete assignment available in Supplemental Figs. 3 and 4)].
Blood Plasma.
Blank plasma used for the study with [14C]AZD9819 included Wistar Han rat (K2-EDTA, male, pooled) and human plasma (K2-EDTA, mixed gender, four-donor pool) prepared at AstraZeneca R&D (Lund, Sweden) and Beagle dog plasma (K2-EDTA, male, three-donor pool) obtained from Harlan-Winkelmann (Borchen, Germany). Blank plasma used for the study with nonradiolabeled AZD9819 included Wistar Han rat plasma (K3-EDTA, male) and Beagle dog plasma (K3-EDTA) obtained from Bioreclamation (Westbury, NY) and human plasma (K3-EDTA, mixed gender, pooled) obtained from BioChemed Services (Winchester, VA).
Chemicals, Biochemicals, and Proteins.
Most were purchased from Sigma-Aldrich (St. Louis, MO), e.g., allopurinol, l-ascorbic acid (vitamin C, catalog number 255564), bovine liver catalase (catalog number C40), reduced l-glutathione (GSH; catalog number G4251), hemoglobin human (catalog number H7379), hydrogen peroxide solution 30 wt.% (catalog number 216763), 18O2 gas (97 atom % 18O, catalog number 490474), 18O-water (97 atom % 18O, catalog number 329878), potassium cyanide (KCN), and sodium azide (NaN3).
Human low-density lipoprotein (LDL) lyophilized powder (catalog number L8292, purity: >95%) was also bought from Sigma-Aldrich. The LDL is composed of 20–25% protein and 75–80% lipid. The lipid portion can be further described as follows: 9% free cholesterol, 42% cholesteryl ester, 20–24% phospholipid, and 5% triglyceride.
Plasma Incubation of [14C]AZD9819.
Plasma samples were incubated at 37°C with 14 μM [14C]AZD9819. As a negative control, the stability of [14C]AZD9819 was investigated in 0.1 M phosphate buffer, pH 7.4, supplemented with 5 mM MgCl2 for 2 hours. Aliquots of samples were taken at 0 and 120 minutes, and the reaction was terminated with an equal volume of prechilled acetonitrile (MeCN)/formic acid (99/1, v/v).
Incubations of AZD9819 with Human LDL.
Deionized water was added to lyophilized LDL, resulting in a 5-mg/ml LDL suspension containing approximately 150 mM NaCl and 0.01% EDTA at pH 7.4. Incubation mixtures were prepared with 500 mM phosphate buffer solution pH 7.4 to reach final concentrations of 15 µM AZD9819 and 1 mg/ml LDL, in the absence/presence of 50 mM GSH or 50 mM ascorbic acid. The total incubation volume was 250 µl. Additional incubations at pH 4.5 and pH 10.5 were conducted for 15 µM AZD9819 with LDL in 100 mM ammonium acetate buffer pH 4.5 and 100 mM glycine buffer pH 10.5, respectively. Negative control experiments were also conducted with blank buffer solutions. In all incubations, AZD9819 (2 mM in methanol) was added last to allow approximately 40-minute preincubation of LDL at room temperature in the presence/absence of GSH or ascorbic acid. All LDL incubations were repeated in triplicate and were terminated after incubation at 37°C for 2 hours by adding twice the volume of MeCN containing 1% formic acid.
Incorporation of 18O from molecular 18O2.
Prepared 15 µM AZD9819 in 250 µl of rat plasma was sealed in a 1-dram glass vial with a rubber septum at room temperature. Two stainless steel syringe needles were inserted through the septum, with one into the plasma mixture for purging and the other above the liquid level for venting. The plasma mixture and the vial were slowly purged with 18O2 for approximately 5 minutes. Some plasma mixture was lost through the needle venting to the air due to foaming. Both needles were removed from the septum, and the vial was then incubated at 37°C for 2 hours. The reaction was stopped with MeCN (approximately 2:1, v:v).
Plasma Incubations of AZD9819 in the Presence of NaN3, Allopurino, KCN, Ascorbic Acid, or Reduced GSH.
AZD9819 at 15 µM was incubated at 37°C in rat or dog plasma in the presence of NaN3 (up to 50 mM); in rat, dog, or human plasma in the presence of 100 µM allopurinol; in rat plasma in the presence of 50 mM KCN; in rat or dog plasma with ascorbic acid added (up to 50 mM); and in rat, dog, and human plasma with GSH added (up to 50 mM). The pH of plasma samples with added GSH or ascorbic acid was verified to be still above pH 7 with pH test paper. As control, 15 µM AZD9819 was incubated in the plasma of respective species. The incubation volume was 250 µl. The incubations were terminated at 2 hours by adding twice the volume of MeCN.
Heat-Inactivated Rat Plasma.
A tube of several milliliters of rat plasma was inactivated at 56°C for 1 hour. In separate vials, 15 µM AZD9819 and 15 µM ROP standard were incubated with 500 µl of inactivated rat plasma at 37°C. AZD9819 and ROP standard were also incubated in untreated rat plasma at the same concentration as respective positive controls. The reaction was terminated at 2 hours by adding an equal volume of MeCN. Additionally, AZD9819 was incubated at 37°C for 2 hours in denatured rat plasma by mixing with an equal volume of MeCN at time zero.
Catalase-Pretreated Plasma.
Two hundred fifty microliters of rat plasma was preincubated with 1000 unit/ml bovine liver catalase at 37°C for 30 minutes. Then, AZD9819 was added at 15 µM, and the incubation was continued for 2 hours before being terminated by adding twice the volume of MeCN. As a control, 15 µM AZD9819 was incubated in untreated rat plasma, including a preincubation at 37°C for 30 minutes without catalase. The effectiveness of the catalase was confirmed by incubation with 500 µM H2O2 in pH 7.4 buffer when the reactivity of the H2O2 was completely eliminated.
Incubation of AZD9819 with Human Hemoglobin in the Absence/Presence of H2O2.
AZD9819 at 15 µM was incubated at 37°C with 100 µM human hemoglobin in 100 mM phosphate buffer pH 7.4, with 100 µM hemoglobin in the presence of 500 µM H2O2, or with 500 µM H2O2 alone in the buffer. After 2 hours, bovine liver catalase was added to the incubation mixture at 1000 unit/ml, followed by an additional 15-minute incubation. As a negative control, 500 µM H2O2 in the buffer was preincubated with 1000 units/ml for 15 minutes before AZD9819 was added. At the end of all incubations, twice the volume of MeCN containing 1% formic acid was added to the incubation mixture.
Radiochromatography and Liquid Chromatography–MS and Tandem Mass Spectrometry of [14C]AZD9819 Incubation Products.
After centrifugation of terminated incubation mixtures to pellet proteins, supernatants were recovered and diluted with one volume of deionized water. Aliquots of 100-μl samples were injected into a liquid chromatography (LC)–radioactivity detector system, comprising an 1100 series LC system (Agilent, Palo Alto, CA) coupled to a Flo-One 500 series on-line radioactivity flow detector with a 500-μl liquid cell (PerkinElmer, Waltham, MA). LC separation used a C18, 3-μm, 150 × 4.6–mm column (ACE, Aberdeen, Scotland, UK). The mobile phases consisted of MeCN:H2O (5:95; A) and MeCN:H2O (95:5; B). Both solvents contained 0.1% formic acid. The LC gradient started at 5% B, increased linearly to 25% B during the first minute and then to 65% B over the next 24 minutes. It was then increased to 100% B at 26 minutes and retained for 3 minutes, then finally decreased to 5% B at 30 minutes. The flow rate was 1 ml/min of LC mobile phase and 3 ml/min of liquid scintillation cocktail.
A LC system of the same type as described earlier was used in line with a Q-ToF Premier mass spectrometer (Waters, Wilmslow, UK) equipped with a LockSpray interface and electrospray probe. The LC-MS and tandem mass spectrometry (MS/MS) analysis used the same type but a smaller LC column (50 × 2.1 mm) and the same LC mobile phases as described earlier with similar but short gradients at a flow rate of 0.25 ml/min. MS/MS was performed at 30-eV collision energy with argon as the collision gas.
LC-UV-MS and MS/MS of Nonradiolabeled AZD9819 Incubation Products.
After centrifugation of terminated incubation mixtures to pellet proteins, aliquots of 10- or 15-µl supernatant samples were injected into a LC-UV-MS system, consisting of an Acquity UPLC system (Waters, Milford, MA) and an LTQ-Orbitrap XL high-resolution mass spectrometer (Thermo Scientific, San Jose, CA). An ACE C18, 3-μm, 150 × 2.1–mm column was used for LC separation. The mobile phases at a flow rate of 0.2 ml/min consisted of water (A) and acetonitrile (B). Both solvents contained 0.1% formic acid. The LC gradient started at 5% B, increased linearly to 25% during the first minute, and then to 65% B over the next 24 minutes. It was then increased to 100% B at 26 minutes and retained for 3 minutes, then finally decreased to 5% B at 30 minutes. The UV wavelength at 268 nm with 6-nm resolution was monitored in acquisition of UV chromatograms, and photodiode array UV spectra of 200–500 nm were also acquired.
LC-MS spectra were acquired at a resolution of 30,000. Collision-induced dissociation (CID) with helium as a collision gas in the linear ion trap, or the instrument manufacturer’s “high-energy collision-induced dissociation” (HCD) with nitrogen as the collision gas in the HCD cell, was used in acquisition of MS/MS spectra by the Orbitrap analyzer at a resolution of 15,000. The normalized collision energy at 45 and 35 of the manufacturer’s respective units was used in HCD and CID experiments. Fragmentation by the HCD is similar to that by the CID in QTof Premier mass spectrometer used for the radiolabeled study, both of which produce more fragments from [M+H]+ ions of AZD9819 or its products. However, the linear ion trap CID was used for most of the experiments in the present study, due to higher signals of the ion trap CID.
Nuclear Magnetic Resonance Spectroscopy.
1H NMR, 13C NMR, two-dimensional heteronuclear single quantum coherence and heteronuclear multiple bond correlation (HMBC) spectra were recorded at 298 K in dimethylsulfoxide-d6 on a Bruker Avance Spectrometer (Billerica, MA) at a proton frequency of 599.7 MHz and carbon frequency of 150.8 MHz. Referencing is reported relative to deuterated solvent signals at 2.500 ppm (proton) and 39.52 ppm (carbon).
X-Ray Crystallography of ROP Synthetic Standard.
Single crystals of appropriate size and quality for crystal structure determination were grown with the free interface diffusion method. Acetonitrile was used as solvent and diisopropyl ether as antisolvent. Crystal data for the ROP synthetic standard are as follows: C25H19F3N6O3, Mr = 508.46 g/mol, colorless, triclinic, P-1 (No. 2), a = 11.3364(4)Å, b = 12.3259(4)Å, c = 18.0013(7)Å, α = 99.870(2)°, β = 95.648(2)°, γ = 107.680(2)°, V = 2330.53(14) Å3, Z = 4, ρcalc = 1.449 g/cm3, µ = 0.115 mm−1, R1 = 0.047, wR2 = 0.154, GOF = 1.08. The data were collected at 200 K on a Bruker ApexII diffractometer with graphite-monochromated MoK(α) radiation and using a 0.22 × 0.20 × 0.04–mm crystal (Rint = 0.048). The crystal structure was determined by direct methods and refined by full matrix least-squares analyses with anisotropic temperature factors for all atoms except hydrogen atoms. Hydrogen atom positions were calculated using known molecular geometries, refined in riding mode with fixed isotropic temperature factors. The fluorine atoms of the −CF3 group in one of the molecules in the asymmetric unit are disordered. Cambridge Crystallographic Data Centre number 1039312 contains the supplemental crystallographic data for the ROP synthetic standard. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre at http://www.ccdc.cam.ac.uk/data_request/cif.
Results
Figure 1 shows radiochromatographic profiles of rat, dog, and human plasma–generated products of [14C]AZD9819 following a 2-hour incubation at 37°C. Chemical structures of [14C]ROP and its deacetylation product are provided in the figure for easy reading. During our investigations, a great number of LC-UV-MS chromatograms demonstrating ROP formation in plasma samples were acquired. We have chosen to present the radiochromatograms here as they convincingly demonstrate that ROP is the sole direct product of the parent compound AZD9819. The deacetyl-ROP that occurred mainly in rat plasma was shown to be an enzymatic hydrolysis product of ROP (described later). Interestingly, we were able to synthesize an authentic standard of ROP before we even knew what the structure was. The choice of synthetic method resulted from preliminary work using a commercial peroxidase in the presence of H2O2 (Coprinus cinereus peroxidase recombinant in an aspergillus; Novozymes, Copenhagen, Denmark) to make a similar oxidation product of an analog compound (details of the analog compound not provided for proprietary reasons). ROP was made from AZD9819 at high yield by reaction with sodium perborate in MeCN:H2O (Materials and Methods: The Synthetic Standard of ROP). The synthetic standard was unambiguously proven to be identical to ROP generated in plasma by a perfect match in LC-UV-MS/MS (retention time, UV spectra, and high-resolution MS/MS spectra showing the accurate mass of fragment ions; Supplemental Fig. 5). It was also verified that the plasma-generated [14C]ROP at our laboratory in Lund, Sweden and nonradiolabeled ROP at our laboratory in Waltham, MA have the same chemical structure by matching all LC-MS/MS fragmentations (Supplemental Fig. 6).

Radiochromatograms showing products formed by incubation of 14 μM [14C]AZD9819 in rat (A), dog (B), and human (C) plasma at 37°C for 2 hours.
Even after a high-purity standard was synthesized, it was still challenging to elucidate the structure. ROP does not share any similar fragmentation with AZD9819 in MS/MS (Supplemental Figs. 5 and 6), and the lack of protons at the center core of the ROP means that the presence of a new oxazole ring cannot be proven directly by NMR. Hence, a number of putative structures (Supplemental Fig. 7), including those we could envisage arose from possible rearrangement mechanisms following an initial oxidation, were proposed to generate hypothetical products to match NMR and MS/MS data (Supplemental Figs. 3–6). The majority of these putative structures deliberately contained an oxazole substructure, because preliminary assignment of a MS/MS fragment ion of ROP observed in biologic samples had hinted at this possibility (Supplemental Fig. 6).
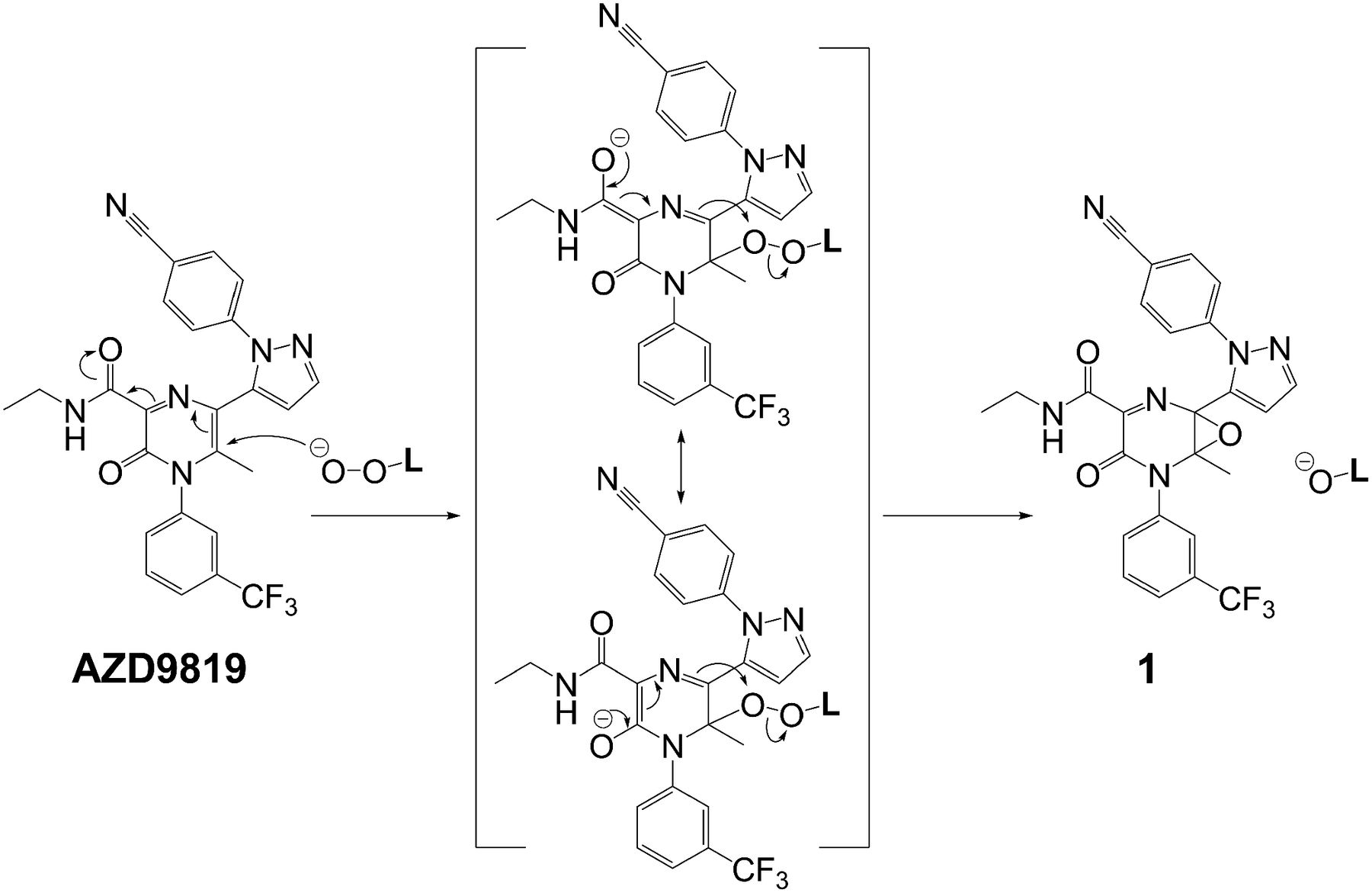
Scheme 1 shows the deduced structure for ROP and the proposed oxidative rearrangement mechanism for its formation by putting all the information together. The cause of the initial oxidation and other details of the mechanism will be discussed later. A key part of the structural elucidation process was the use of predicted 13C chemical shifts by ACD/Labs software (Toronto, ON, Canada), and a comparison is shown in Table 1 illustrating the change in the chemical shift of carbon atoms that map from AZD9819 to ROP (Scheme 1) and how well they match predicted values. HMBC NMR correlation data were also useful. For example, Fig. 2 shows a portion of the HMBC spectrum of the ROP synthetic standard. The correlations observed, specifically between H33 and the carbonyl C34 of the acetyl group, prove the position of the acetyl group in ROP and led us to propose the transfer of this group in the mechanism (4 to 5 in Scheme 1). We were subsequently able to obtain an X-ray structure from a single crystal of the synthetic standard to further corroborate the structural identification (Fig. 3).

Proposed mechanism for the lipid peroxide–dependent oxidation of the pyrazinone-carboxamide core of AZD9819 and subsequent rearrangement.
Comparison of carbon NMR chemical shifts of selected equivalent carbons as numbered in Scheme 1 in AZD9819 and ROP comparing with predicted shifts (ACD/Labs carbon NMR predictor 2012 build 60488)
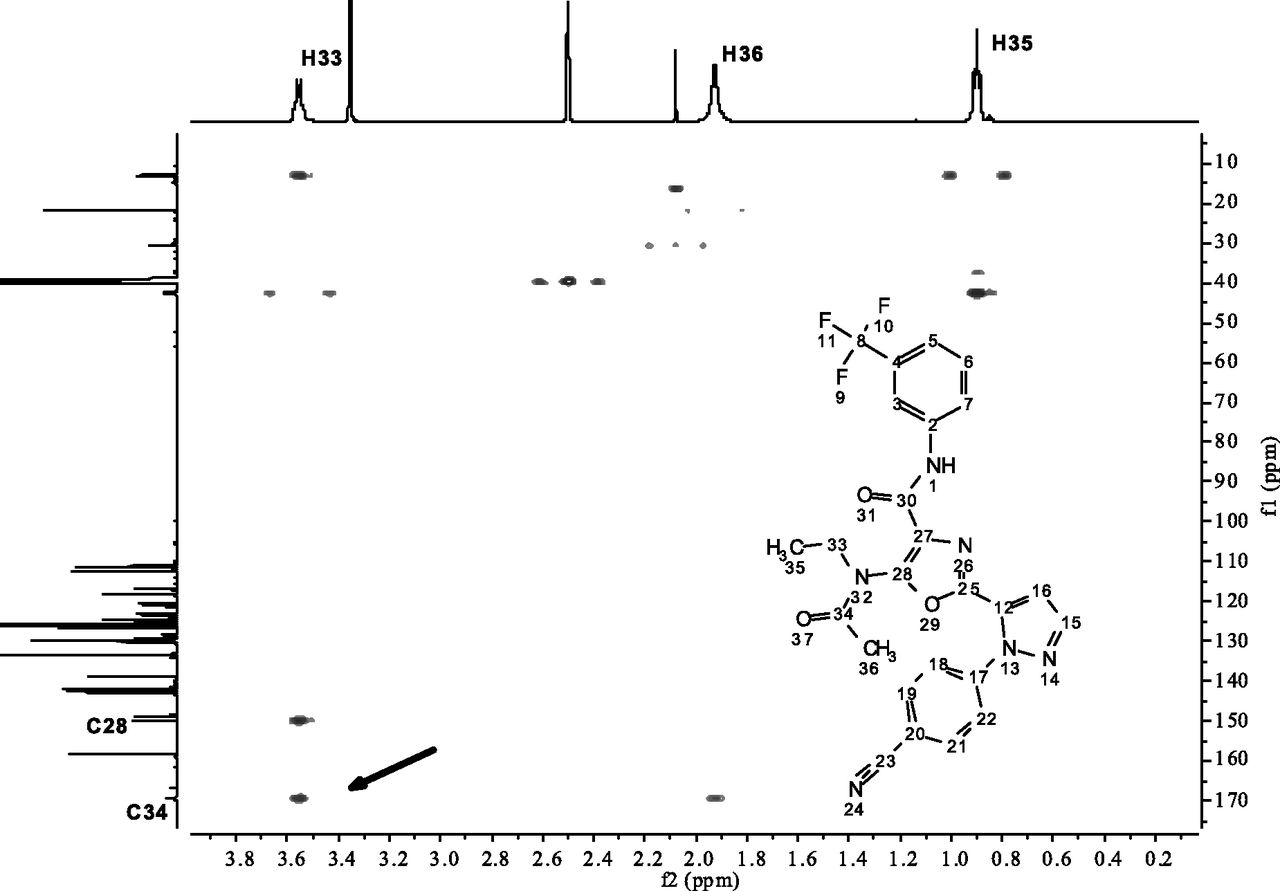
HMBC two-dimensional NMR of the ROP synthetic standard. The highlighted correlation suggests the migration of the acetyl group from 4 to 5 in Scheme 1.

X-ray crystallography molecular structure of ROP synthetic standard (50% probability ellipsoids). One of the molecules in the asymmetric unit is shown (Z′ = 2).
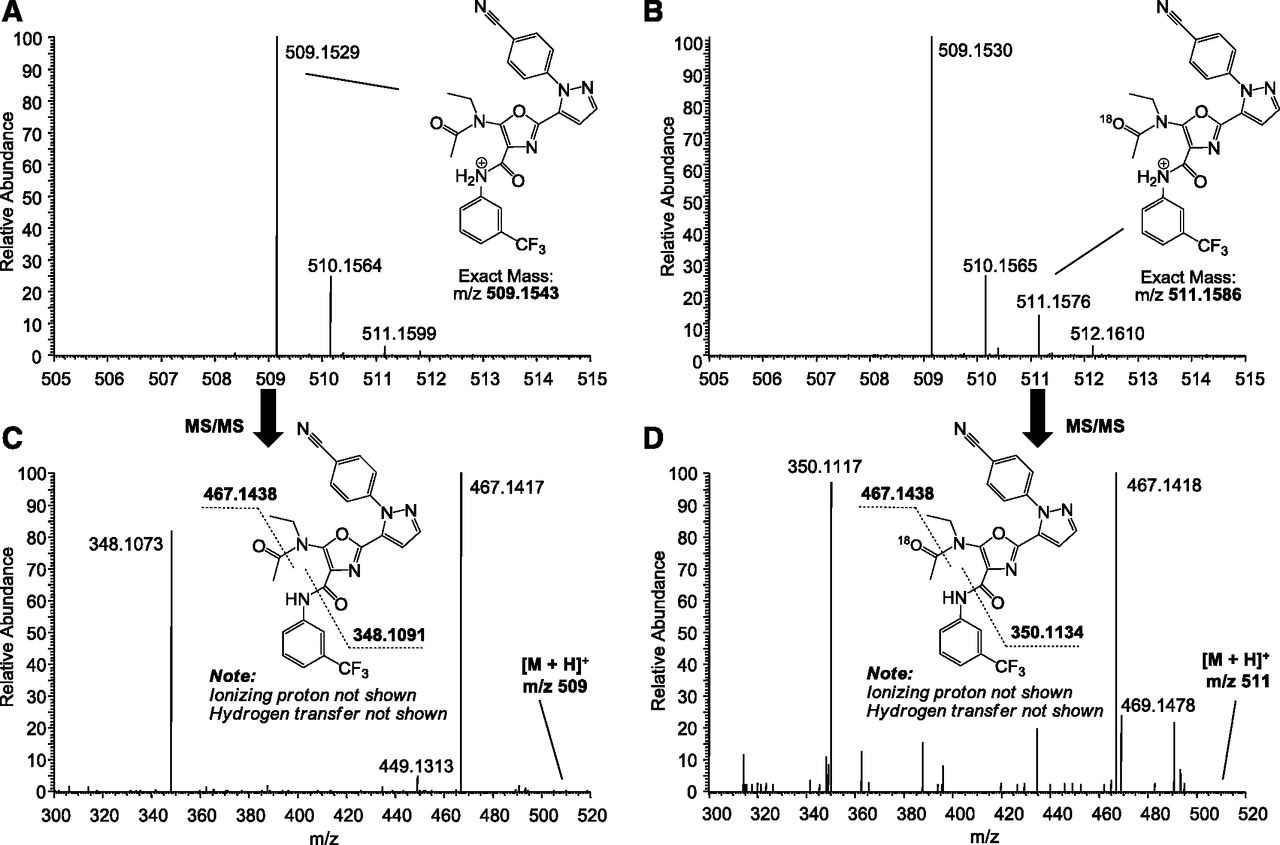
Figure 4 illustrates that an 18O atom from molecular 18O2 is incorporated to the location predicted by the proposed oxidative rearrangement mechanism. In the LC-MS spectrum of rat plasma–generated ROP (Fig. 4A), the naturally occurring isotopic peak at m/z 511 (i.e., 2 Da higher than the monoisotopic peak at m/z 509) has a low relative abundance of less than 5%. When 18O2 was added to the rat plasma incubation of AZD9819, 18O-incorporated [M+H]+ ions at m/z 511 were observed by a clear increase in the relative abundance of the m/z 511 peak (Fig. 4, B vs. A). MS/MS fragmentation of 18O-[M+H]+ ions (m/z 511) and the monoisotopic [M+H]+ ions (m/z 509) are compared in Fig. 4, D and C. Two major fragment ions occur in both spectra. The fragment ion formed by the cleavage of the acetyl amide bond remains unchanged at m/z 467.14 after the incorporation of an 18O atom, whereas the fragment ion formed by the neutral loss of a 3-(trifluoromethyl)aniline shifts by 2 Da from m/z 348.11 to 350.11 (Fig. 4, D vs. C). This indicates that the incorporated 18O is indeed located at the acetyl carbonyl as expected by the proposed mechanism (Scheme 1). It is worth noting that naturally occurring isotopic ions at m/z 511 (Fig. 4A) consist of a mixture of one 13C + one 15N atom, two 13C or two 15N atoms, or one 18O atom with random distribution across the whole molecule. This would result in both 1- and 2-Da shifts to a higher m/z for both fragments (Supplemental Fig. 8).

LC-MS spectra of protonated molecular ion regions showing rat plasma–generated ROP (A) and the incorporation of an 18O atom from 18O2 added to the incubation (B). LC-MS/MS CID product ion spectra acquired for [M+H]+ ions at m/z 509 (C) and 18O-[M+H]+ ions at m/z 511 (D), indicating 18O incorporation at the location predicted by the proposed mechanism in Scheme 1.
ROP was not produced by cytochrome P450 in liver, as it was absent from incubations of AZD9819 in rat, dog, and human hepatocytes (data not shown). In searching for the cause of the oxidation of AZD9819 in plasma, a number of experiments applying different reagents or experimental conditions to the incubations in plasma were performed and are summarized in Table 2. The percentages in the table were estimated from LC-UV peak areas with consideration of the different UV response of ROP versus AZD9819 (with an absorbance ratio of approximately 1.5 at 268 nm). Additionally, it was assumed that deacetyl-ROP observed mainly in rat plasma has the same UV absorbance as ROP. The percentage values in Table 2 are intended for qualitative comparison and, along with the other experiments described here, helped guide us to the cause of the oxidation.
Qualitative comparison of the effect of different agents or experimental conditions on the formation of ROP from AZD9819
All incubations and controls are conducted at 37°C for 2 hours unless specified otherwise.
The formation of ROP was not a simple chemical degradation process, because ROP was not produced in any of the control incubations of AZD9819 in blank buffer solutions. It was also not a photo-oxidative process, because the use of an amber vial or wrapping a vial in aluminum foil gave the same yield of ROP as the control incubation in a clear vial under ambient light (Table 2).
We considered a few possible enzymatic causes. Peroxidases are known to catalyze epoxidation of C=C bonds by peroxides such as H2O2 (Ortiz de Montellano, 1992; Adam et al., 1999). Indeed, as stated earlier, we made a similar oxidation product of an analog compound using a commercial peroxidase. However, the oxidation of AZD9819 in plasma is not inhibited by sodium azide (NaN3; Table 2), a known inhibitor of peroxidases (Ortiz de Montellano, 1992; Pozdnyakova et al., 2013). We also excluded xanthine oxidase (XO). Sharma et al. (2011) have reported a XO-mediated oxidation of a quinoxaline derivative in rodent plasma involving incorporation of an 18O atom from H218O but not from molecular 18O2. However, the addition of the XO inhibitor allopurinol (Sharma et al., 2011) to plasma did not inhibit formation of ROP (Table 2). Moreover, ROP incorporated an 18O from molecular 18O2 (Fig. 4) but not from H218O (data not shown) as would be expected. Additionally, we ruled out the involvement of hemoglobin. Literature reports on hemoglobin-catalyzed epoxidation of styrenes (Ortiz de Montellano and Catalano, 1985; Tschirret-Guth and Ortiz de Montellano, 1996) and retinoic acid (Iwahashi et al., 1985) offered a possible cause if hemolysis had occurred and released hemoglobin into the plasma. In both cases, the reaction was effectively inhibited by cyanide ions (Iwahashi et al., 1985; Ortiz de Montellano and Catalano, 1985). However, in our case, KCN enhanced rather than inhibited ROP formation (Table 2). Furthermore, incubation of 15 µM AZD9819 with 100 µM human hemoglobin did not produce any ROP, and the incubation of AZD9819 in 100 µM hemoglobin in the presence of 500 µM H2O2 did not catalyze the reaction (data not shown).
The formation of ROP from AZD9819 was most likely an ex vivo and in vitro phenomenon. During preclinical and clinical development, metabolite identification was conducted in rat/dog plasma, urine, feces, and bile samples collected after an i.v. or oral dose of [14C]AZD9819 and in human plasma and urine samples collected following inhaled doses of AZD9819. Among all in vivo samples, we did not observe any ROP. Furthermore, complete recovery of total radioactivity in preclinical mass balance studies after a dose of [14C]AZD9819 (virtually 100% in rat, >93% in dog) suggested it was unlikely that ROP or its intermediates were formed but then bound to macromolecules in vivo.
The effect of temperature on ROP formation was revealing (Table 2). The drop in yield from 37°C to room temperature might not be as great as would be expected for an enzymatic reaction, and ROP formation was not completely eliminated, even on ice. Inactivation of rat plasma at 56°C for 1 hour did not prevent the ROP formation from AZD9819 (Table 2) but abolished formation of deacetyl-ROP from ROP synthetic standard, suggesting deacetyl-ROP is an enzymatic product of ROP (Supplemental Fig. 9). In contrast, denaturing the plasma with MeCN did eliminate ROP formation (Table 2). Collectively, these observations suggested the likely involvement of heat-resistant proteins. However, we did not realize that these could be lipoproteins until lipid peroxides emerged to be the probable cause from subsequent experiments.
ROP did not appear to be produced by H2O2 oxidation in plasma, because the incubation of 15 µM AZD9819 with up to 500 µM H2O2 in pH 7.4 buffer only produced a trace amount of ROP (less than 1% of parent; data not shown). Moreover, pretreatment of rat plasma with bovine liver catalase at 1000 units/ml, which would decompose any H2O2 present in the plasma, did not abolish the oxidation of AZD9819 (Table 2). We also ruled out the formation of ROP by hydroxyl radical or hydroperoxyl radical from H2O2, because ROP was not formed by the Fenton reaction of AZD9819 with 100 µM H2O2 and 100 µM ferrous ion (Fe2+) in a pH 4.5 buffer (within the optimal pH 3–6 range of the Fenton reaction) (Data not shown). Intriguingly, during these experiments, we made the fortuitous discovery that ROP could be produced from AZD9819, albeit alongside other oxidative products, using a very high concentration of H2O2 (90 mM) in pH 7.4 phosphate buffer (Supplemental Fig. 10). This hinted that, even if H2O2 was not involved in ROP formation in plasma, perhaps other peroxides might be.
Ascorbic acid (one form of vitamin C) was tested to determine whether a radical mechanism could be involved. Inhibition was modest at 1 mM, but significant at 50 mM (Table 2). We were unaware that ascorbic acid is “an outstanding antioxidant in blood plasma” against lipid peroxidation until, during the manuscript preparation, we learned this from two highly cited papers that will be discussed later (Frei et al., 1988, 1989).
In an effort to trap the epoxide presumed in our mechanism, GSH was added to rat plasma at 5 mM, resulting in a clear decrease in ROP (Table 2). Importantly, GSH conjugates were not found, but the disappearance of parent was also attenuated (data not shown). This suggests that GSH probably prevents the reaction at the first epoxidation stage. Additional experiments with 50 mM GSH in plasma showed even greater inhibition on both ROP formation (Table 2) and AZD9819 depletion (data not shown). GSH is an important antioxidant in biologic systems, preventing damage by reactive oxygen species such as free radicals and peroxides, in addition to being a strong nucleophile that reacts well with soft electrophiles. GSH-dependent selenoperoxidases catalyze two-electron reduction of lipid peroxides (LOOH) to inert lipid alcohols (LOH), whereas two reduced GSH molecules become one oxidized GSSG (Girotti, 1998).
Collectively, these observations led us to consider lipid peroxides as the probable cause for the oxidation of AZD9819 in plasma. Moreover, a lipid peroxide–mediated oxidation would incorporate molecular oxygen during the lipid peroxidation chain propagation (Frei et al., 1989; Girotti, 1998) and would be consistent with our observation on the involvement of 18O2 (Fig. 4).
Finally, we succeeded in generating ROP using human LDL (1 mg/ml) in pH 7.4 buffer (Fig. 5B). The level of ROP formation was diminished in the presence of 50 mM GSH or ascorbic acid (Fig. 5, C and D). Percentages of ROP formation derived from LC-UV peak areas and UV signal response ratio are compared in Fig. 5F.

LC-MS accurate-mass extracted-ion chromatograms following a 2-hour incubation of 15 µM AZD9819 at 37°C in phosphate buffer pH 7.4 (A), 1 mg/ml human LDL in phosphate buffer pH 7.4 (B), B + 50 mM GSH (C), B + 50 mM ascorbic acid (D), and 1 mg/ml human LDL prepared in glycine buffer pH 10.5 (E). Also displayed are yields of ROP with different incubations (F), according to LC-UV peak areas and relative UV signal response of two compounds. Error bars are plotted in the standard deviation of triplicate incubations.
Discussion
The inhibitive effects of ascorbic acid and GSH on AZD9819 oxidation in both the LDL and plasma incubation provided evidence supporting the involvement of lipid peroxides in oxidation. Frei at al. (1988, 1989) have previously stated that ascorbate is the only plasma antioxidant that can completely protect plasma lipids against detectable peroxidative damage induced by aqueous peroxyl radicals. Additionally, they have demonstrated that protein thiol groups are second in line for antioxidant defense, whereas naturally occurring GSH in isolated plasma is present only at a very low concentration (<0.5 µM) (Frei et al., 1988).
Lipid peroxide–mediated oxidation explains why ROP was only formed ex vivo and in vitro, since a number of scavengers of lipid peroxyl radicals and lipid peroxides maintain a very low level of lipid peroxides in a living biologic system (Frei et al., 1988, 1989; Girotti, 1998). However, those scavengers can be depleted ex vivo, allowing the buildup of lipid peroxides by chain propagation. Lipid peroxide–mediated oxidation also rationalizes why protein precipitation by acetonitrile abolishes the ROP formation in acetonitrile-denatured plasma (Table 2). Frei et al. (1988) reported that, after the depletion of ascorbate in plasma, peroxidation occurred first to lipids transported in lipoproteins, whereas nonesterified fatty acids were still protected from oxidation by binding to plasma albumin. In the LDL incubation without plasma albumin, acetonitrile containing 1% formic was used as a presumably better quench solution. Differing levels of pre-existing lipid peroxides would rationalize our observation of variations in ROP formation in different plasma samples of the same species, e.g., number of free/thaw cycles, length of storage time at –18°C (data not shown). Future work could include quantitative analysis of lipid peroxides, e.g., to study the relationship between the level of lipid peroxides and the ROP formation, and to investigate factors affecting the level of peroxides in plasma samples. Finally, ROP formation via epoxidation of AZD9819 in plasma is not contradictory to the chemical preparation of ROP using sodium perborate, since sodium perborate functions mainly as a convenient source of mildly alkaline hydrogen peroxide and is a known agent for epoxidation of a wide range of alkenes while also being a known agent of heteroatom oxidation (Mckillop and Sanderson, 2000; Bortolini et al., 2009).
The level of ROP generated by LDL was enhanced severalfold when incubated at pH 10.5 relative to that at pH 7.4 (Fig. 5, E vs. B; Fig. 5F). Furthermore, ROP was absent from the LDL incubation in pH 4.5 buffer (data not shown), consistent with the empirical use of acidification to prevent ex vivo oxidation of AZD9819 in plasma samples. No ROP was formed when AZD9819 was incubated in the individual buffer solutions as a control. The calculated pKa (ACD/Labs pKa Database 2012) for an allylic hydroperoxide of a polyunsaturated fatty acid is approximately 11.3, and thus the pH dependence suggests that a nucleophilic epoxidation of the C=C bond in the pyrazinone ring of AZD9819 (Scheme 2) is highly plausible. It is known that nucleophilic epoxidation by hydroperoxide can occur at alkenes that are conjugated with electron-withdrawing groups (Apeloig et al., 1983).

Postulated mechanism for nucleophilic epoxidation of AZD9819 by lipid hydroperoxides.
Interestingly, the addition of 50 mM KCN to the LDL incubation at pH 7.4 approximately doubled the formation of ROP [data not shown, but similar to that observed in rat plasma (Table 2)]. A cyanide conjugate with [M+H]+ at m/z 494.1547 was detected in the presence of 50 mM KCN in both the LDL and rat plasma incubations of AZD9819. The measured accurate mass in LC-MS indicated that the cyanide conjugate had exactly the same elemental composition as that of a HCN adduct of deacetyl-ROP. However, the LC-MS/MS fragmentation does not support the same skeleton structure as deacetyl-ROP (spectra not shown). We assume that CN− adds to the imine of intermediate 1, ultimately leading to the loss of HCN and formation of oxazole as a mechanism analogous to that when HO− adds to the intermediate in Scheme 1. Additionally, an unknown rearrangement mechanism after the CN− addition might have led the formation of the rearranged cyanide conjugate detected at m/z 494.1547.
The hydration of the imine in our proposed mechanism is necessary to change the rigid sp2 carbon to a more flexible sp3 center, thereby allowing intramolecular nucleophilic attack of the carboxamide oxygen on the epoxide (1 to 2, Scheme 1). We also note that an oxygen anion is naturally formed for the proposed ring closure (2, Scheme 1). Previously reported mechanisms for P450-mediated oxidation of a pyrazinone ring are related in that they involve either hydrolysis or GSH addition to an epoxide (Singh et al., 2003; Subramanian et al., 2003; Zhuo et al., 2010), presumably catalyzed by epoxide hydrolases or glutathione S-transferases present in liver (Parkinson, 2001). These led to the pyrazinone ring opening by the breakage of one or both C–N bonds, resulting in multiple products. Our proposed mechanism, involving a nonenzymatic epoxidation of AZD9819 followed by imine hydration and subsequent elimination of a water molecule (Scheme 1), leads to a single rearranged product and is therefore consistent with the radiochromatographic data (Fig. 1).
The final rearrangement step proposed for intermediates 4 to 5 in Scheme 1 can be viewed as the intramolecular transfer of the negative charge from an apparently high energy anion to a reasonably low energy anion and therefore the formation of a more stable species. The negative charge in 5 can be delocalized by adjacent carbonyl and phenyl groups, whereas the charge in 4 would be delocalized only by the oxazole. Interestingly, intermediate 4 was briefly assumed to be the likely structure of ROP before the characterization of ROP by HMBC two-dimensional NMR, as shown in Fig. 2. At that time, we had to postulate an acetyl migration rearrangement in gas-phase MS/MS fragmentation to interpret a major fragment ion by the neutral loss of a 3-(trifluoromethyl)aniline (m/z 348.11). Afterward, the HMBC data made us quickly realize that the rearrangement of acetyl migration had already happened in the formation of ROP.
The deacetyl-ROP that occurred mainly in rat plasma has been shown to be an enzymatic hydrolysis product of ROP (Supplemental Fig. 9A). It is known that hydrolysis of carboxamide in plasma can be mediated by carboxylesterases (Parkinson, 2001). The species difference in deacetyl-ROP between rat and dog/human (Fig. 1, A vs. B and C) can be readily rationalized by the differences in carboxylesterase expression and the hydrolase activity in plasma (Bahar et al., 2012).
To the best of our knowledge, this is the first reported case of epoxidation of a pharmacological agent by lipid peroxides in plasma. We acknowledge that epoxidation by lipid peroxides is neither a new concept nor a new phenomenon. It is known that lipid epoxides can be generated as secondary oxidation products of lipid peroxidation (Scheick and Spiteller, 1993; Chen and Chung, 1996). Scheick and Spiteller (1993) reported the epoxidation at enol ethers R1-O-CH=CH-R2 of a number of plasmalogens by reaction with a lipid hydroperoxide that was produced from linoleic acid by tomato homogenate. Incorporation of 18O from molecular 18O2 occurred in the final trapped products derived from the epoxides of a number of plasmalogens. Chen and Chung (1996) reported that two linoleic acid–derived hydroperoxides were capable of epoxidizing a lipid peroxidation product, 4-hydroxy-2-nonenal, to a mutagenic epoxy aldehyde. Lipid hydroperoxides produced during the peroxidase cycle are also known to be capable of epoxidizing polycyclic aromatic hydrocarbon as an alternative pathway to P450-mediated activation of polycyclic aromatic hydrocarbon to mutagenic epoxides (Reed, 1987; Parkinson, 2001). Additionally, non–P450-catalyzed epoxidation of the terminal furan of aflatoxin B1 (a hepatocarcinogen and hepatotoxin produced by fungal metabolism) was shown to be dependent on the preoxidation of arachidonic acid by prostaglandin H synthase (Battista and Marnentt, 1985; Reed, 1987; Parkinson, 2001). The feasibility of epoxidation by lipid hydroperoxides has therefore been demonstrated in the literature.
It seems unlikely that AZD9819 and related compounds are the only xenobiotic analytes susceptible to lipid peroxide–dependent oxidation. In the present study, we have demonstrated two practical, effective means of preventing the oxidation: lowing the pH to stop the nucleophilic reaction of epoxidation by lipid hydroperoxides, or adding the antioxidant ascorbic acid to inhibit the radical reaction of lipid peroxidation. We hope that knowledge of this new possible cause of analyte instability in plasma samples and the means of prevention will prove useful for other scientists in biotransformation and bioanalysis.
Acknowledgments
The authors thank Dr. Neal Castagnoli of Virginia Tech (Blacksburg, VA) and Dr. Richard Thompson, Dr. Matthew Perry, and Dr. Per-Ola Norrby of AstraZeneca at Mölndal, Sweden for helpful discussions. The authors thank their chemistry colleagues Peter R. Hansen for original synthesis of AZD9819, Grigorios Nikitidis for the scaled-up synthesis, and Tobias Persson for the synthesis of [14C]AZD9819 at AstraZeneca at Lund in Sweden. The authors thank Ann Jönsson and Kristina Olsmar for the bioanalytical stability testing, and Helena Nilsson and Dr. Per Brunmark for LC-MS work on in vitro metabolites at AstraZeneca (Lund, Sweden). The authors also thank a number of biotransformation experts from biotech and other pharmaceutical companies for stimulating discussions regarding partial results and a partial mechanism presented at two conferences.
Authorship Contributions
Participated in research design: Gu, Lewis, Wells, Hosagrahara, Johnsson, Hallström.
Conducted experiments: Gu, Lewis, Wells, Svensson, Johnsson, Hallström.
Contributed new reagents or analytic tools: Wells.
Performed data analysis: Gu, Lewis, Svensson, Johnsson, Hallström.
Wrote or contributed to the writing of the manuscript: Gu, Lewis, Wells, Svensson, Hosagrahara, Johnsson, Hallström.
Footnotes
- Received April 26, 2015.
- Accepted July 22, 2015.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AZD9819
- 6-(1-(4-cyanophenyl)-1H-pyrazol-5-yl)-N-ethyl-5-methyl-3-oxo-4-(3-(trifluoromethyl)phenyl)-3,4-dihydropyrazine-2-carboxamide
- CID
- collision-induced dissociation
- GSH
- l-glutathione
- HCD
- high-energy collision-induced dissociation
- HMBC
- heteronuclear multiple bond correlation
- KCN
- potassium cyanide
- LC
- liquid chromatography
- LDL
- low-density lipoprotein
- MeCN
- acetonitrile
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- P450
- cytochrome P450
- ROP
- rearranged oxidation product
- XO
- xanthine oxidase
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}