


Abstract
Thrombin is a serine protease that plays a key role in the blood coagulation cascade. Compound I [2-[6-chloro-3-[(2,2-difluoro-2-pyridin-2-ylethyl)amino]-2-oxopyrazin-1(2H)-yl]-N-[(3-fluoropyridin-2-yl)methyl]acetamide] is a potent, selective, and orally bioavailable thrombin inhibitor that is being studied as a possible anticoagulant. Biotransformation studies in rats revealed that 84% of an i.v. dose of I was excreted in the form of two metabolites. Both metabolites were formed by metabolic activation of the pyrazinone ring in I and subsequent rearrangement leading to two novel dihydro-imidazole and imidazolidine derivatives. The structures of these metabolites and their mechanism of formation were elucidated by additional use of two 13C single labels in the pyrazinone ring of I in combination with mass spectrometry and NMR techniques. The metabolite structures described here illustrate the rich metabolic chemistry of the amino-pyrazinone heterocycle.
Thrombin is a serine protease that plays multifarious roles in the complex blood coagulation cascade (Goldsack et al., 1998; Mann et al., 2003). Existing anticoagulation therapy (Majerus and Tollefsen, 2001; Weitz and Hirsh, 2001) primarily consists of low-molecular-weight heparin and warfarin. Both therapeutic agents suffer from significant limitations: low-molecular-weight heparin requires either continuous intravenous infusion or subcutaneous injection, and warfarin has a delayed onset of action, narrow therapeutic index, and a high potential for drug-drug interactions. Hence, a small-molecule thrombin inhibitor represents an alternative target in the search for orally administered anticoagulants that could offer a safer alternative to existing therapies for the treatment and prevention of thromboembolic disorders (Vacca, 2000; Burgey et al., 2003b).
Compound I [2-[6-chloro-3-[(2,2-difluoro-2-pyridin-2-ylethyl)-amino]-2-oxopyrazin-1(2H)-yl]-N-[(3-fluoropyridin-2-yl)methyl]acetamide] is a potent, selective and orally bioavailable thrombin inhibitor from the amino-pyrazinone acetamide series (Burgey et al., 2003a,b). In this study, we provide details of the in vivo biotransformation of I in rats. The structures and mechanisms leading to novel dihydro-imidazole and imidazolidine derivatives have been delineated by MS1 and NMR analysis and the additional use of two singly labeled 13C analogs of I.
Materials and Methods
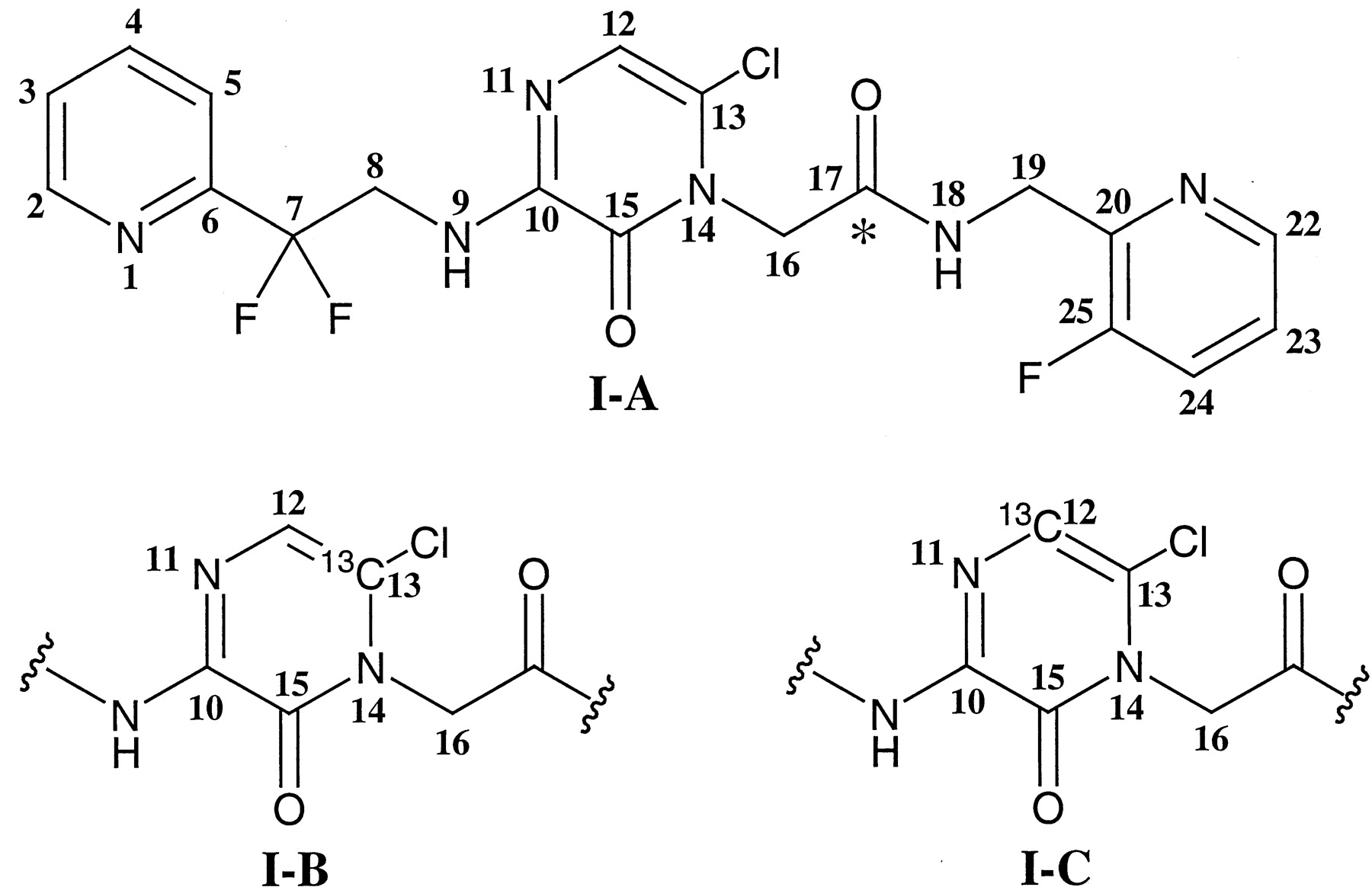
Chemicals. Compound I-A and its 14C analog, shown in Fig. 1, were synthesized at Merck Research Laboratories (West Point, PA) (Burgey et al., 2003). The 14C label was incorporated on the carbon 17 (see Fig. 1 for nomenclature) having a specific activity of 47.7 μCi/mg and a radiochemical purity of >98% as determined by HPLC. Compounds I-B and I-C are structurally identical to compound I-A except for a mono 13C label at carbon labeled 13 or 12 (Fig. 1), respectively, of the pyrazinone ring. Both I-B and I-C were prepared at Merck Research Laboratories (Labeled Compound Synthesis group, Rahway, NJ) and were determined to have a purity of >99% by NMR. CD3OD (99.96% D) was obtained from Isotec Inc. (Miamisburg, OH). All other chemicals were of analytical grade and were obtained from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific Co. (Pittsburgh, PA).

Structure of compoundI.
Position of the 14C label for the radiolabeled I is shown with an asterisk (I-A). Compound I with 13C label at carbon 13 (I-B) and carbon 12 (I-C) of the pyrazinone portion of I.
Animal Studies and Sample Collection. For metabolite profiling, [14C]-labeled I was administered i.v. at a pharmacologically relevant dose (Burgey et al., 2003b) of 5 mg/kg (10 mg/ml in DMSO) to bile duct-cannulated male Sprague-Dawley rats (n = 3). Specimens of bile and urine were collected over ice at different time points for 48 h postdosing to quantify the in vivo metabolites.
For structural characterization by NMR, M1 and M2 from I-A, I-B, and I-C were isolated either from rat bile or urine. For M1 and M2 from I-A, a 50 mg/kg i.v. dose of I was administered to bile duct-cannulated male Sprague-Dawley rats (n = 4). The dosing solution was prepared by mixing the 14C and 12C forms of I-A in an approximately 1:40 ratio as a 50 mg/ml solution in DMSO. Bile and urine were collected on ice at 0- to 4-h or 0- to 8-h and 8- to 24-h time points. The bile and urine from different animals were pooled and stored at -70°C until analyzed.
For structural characterization of M1 and M2 obtained from I-B or I-C, a 20 mg/kg i.v. dose was administered to bile duct-cannulated male Sprague-Dawley rats (n = 2). Dosing solutions were prepared by mixing [14C] I and I-B or I-C in an approximately 1:9 ratio as a 20 mg/ml solution in DMSO.
Metabolite Profiling. The HPLC system consisted of an autosampler (SYS-S200; PerkinElmer Instruments, Norwalk, CT) and a Rheos HPLC pump (Flux Instruments, Basel, Switzerland) attached to a C18 analytical column (Hypersil BDS 4.6 × 250 mm, 5 μm, ThermoHypersil, Cheshire, UK). The mobile phases consisted of 0.1% formic acid (solvent A) and acetonitrile (solvent B). A flow rate of 1 ml/min and the following solvent gradient were used: from 0 to 21 min 5 to 35% B, 21 to 25 min 35 to 80% B. The HPLC effluent was split 9:1 with the larger proportion diverted to a radiometric flow detector (500TR flow scintillation analyzer; PerkinElmer Life Sciences, Boston, MA) and the remainder delivered to a mass spectrometer (conditions given below) that allowed simultaneous quantification and characterization of radioactive components.
Mass Spectrometry. Mass spectral analysis was performed on an ion-trap mass spectrometer (LCQ Classic; Thermo Finnigan, San Jose, CA) equipped with an electrospray source operated in the positive ion mode. The electrospray-ionizing voltage was 4.5 kV, the heated capillary temperature was approximately 200°C, and the collision energies were optimized for each experiment.
Mass spectra on isolated samples (described below) of M1 and M2 from I-B and I-C also were recorded on a quadrupole time-of-flight mass spectrometer (Q-TOF2, Micromass UK Ltd., Manchester, UK) equipped with a Z-spray electrospray ionization source operated in the positive ion mode. The mass spectrometer was tuned to a resolution of 8,000 and calibrated over the m/z range 100 to 1,000 using a 0.02% solution of phosphoric acid (in 10% methanol). An aliquot of each of the isolates of M1 and M2 (from I-B and I-C) was introduced using a 50-mm C18 column (Jupiter, 2 × 50 mm, 5 μm; Phenomenex, Torrance, CA), running a linear gradient from 90% A to 10% A in 5 min at a flow rate of 200 μl/min. The accurate mass measurement was performed with the LockSpray technique using the tuning solution as a real-time calibrant.
Metabolite Isolation for NMR Analysis. Isolations were carried out using two successive chromatographic separations on a quaternary pump HPLC system (HP 1100; Agilent Technologies, Palo Alto, CA). Samples were maintained at 5°C in the autosampler until analyzed. The mobile phase consisted of 25 mM aqueous potassium phosphate buffer at pH 7.4 (solvent A) and acetonitrile (solvent B) and was delivered at a constant flow rate of 4 ml/min. The first purification was performed on a semipreparative reverse-phase column (Hypersil BDS 10 × 250 mm, 5 μm) under the following solvent gradient conditions: 0 to 0.5 min 8% B, 0.5 to 20 min 8 to 28% B, 20.1 to 21.0 min 90% B, 21.1 to 25.0 min 8% B. The HPLC effluent was split in a 1:4 ratio with the larger fraction diverted to a fraction collector (set to collect 0.2-min fractions from 8 to 12 min after sample injection) precooled with ice. The smaller flow was diverted to a UV detector (λ = 260 nm) in line with a flow scintillation analyzer. The 0- to 4-h urine or bile samples were thawed, freeze-dried, reconstituted in 5 ml of buffer (solvent A), and injected in 500-μl aliquots. The fractions containing metabolites were pooled, freeze-dried, and reconstituted in 2.2 ml of buffer for another round of purification. The second HPLC purification was carried out with the same setup under isocratic conditions, 83% A/17% B at 1 ml/min flow rate, using an analytical C8 column (Prodigy 4.6 × 250 mm, 5 μm; Phenomenex). The isolates from first HPLC step were injected in 200-μl aliquots and the fraction collector was set to collect 0.33-min fractions from 7 to 15 min after sample injection. Fractions containing metabolite(s) then were pooled and freeze-dried for NMR analysis.
NMR Analysis. Isolated M1 (ca. 300 μg) and M2 (ca. 500 μg) from I-A were dissolved in 160 μl of dry-ice chilled CD3OD and transferred to a 3-mm NMR tube. All NMR experiments were performed at ≤ -20°C on a 500- or 600-MHz Inova spectrometer (Varian, Inc., Palo Alto, CA) equipped with a 3-mm inverse detection probe or a 3-mm broad band direct-observe probe. 1H, proton-decoupled 13C, 1H-13C gHMQC, and 1H-13C gHMBC data sets were collected.
Purified M1 and M2 from I-B and I-C, ca. 50 to 100 μg, also were dissolved in 160 μl of chilled CD3OD and transferred to a 3-mm tube. gHMBC data were collected on a 600 MHz Inova spectrometer equipped with a 5-mm inverse detection probe. The proton-decoupled 13C NMR data were collected either on a 400- (with a 5-mm broad band direct-observe probe) or 500-MHz spectrometer (with a 3-mm broad band direct-observe probe).
M3 Synthesis. Triethylamine (77 ml, 550 mmol) was added to a suspension of glycine HCl salt (38.4 g, 276 mmol) in methylene chloride (300 ml), and the mixture was stirred at room temperature for 1 h. Ethyl chlorooxoacetate (30 ml, 275 mmol) was then introduced, and the resulting mixture was stirred at room temperature for 18 h. Water (25 ml) was added and layers were separated. Organic layer was dried over MgSO4 (5 g), filtered, and concentrated to afford ethyl N-(ethylcarboxymethyl)oxamate (42.2 g, 75%).
A solution of ethyl N-(ethylcarboxymethyl)oxamate (203 mg, 1 mmol), 2-(2-amino-1,1-difluoroethyl)pyridine (316 mg, 1 mmol) (Burgey et al., 2003b), triethylamine (0.5 ml), and 2-propanol (3 ml) was stirred at room temperature for 18 h then purified by flash chromatography on silica gel (30% ethyl acetate in hexane) to give a white solid (110 mg, 35%), which was dissolved in methanol (5 ml) and 1 M NaOH solution (5 ml). After stirring at room temperature for 2 h, solvent was removed under vacuum to give a light yellow solid. Dimethylformamide (2 ml) was added and a solution was formed. To this solution, 1,2-dichloroethane (191 mg, 1 mmol), 1-hydroxybenzotriazole hydrate (159 mg, 1 mmol), triethylamine (1 ml), and 2-(aminomethyl)-3-fluoropyridine (50 mg, 0.25 mmol) (Burgey et al., 2003b) were added, and the mixture was stirred at room temperature for 16 h. Reverse-phase HPLC purification (Zorbax RX C18, MeCN:water:trifluoroacetic acid = 12:88:0.1) yielded M3 (66 mg, 66%) as a white solid.
Results
Excretion Profiles in Urine and Bile. Metabolites M1 and M2 in rat urine and bile together accounted for 84% of the dose of I. Metabolite M3, excreted in bile, constituted 2% of the dose, whereas excretion of unchanged parent drug accounted for 1% of the dose. The remaining components, about 10% of the dose, observed in rat bile consisted of glutathione and cystein-yl-glycine adducts of compound I. The excretion profile of I in rat urine and bile is summarized in Table 1.
Major metabolites of I in urine and bile of rats after i.v. administration of 14C-labeled I at a 5 mg/kg dose (n = 3).
The listed retention times are from the HPLC method used for metabolite profiling (detailed under Materials and Methods)
Metabolite Identification
M1. A full-scan mass spectrum of M1 (summarized in Table 2), showed a protonated molecule, [M+H]+, at m/z 467 and an isotope pattern indicating loss of the chlorine atom. The product ion spectrum of m/z 467 showed an abundant peak at m/z 423, 44 Da less than the parent ion, consistent with the neutral loss of CO2.
Summary of MS data for metabolites M1 and M2 derived from I-A to I-C
The 1H 1-D spectra of the free base forms of I-A and M1 are shown in Fig. 2, A and B, respectively, and the corresponding NMR data are listed in Table 3. Comparison of 1H NMR spectra of I-A and M1 revealed that the pyridine and fluoro-pyridine moieties were intact in M1. The methylene (H19) appearing as a singlet at 4.63 ppm in I-A also remained essentially unchanged in M1 (Fig. 2B). The geminal protons of the glycine methylene (H16) appearing as a singlet at 4.95 ppm in I-A appeared as nonequivalent doublets at 3.83 and 4.25 ppm with an AB scalar coupling pattern (2JHH = 17.1 Hz) in M1. Similarly, the methylene (H8) next to the geminal difluoride resonated at 4.29 ppm as a triplet (3JHF = 13.9 Hz) in I-A and as nonequivalent multiplets at 4.10 and 4.25 ppm in M1. The vinylic proton (H12) of the pyrazinone ring appearing at 6.84 ppm in I-A was absent in the M11H spectrum. The M1 proton data, therefore, point to alteration of the pyrazinone ring and the presence of two nonequivalent methylenes with the stated J-couplings to indicate formation of a chiral element in the metabolite.

1H spectrum of I-A (A), M1 from I-A (B), and M2 from I-A (C).
Identifiable 1H and 13C chemical shifts for I and its metabolites.
All δ values are referenced to the CD2HOD resonances at δH 3.33 ppm and at δC 47.85 ppm. NMR sample temperatures for I, M1 and M2 were 0°, −30°, and −20°C, respectively, and 25°C for the M3 synthetic standard. Unless noted otherwise, the listed coupling constants are from JHH scalar coupling interactions and all carbon peaks are singlets. Splitting pattern notation: s, singlet; d, doublet; t, triplet; m, multiplet. a From gHMQC data; signal to noise for the C8 and C24 resonances in the 13C 1-D spectrum were inadequate to accurately measure JCF couplings.
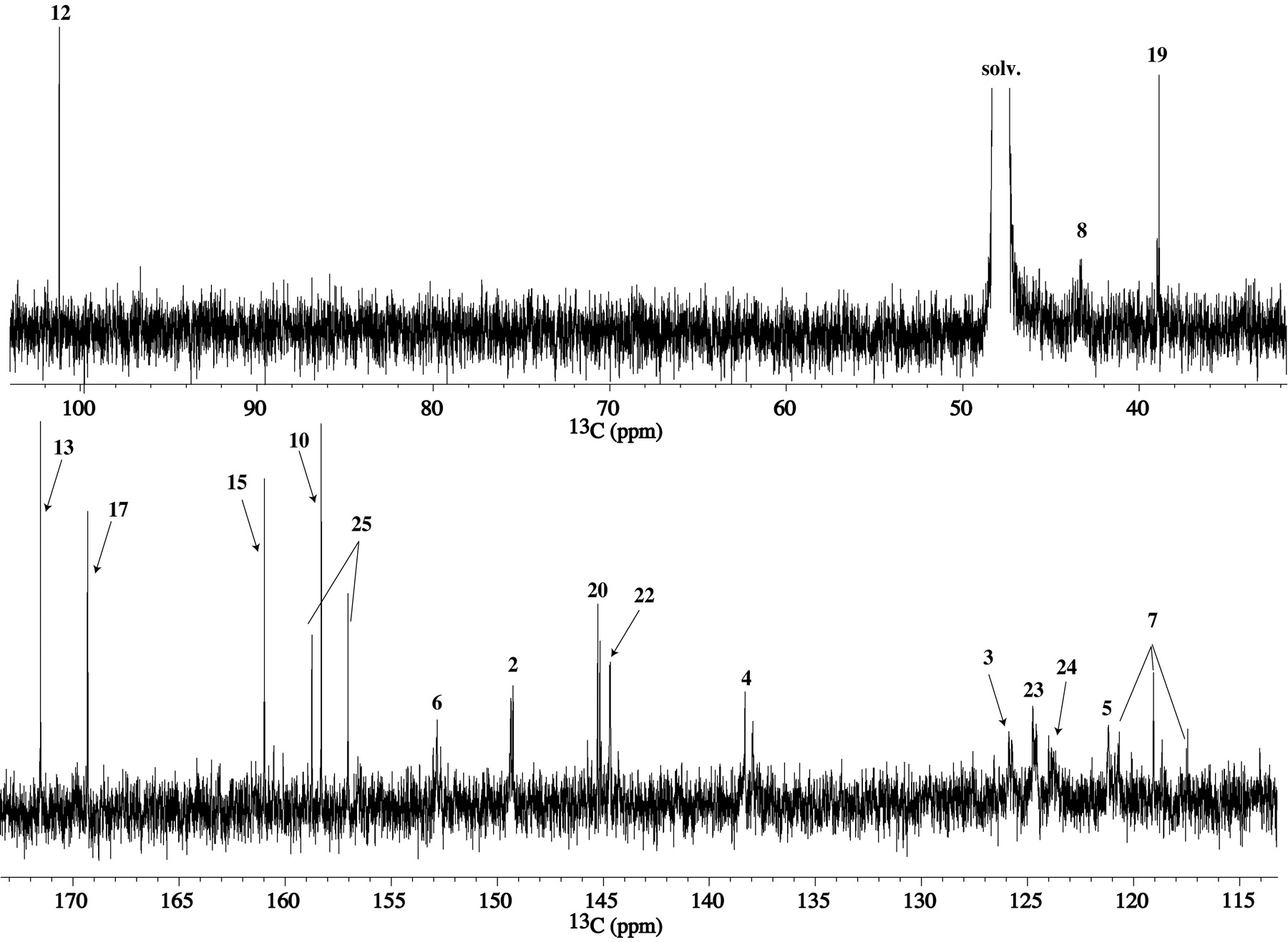
The proton-decoupled 13C 1-D and gHMQC spectra of M1 from I-A are reproduced in Figs. 3 and 4A, respectively. All the resonances/correlations expected for the pyridine (C2-C5), geminal difluoride (C7), methylene (C19), and fluoro-pyridine (C20-C25) moieties were observed in the 13C 1-D and/or gHMQC spectra. The carbon resonances for methylenes 8 and 16 shifted from 44.4 and 47.3 ppm in I-A to 45.5 and 43.2 ppm, respectively, in M1. The carbon resonances for C8 and C16 were below the noise level in the 13C 1-D, most likely due to their long longitudinal relaxation times. The pyrazinone vinylic (C12) and chloro (C13) carbons that appeared at 119.9 and 118.6 ppm, respectively, in I-A were not present in the same region of the 13C spectrum in M1.

Proton-decoupled 13C spectrum ofM1fromI-A.

gHMQC (A) and selected region of the gHMBC (B) spectrum of M1 fromI-A.
From the M113C 1-D and gHMQC data, there were five nonprotonated and nonfluorinated carbon peaks at δC 101.2, 158.3, 161.0, 169.3, and 171.5 ppm that remained to be assigned. A selected region of the gHMBC spectrum of M1 from I-A is shown in Fig. 4B. HMBC correlations are seen from the methylene 8 protons (δH 4.10, 4.25 ppm) to δC 158.3 ppm and to the CF2 carbon (C7) at 119.1 ppm. The methylene protons (δH 3.83, 4.25 ppm) at position 16 show HMBC correlations to the carbon peaks at 101.2, 161.0, and 169.3 ppm. An HMBC correlation is also seen to δC 169.3 ppm from H19 (4.61 ppm), and hence the 169.3 ppm peak is assigned to the carbonyl carbon 17. No HMBC correlations are seen to δC 171.5 ppm. The peak at δC 101.2 ppm is suggestive of a quaternary carbon with attached heteroatoms and the peak at 171.5 ppm would be consistent with a -COOH carbon whose presence also was indicated by the MS data. The MS and NMR data for M1 derived from 13C-labeled I (I-B and I-C) helped resolve the connectivity and assignments of the as-yet unassigned peaks in the carbon spectra.
A full-scan, high-resolution mass spectrum of M1, derived from I-B, showed an [M+H]+ ion at m/z 468.1326, which corresponds to a molecular formula of 13CC18H18F3N6O5+ (a deviation of 0.2 ppm from the calculated mass) and indicates loss of the chlorine atom. The product ion spectrum of m/z 468.1326 ion showed an abundant peak at m/z 423.1397, 44.9929 Da less than that of the parent indicating the neutral loss of a molecule of 13CO2 (4 ppm deviation from the calculated molecular weight). The 1H 1-D spectrum of M1 from I-B (not shown) was identical to that reproduced in Fig. 2B. The 13C 1-D spectrum of M1 from I-B, displayed in Fig. 5A, shows that the 13C label originating at the chloro-carbon (C13) of the pyrazinone ring in I-B is observed at δC 171.5 ppm. The MS and 13C NMR data for M1 from I-B reaffirm the assignment of the peak at 171.5 ppm to a carboxylic acid carbon. The gHMBC spectrum of M1 from I-B (not shown) did not show any correlations to the 13C-labeled carbon at 171.5 ppm.

Proton-decoupled 13C spectrum ofM1fromI-B(A), and proton-decoupled 13C (B) and gHMBC (C) spectra ofM1fromI-C.
A full-scan, high-resolution mass spectrum of M1 derived from I-C showed an [M+H]+ ion at m/z 468.1332 that corresponds to a molecular formula of 13CC18H18F3N6O5+ (a deviation of 1.5 ppm from the calculated mass). The product ion spectrum of m/z 468.1332 shows an abundant peak at m/z 424.1445, 43.9887 Da less than that of the parent ion indicating the neutral loss of a CO2 (25 ppm deviation from the calculated mass). The 1H chemical shifts for M1 from I-C (spectrum not shown) were identical to those shown in Fig. 2B. The methylene protons at position 16 with δH 3.83 and 4.25 ppm show additional J couplings of 2.3 and 3.5 Hz, respectively, that are derived from the through-bond proximity of its geminal protons to the 13C label. The proton-decoupled 13C 1-D and the gHMBC spectra of M1 from I-C are displayed in Fig. 5, B and C. The 13C label from the vinylic carbon (C12) of the pyrazinone ring in I-C shows up at δC 101.2 ppm in M1. A strong HMBC correlation is observed from methylene protons 16 to the 13C labeled carbon at 101.2 ppm (Fig. 5C).
The M1 structure shown in Fig. 6 is inferred from the combined MS and NMR data for the M1 metabolite derived from I-A to I-C. The pyrazinone ring in I appears to undergo bioactivation and then rearranges to form a 2-hydroxy-5-oxo-2,5-dihydro-1H-imidazole-2-carboxylic acid moiety (Fig. 6). The quaternary carbon (C12) at δC 101.2 ppm represents the newly formed chiral center, which renders the geminal protons in methylenes 8 and 16 diastereotopic. The carboxylic acid at C13 (δC 171.5 ppm) derived from the chloro-carbon of pyrazinone in I is attached to the quaternary carbon. The δC values at 161.0-ppm and 158.3-ppm peaks are assigned to the amide carbonyl carbon 15 and the amidine carbon 10 of the dihydro-imidazole, respectively.

Structures of metabolitesM1, M2, andM3.
M2. A full scan MS of M2 (Table 2) showed features similar to those of M1. Thus, M2 exhibited a protonated molecule, [M+H]+, at m/z 467 and an isotope pattern indicating the absence of the chlorine atom. The product ion spectrum of m/z 467 showed a prominent peak at 423, a loss of 44 Da from the parent ion, consistent with the neutral loss of CO2.
The 1H 1-D spectrum of the M2 from I-A is shown in Fig. 2C, and the corresponding NMR data are listed in Table 3. The 1H spectral features were similar to those of M1 (Fig. 2B). All the resonances from the pyridyl moieties were present in the M21H spectrum. The vinylic proton in the pyrazinone ring (H12 at 6.84 ppm) of I-A (Fig. 2A) was absent in M2. The geminal protons from methylene 8 appear as AB multiplets at 3.28 and 3.42 ppm, and those from methylene 16 also resonate as AB doublets (2JHH = 16.7 Hz) at 3.65 and 3.98 ppm. The presence of two nonequivalent methylenes indicates the presence of a chiral element in the M2 metabolite, and together with the other spectral evidence, suggests modification of the pyrazinone ring.
The 13C 1-D and gHMQC spectra of M2 from I-A are displayed in Figs. 7 and 8A, respectively, and indicate that all the protonated carbons from the pyridine moieties (C2-C6, C20-C25), CF2 (C7), and methylene 19 are intact in M2. The carbon chemical shifts for methylenes 8 and 16 appear at δC 44.5 and 42.8 ppm, similar to that observed in M1. The vinylic carbon (δC 119.9 ppm) and chloro-carbon (δC 118.6 ppm) peaks of the pyrazinone ring in I-A are absent in the M213C spectrum.

Proton-decoupled 13C spectrum ofM2fromI-A.

gHMQC (A) and selected region of the gHMBC (B) spectrum ofM2fromI-A.
As in the case of M1, M2 exhibited five nonprotonated carbon peaks at δC 85.7, 160.6, 160.8, 168.3, and 168.9 ppm that remained to be assigned. A gHMBC spectrum of M2 from I-A showing the HMBC correlations originating from methylene protons (at positions 8, 16, and 19) is presented in Fig. 8B. Methylene protons at positions 16 (δH 3.65, 3.98 ppm) and 19 (δH 4.58 ppm) show a common HMBC correlation to δC at 168.3 ppm. Hence, the δC 168.3-ppm peak is assigned to the amide carbonyl C17. Unlike the situation in M1, methylenes 8 (δH 3.28, 3.42 ppm) and 16 both show HMBC correlations to the peak at δC 85.7 ppm. HMBC peaks also are seen from methylene 8 to the geminal difluoride carbon (C7) at 119.9 ppm and from methylene 16 to the 160.8-ppm peak. The peak at δC 85.7 ppm suggests the presence of a quaternary carbon with attached heteroatoms, and the peak at 168.9 ppm would be consistent with a -COOH carbon. The peaks at δC 160.6 and 160.8 ppm would be consistent with amide carbonyl carbons.
A full-scan, high-resolution mass spectrum of M2 derived from I-B showed a protonated molecular species at m/z 468.1160. The parent ion of M2 appeared to fragment readily in the Z-spray ionization source, precluding accurate mass and elemental composition assignment. However, the product ion spectrum of m/z 468.1160 showed a major peak at 424.1448, which corresponds to a molecular formula of 13CC17H18F3N6O3+ (a deviation of 0.2 ppm from the calculated mass), indicating the neutral loss of CO2. A full-scan, high-resolution MS of M2 from I-C showed a protonated molecule at m/z 468.1361 and ready fragmentation in the ionization source. The product ion spectrum of m/z 468.1361 demonstrated an abundant ion at m/z 424.1434 which corresponds to a molecular formula 13CC17H18F3N6O3+ (within 3.1 ppm of the calculated mass) indicating a neutral loss of CO2. M2 derived from either I-B or I-C failed to show a neutral loss of 13CO2; hence, neither the chloro-carbon (C13) nor the vinylic-carbon (C12) of the parent pyrazinone ring ended up as the carboxylic acid group in M2.
In the 1H 1-D spectrum of M2 derived from I-B (not shown), additional JCH couplings derived from the 13C label are seen for the methylene protons 16 (3.1 Hz at 3.65 ppm and 3.5 Hz at 3.98 ppm). Otherwise, the 1H 1-D spectra for M2 derived from either I-B or I-C were identical to that of M2 derived from I-A (Fig. 2C). The 13C 1-D of M2 from I-B, displayed in Fig. 9A, showed that the 13C label originating at the chloro-carbon (C13) of the pyrazinone ring in I-B is observed at δC 160.8 ppm. The gHMBC spectrum of M2 from I-B is shown in Fig. 9B. A strong HMBC correlation is seen from the geminal protons of methylene 16 (δH 3.65, 3.98 ppm) to the 13C-labeled carbon at 160.8 ppm. The other HMBC correlations (Fig. 9B) are from H16 to natural abundance 13C carbons at δC 85.7 and 168.3 (C17) ppm.

Proton-decoupled 13C (A) and gHMBC (B) spectra of M2 from I-B ; proton-decoupled 13C spectrum of M2 from I-C (C).
The 13C 1-D spectrum of M2 from I-C is shown in Fig. 8C. The 13C label originating from the vinylic-carbon (C12) of the pyrazinone ring in I-B appears at 160.6 ppm. The gHMBC spectrum of M2 from I-C (not shown) did not display any correlations to the 13C label-derived carbon.
The MS and NMR data for M2 derived from I-A to I-C show that, unlike the case in M1, the 13C labels in the parent pyrazinone ring end up neither as a quaternary carbon (δC 85.7 ppm) nor as a carboxylic acid carbon (δC 168.9 ppm). The 13C labels appear at 160.6 and 160.8 ppm, with the latter 3 bonds from the C16 methylene protons. The M2 structure shown in Fig. 6 is deduced from the combined MS and NMR data. The pyrazinone ring in I appears to be metabolically activated and then rearranges to form a 4,5-dioxoimidazolidine-2-carboxylic acid moiety. The quaternary carbon (C10) at 85.7 ppm represents the newly generated chiral center that leads to nonequivalent geminal protons in methylenes 8 and 16. The carboxylic acid (δC 168.9 ppm, C15) is attached to the chiral center carbon (C10). The two 13C label-derived carbons from I-B and I-C end up as amide carbonyls; the δC 160.6 and 160.8 ppm peaks in the 13C 1-D spectrum (Fig. 7) are assigned to C12 and C13, respectively, of the imidazolidine ring in M2.
M3. M3 was identified by comparing its HPLC retention time and liquid chromatography-tandem MS profile with that from a synthetic standard. The 1H and 13C chemical shifts of the M3 synthetic standard are listed in Table 3. It is notable that the δCs for the amide carbonyl (C10 and C15) in M3 are 160.8 and 160.6 ppm, identical to those for C12 and C13 in M2.
Discussion
Metabolism was the major route of elimination for compound I in rats; only 1% of the dose was excreted intact in bile and urine (Table 1). Metabolism of the pyrazinone ring leading to formation of M1 and M2 was the major metabolic pathway, accounting for 84% of the IV dose. M3 constituted 2% of the dose and was detected only in rat bile. Overall the recovery of I was essentially complete, with the remainder of the dose excreted as either glutathione adducts or their degradation products that remain to be characterized.
The method used to isolate M1 and M2 was critical in maximizing their stability. M1 and M2 degraded fairly rapidly under acidic or basic HPLC conditions. Maintaining a pH of 7.4 was important to preserve sample integrity during the isolation procedures.
Both M1 and M2 are novel, complex rearrangement products derived from metabolic activation of the pyrazinone ring in I. The six-membered pyrazinone ring in I is converted to either a 2-hydroxy-5-oxo-2,5-dihydro-imidazole-2-carboxylic acid moiety in M1 or a 4,5-dioxoimidazolidine-2-carboxylic acid moiety in M2. The use of 1H and 13C NMR spectroscopy, in combination with 13C labels at the affected carbons of the pyrazinone ring, provided a valuable insight into the structure and the mechanism of formation of M1 and M2. The use of single 13C labels was especially helpful since both M1 and M2 were devoid of protons in the central portion of the molecule.
The proposed structure for M1, based on the MS and NMR data for M1 from I-A, was confirmed by use of the 13C labels in the pyrazinone ring. However, there were three possible structures (Fig. 10) for M2 that satisfied the MS and NMR data for M2 derived from I-A. The 13C labels from C-13 (in I-B) and C-12 (in I-C) resonate at δC 160.8 and 160.6 ppm, respectively. HMBC correlations are seen from H16 to δC 160.8 ppm (assigned to C13), a constraint that can be satisfied by all three structures in Fig. 10. A δC of 160.6 ppm is possible for an amide carbonyl carbon (C12 in Fig. 10, A and B) as well as an amidine carbon (C12 in Fig. 10C). However, no HMBC correlations were seen from H8 to C12 in M2 derived from I-C (or I-A); therefore, the structures in Fig. 10, B and C were ruled out for M2.

PossibleM2structures based on MS and NMR data forM2derived fromI-A.
A possible scheme for the formation of M1, M2, and M3 from I is shown in Fig. 11. The proposed mechanism involves epoxide formation (A) across the C12-C13 double bond in the pyrazinone ring of I followed by hydrolysis to form diol B. The chlorohydrin moiety of diol B then loses a molecule of HCl and, after a second oxidative step, results in the pyrazine-2,3,5-4H-trione (C).

Proposed mechanism for formation ofM1, M2, andM3metabolites fromI.
Addition of water at either carbon 13 or carbon 15 that flanks the nitrogen 14 of the pyrazine-trione leads to ring-contracted metabolites M1 and M2. Thus, when the carbonyl carbon 13 of the pyrazine trione C undergoes hydration, amino geminal-diol D results. The six-membered ring in D exists in equilibrium with its open-ring form E. Cyclization of E then leads to the five-membered dihydro-imidazole derivative M1. Also, formation of M3 can be rationalized as a byproduct resulting from hydrolysis of E.
On the other hand, when the carbonyl carbon 15 of pyrazine-trione C is hydrated, the amino geminal-diol F is formed. The six-membered ring in F likely exists in equilibrium with the amidine carboxylic acid derivative G. Ring contraction of this species results when the electron lone pair from nitrogen 14 in G attacks the amidine carbon 10 and tautomerizes to form the imidazolidine ring in M2.
A possible variation of this proposed mechanism would be a benzil-benzylic acid like rearrangement of intermediates D and F giving M1 and M2 directly; however, formation of M3 would still require two hydrolytic steps.
The metabolic activation and subsequent fate of a related pyrazinone derivative has been reported previously (Singh et al., 2003). In that report, an analog of I in which the chlorine was replaced by a methyl group was shown to undergo metabolism on the pyrazinone ring but without resulting in rearrangement products like M1 and M2. An intermediate analogous to B (in Fig. 11) was proposed. Since the intermediate lacked a chlorine group, the ring opened with an amide nitrogen serving as the leaving group. Subsequent cyclization on the nitrogen analogous to 9 in B gave the observed imidazole.
To the best of our knowledge, there is no literature precedence to the trione-imine C or its hydrated intermediates D and F. However, tetraketo-piperazines have been shown to readily undergo ring contraction in the presence of alcohols to give 4,5-dioxoimidazolidine-2-ol-carboxylic acid esters (Hoare and Yates, 1981). When ethylene glycol was used as the alcohol, a substantial amount of an oxamide, analogous to M3, was produced.
Following an oral administration of a 100 mg/kg dose of [14C]-I to rats, the maximum level of radioactivity that became irreversibly bound to plasma or liver proteins was 6- to 10-fold lower than with a corresponding dose of the methyl analog (data on file, Merck Research Laboratories). This lower level of covalent binding with I appears to correspond to the observed differences in the biotransformation pathways of the two compounds. Thus both agents have the potential to form epoxide intermediates, which lead to products of intramolecular rearrangement; however, the methyl analog also forms a reactive electrophilic methide-imine intermediate which would be expected to alkylate proteins.
To conclude, the metabolite structures characterized here appear to represent the first examples of the biotransformation of a substituted pyrazinone to dihydro-imidazole and imidazolidine derivatives and illustrate the rich metabolic chemistry of the amino-pyrazinone heterocycle.
Acknowledgments
We thank G. Gatto for preparation of the 14C-labeled I; and F. A. Deluna, S. Ciccotto, S. Fauty, T. Killino, T. Pittman, and A. Taylor for animal studies. We also thank Drs. T. A. Baillie and B. Wong for many helpful discussions and editorial comments on the manuscript.
Footnotes
-
↵1 Abbreviations used are: MS, mass spectrometry; HPLC, high-performance liquid chromatography; DMSO, dimethyl sulfoxide; 1-D, one-dimensional; gHMQC, gradient heteronuclear multiple quantum coherence; gHMBC, gradient heteronuclear multibond coherence.
-
Current address for M.V.S.-E.: Amgen Inc., One Amgen Center Drive (Mail Stop 5-2-A), Thousand Oaks, CA 91320
- Received July 7, 2003.
- Accepted August 19, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}