


Abstract
Quantitative characterization of UDP-glucuronosyltransferase (UGT) enzymes is valuable in glucuronidation reaction phenotyping, predicting metabolic clearance and drug-drug interactions using extrapolation exercises based on pharmacokinetic modeling. Different quantitative proteomic workflows have been employed to quantify UGT enzymes in various systems, with reports indicating large variability in expression, which cannot be explained by interindividual variability alone. To evaluate the effect of methodological differences on end-point UGT abundance quantification, eight UGT enzymes were quantified in 24 matched liver microsomal samples by two laboratories using stable isotope-labeled (SIL) peptides or quantitative concatemer (QconCAT) standard, and measurements were assessed against catalytic activity in seven enzymes (n = 59). There was little agreement between individual abundance levels reported by the two methods; only UGT1A1 showed strong correlation [Spearman rank order correlation (Rs) = 0.73, P < 0.0001; R2 = 0.30; n = 24]. SIL-based abundance measurements correlated well with enzyme activities, with correlations ranging from moderate for UGTs 1A6, 1A9, and 2B15 (Rs = 0.52–0.59, P < 0.0001; R2 = 0.34–0.58; n = 59) to strong correlations for UGTs 1A1, 1A3, 1A4, and 2B7 (Rs = 0.79–0.90, P < 0.0001; R2 = 0.69–0.79). QconCAT-based data revealed generally poor correlation with activity, whereas moderate correlations were shown for UGTs 1A1, 1A3, and 2B7. Spurious abundance-activity correlations were identified in the cases of UGT1A4/2B4 and UGT2B7/2B15, which could be explained by correlations of protein expression between these enzymes. Consistent correlation of UGT abundance with catalytic activity, demonstrated by the SIL-based dataset, suggests that quantitative proteomic data should be validated against catalytic activity whenever possible. In addition, metabolic reaction phenotyping exercises should consider spurious abundance-activity correlations to avoid misleading conclusions.
Introduction
Understanding the relative contributions of different enzymes toward the metabolism of therapeutic drugs, including conjugative pathways, is of considerable value to the pharmaceutical industry and its regulatory agencies (Milne et al., 2011), with relevant applications in metabolic reaction phenotyping and prediction of drug-drug interactions (Miners et al., 2010). In particular, reaction phenotyping for drug-metabolizing enzymes using correlation approaches is crucially dependent on robust analytical methods used to measure both activity and expression levels in individual samples (Zientek and Youdim, 2015). However, despite recent efforts aimed at developing assays to characterize the abundance and activity of drug-metabolizing enzymes, with considerable success especially in the case of cytochrome P450 enzymes (Walsky and Obach, 2004; Gröer et al., 2014), this level of understanding is still hindered by the lack of standard and consistent methods for quantifying uridine-5′-diphospho-glucuronosyltransferase (UGT) expression and function (Guillemette et al., 2014). Correlations of enzyme abundances and activity were previously demonstrated for several cytochrome P450 enzymes (Snawder and Lipscomb, 2000; Olesen and Linnet, 2001) and some UGT enzymes (mainly UGTs 1A1, 1A6, 1A9, and 2B7) (Jones et al., 2012; Sato et al., 2012; Knights et al., 2016), with a variety of substrates and different levels of correlation. However, specificity of substrates for many UGT isoforms has yet to be demonstrated.
Several studies published recently reported different proteomic methodologies driven by advances in LC-MS technology (Fallon et al., 2008; Ohtsuki et al., 2012; Achour et al., 2014a; Prasad et al., 2014; Vildhede et al., 2014; Harwood et al., 2015; Fallon et al., 2016). These methodologies focused on obtaining expression values from a range of mammalian tissues/organs (e.g., liver, intestine, kidneys) and in vitro systems (e.g., hepatocytes, Caco-2 cell lines), with a view to providing systems data for in vitro-in vivo extrapolation (IVIVE) of pharmacokinetic profiles using computerized physiologically based pharmacokinetic (PBPK) models (Rostami-Hodjegan, 2012; Bosgra et al., 2014). With advantages and limitations of such methodologies (Al Feteisi et al., 2015a), selection is usually influenced by several factors, mainly related to the economics of such investigations and their intended applications (Al Feteisi et al., 2015b).
However, recently reported disparities in abundance have highlighted the need to explore the effects of intermethodology and interlaboratory differences on end-point measurement of proteins involved in drug absorption, distribution, metabolism, and excretion (ADME). For example, the abundance of hepatic organic anion transporting polypeptide 1B1 showed a wide variation (up to 10-fold) in sets of nonmatched samples measured using two different methodological workflows (Prasad et al., 2014; Vildhede et al., 2014). Meta-analyses of abundance measurements of drug-metabolizing P450 enzymes (Achour et al., 2014b), UGT enzymes (Achour et al., 2014c), and drug transporters (Badée et al., 2015) revealed up to 600-, 250-, and 100-fold differences in collated data, respectively, with a large level of interstudy heterogeneity (Higgins and Thompson’s index of up to 99%), suggesting that variability in data used in pharmacokinetic extrapolation and simulation cannot be attributed to biologic interindividual variation in expression alone.
The effects of several steps of proteomic analysis on the outcome of quantification in these methodological workflows have been investigated, with such factors as solubilization of membrane proteins (Balogh et al., 2013) and selection of peptide standards (Harwood et al., 2016a) being suggested to influence end-point measurement. Sample preparation can also contribute to this variability, with proteins either quantified directly in whole cell/tissue lysate (Weiß et al., 2015; Wiśniewski et al., 2016) or in enriched subcellular fractions (Schaefer et al., 2012; Gröer et al., 2013). In addition, differences in proteolytic strategies, efficiency of protein and peptide recovery and LC-MS analysis of peptides have also been suggested to introduce bias into abundance measurements (Chiva et al., 2014; Harwood et al., 2015). Other studies suggested that different analytical methods used in the same laboratory setting can generate reasonably consistent measurements (Qiu et al., 2013; Prasad and Unadkat, 2014).
Differentiating between true interindividual variability and methodological differences introduced by the adopted measurement technique is becoming increasingly important to improve the reliability of PBPK models to produce better population predictions related to drug therapy. Consequently, cross-laboratory and cross-methodology studies that investigate the ultimate effects of methodological differences are required, especially those that can relate abundance differences to effects on predicting activity. Therefore, this study aimed to compare the abundances of eight UGT enzymes measured in a set of matched human liver microsomal samples using two different proteomic strategies by two independent laboratories, with assessment against the catalytic activity of seven of these UGT enzymes.
Materials and Methods
Materials and Reagents
Selective UGT substrates, metabolite standards, internal standards, and other materials were obtained from commercial sources or biosynthesis of metabolite standards with concentration determination by quantitative NMR. β-Estradiol, β-estradiol-3-glucuronide, chenodeoxycholic acid (CDCA), trifluoperazine, propofol, zidovudine (AZT), alamethicin, bovine serum albumin (product no. A7906), diclofenac, and UDP-glucuronic acid trisodium salt were supplied by Sigma Chemical Co. (St. Louis, MO). R/S-oxazepam was obtained from US Pharmacopeia (Rockville, MD). AZT-5′-glucuronide, [13C6]AZT-5′-glucuronide, 5-hydroxytryptophol, trifluoperazine-N-glucuronide, [D3]trifluoperazine-N-glucuronide, and S-oxazepam glucuronide were from Cerilliant (Austin, TX). Propofol-O-glucuronide was obtained from Santa Cruz Biotechnology (Dallas, TX), and 1-napthyl-β-O-glucuronic acid was obtained from Sequoia Research Products (Pangbourne, United Kingdom). 5-Hydroxytryptophol-O-glucuronide was obtained by biosynthesis and characterized as previously described (Walsky et al., 2012). CDCA-24-glucuronide was obtained from Carbosynth (Compton, UK). All other reagents and solvents used were from standard suppliers and were of reagent or HPLC grade with all purities as defined by the manufacturer. Materials and chemicals used for the quantification of UGT abundances were as described previously (Fallon et al., 2013b; Achour et al., 2014a).
Individual human liver microsomal (HLM) samples used by all participating laboratories were obtained from BD Biosciences (Woburn, MA) and characterized for total protein content, UGT enzyme abundance, and catalytic enzyme activity as described below. Supplemental Table 1 shows brief demographic and clinical information about the sample donors (n = 60).
Quantification of UGT Enzymes
In-house methods for the quantification of eight UGT enzymes (UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, and 2B15) were applied independently by two laboratories: University of North Carolina at Chapel Hill (n = 60) and the University of Manchester, UK (n = 24). Details of methodologies used by these laboratories are provided in the respective reports (Fallon et al., 2013b; Achour et al., 2014a). Differences in methodological workflows are highlighted in Table 1. The individual samples analyzed by each methodology are shown in Supplemental Table 1.
An outline of the methodology criteria constituting the quantitative proteomic workflows specific to each method for the quantitative analysis of the set of UGT enzymes
Quantification of UGT Enzymes Using Stable Isotope-Labeled Peptide Standards
Sample Preparation of Human Liver Microsomes.
In-solution sample preparation was performed as reported previously (Fallon et al., 2008, 2013a,b). Briefly, sample protein content was assessed using bicinchoninic acid assay and samples (n = 60) were diluted 1:20 in 50 mM ammonium bicarbonate. Double-labeled SIL tryptic extension peptides representing UGT1A1, UGT1A9, and UGT2B7 (1 pmol) as well as β-casein (0.5 μg) were added to the samples to help assess digestion efficiency. Proteins were denatured and disulfide bridges reduced using dithiothreitol (40 mM, 60°C, 40 minutes), followed by carbamidomethylation with iodoacetamide (135 mM, room temperature, dark, 30 minutes). Digestion was carried out in-solution with trypsin (5% w/w, 37°C, 4 hours) with mixing at 300 rpm using an IsoTemp Thermal Mixer (Thermo Fisher Scientific, Waltham, MA). The digestion reaction was stopped by adding 75 µl of ice cold acetonitrile. SIL internal standards were then added to each sample at 1 pmol/peptide. This was followed by evaporation and reconstituting in slightly modified mobile phase A (0.1% formic acid/acetonitrile; 98/2) (i.e., containing 2% rather than 1% acetonitrile) for analysis. After thorough mixing, samples were centrifuged at 13,300 × g, and the supernatant was transferred to deactivated vial inserts for LC-MS/MS analysis.
Preparation of Standards.
Selection of proteotypic peptides was carried out as previously reported (Fallon et al., 2008, 2013a; Kamiie et al., 2008). The peptides were tryptic, as well as proteotypic, and contained no linkers or added moieties, such that they could be used directly as standards for quantification. Further considerations based on in silico predictions before selection of peptides were performed to estimate relative hydrophobicity and electrospray efficiency to optimize the selected list. The final selected SIL internal peptide standards list consisted of two peptides per UGT enzyme (Fallon et al., 2013b), and these were obtained from Thermo Biopolymers (Ulm, Germany). Standards were added after digestion of samples at 1 pmol of each peptide.
LC-MS/MS Quantification of UGT Enzymes.
Two HLM samples, one containing a high amount of UGT1A1 and the other a low amount, were analyzed as controls with sample batches. Multiple reaction monitoring (MRM) targeted analysis was used for the quantification on a nanoACQUITY binary pump system (Waters, Milford, MA) coupled to a SCIEX QTRAP 5500 hybrid mass spectrometer operated using Analyst 1.5 software (SCIEX, Framingham, MA). A Waters BEH130 C18 analytical column (150 µm internal diameter × 100 mm) was used, with a flow rate of 2 µl min−1 and a gradient of 0%–42% mobile phase B (acetonitrile) over 24 minutes for seven batches, including quality control samples, and 0%–35% mobile phase B for two batches. The low flow rate and high pressure/narrow diameter of the column meant that injection of only 0.8 µg (in 2 µl) of the sample elicited a highly sensitive response from the system (electrospray interface). MRM schedules were designed using Skyline 1.1 (MacCoss Laboratory Software, Seattle, WA); the best two transitions were used and optimization of collision energies was carried out. Concentrations were calculated from area ratios of endogenous (unlabeled) peptide to heavy labeled peptide. One peptide was selected for each UGT isoform for reporting the abundance.
Quantification of UGT Enzymes Using a Quantitative Concatemer Standard
Sample Preparation of Human Liver Microsomes.
Sample preparation and LC-MS analysis were performed as described previously (Russell et al., 2013; Achour et al., 2014a). Briefly, protein concentrations in samples were estimated based on Bradford assay. An in-gel sample preparation and protein digestion method was used with solubilization with 5% w/v SDS. Microsomal samples (n = 24) and QconCAT standard protein from Escherichia coli culture were diluted 1:10 in SDS loading buffer and loaded on 10% w/v SDS-PAGE gels and run in duplicate; bands were then visualized using Coomassie brilliant blue staining. Gel bands were excised between 45 and 65 kDa, cut to small cubes (2 × 2 mm) and destained. Protein disulfide bonds were reduced using dithiothreitol (10 mM, 50°C, 30 minutes) and alkylated using iodoacetamide (55 mM, room temperature, dark, 1 hour). Proteolysis was carried out in 25 mM ammonium bicarbonate buffer using lysyl endopeptidase (1% w/w, 30°C, 4 hours) followed by trypsin (1% w/w, 37°C, 18 hours). Peptide digests were collected and gel particles were washed with two concentrations of acetonitrile (5% and 50% v/v) to collect remaining peptides. Combined peptide elutions were vacuum concentrated and acidified (3% v/v acetonitrile, 0.1% v/v formic acid), then transferred into LC-MS vials.
Design and Preparation of Standard.
The standard used in this part of the study was prepared in-house as previously reported (Russell et al., 2013; Achour et al., 2015). The QconCAT standard contained two signature peptides for each UGT enzyme. These peptides were selected based on experimental and theoretical considerations (Pratt et al., 2006; Kamiie et al., 2008; Russell et al., 2013). The QconCAT gene construct (plasmid) was expressed in stable-isotope enriched media (for 13C-labeled arginine and lysine) using a protease knockout E. coli strain JM109 (DE3) and purified in inclusion bodies. Enrichment of isotopically labeled protein was assessed using LC-MS and shown to be ≥95% (Achour et al., 2015). Sample preparation of expressed isotopically labeled QconCAT was the same as microsomal samples.
LC-MS/MS Quantification of UGT Enzymes.
Targeted MRM proteomic quantification of UGT enzymes was carried out on a nanoACQUITY nano-HPLC system (Waters, Manchester, UK) coupled to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific) operated using Xcalibur version 2.0.6 Service Pack 1 (Thermo Fisher). A Waters HSS T3 C18 analytical column (75 µm internal diameter × 100 mm) was used for HPLC analysis. Peptides (1 µl, 10 fmol) were eluted at a 0.3 µl min−1 flow rate with a gradient of 3%–50% acetonitrile over 40 minutes. MRM assays were designed and elution profiles analyzed using Skyline 1.4.0.4222.
UGT Enzyme-Selective Glucuronidation Assays
Selective substrate glucuronidation activity was determined by Pfizer (Groton, CT) for individual HLM donors using UGT-selective substrate assays essentially as previously described (Walsky et al., 2012) or employing newly optimized assays for CDCA and S-oxazepam glucuronidation. Substrates were incubated at the apparent maximal velocity (Vmax) to obtain velocities reflecting individual differences in enzyme expression level. Briefly, alamethicin-activated enzyme mixtures (in triplicate) for individual HLM donors (n = 59) were prepared containing 0.025 mg ml−1 HLM (determined by bicinchoninic acid assay) in 100 mM Tris-HCl buffer (pH 7.5 at 37°C), MgCl2 (5 mM), alamethicin (10 μg ml−1), and 2% (w/v) bovine serum albumin, preincubated on ice for 15 minutes. Individual enzyme mixtures also contained final substrate concentrations of 800 µM β-estradiol (UGT1A1), 500 µM CDCA (UGT1A3), 400 µM trifluoperazine (UGT1A4), 3000 µM 5-hydroxytryptophol (UGT1A6), 500 µM propofol (UGT1A9), 1500 µM AZT (UGT2B7), and 1000 µM R/S-oxazepam (UGT2B15). By using a Hamilton Star (Hamilton, Reno, NV) customized method, aliquots of the enzyme premixture (180 µl) were delivered to a 96-well glass insert incubation plate maintained at 37°C and preincubated for 5 minutes at 37°C to reach incubation temperature. After the preincubation period, reactions were initiated with the addition of 20 µl UDP-glucuronic acid (5 mM final concentration; 0.2 ml final incubation volume). Incubations for determination of β-estradiol-3-glucuronide, trifluoperazine-N-glucuronide, 5-hydroxytryptophol-O-glucuronide, propofol-O-glucuronide, and zidovudine-5′-glucuronide were quenched after 20–60 minutes with organic solvent containing internal standards, centrifuged, and supernatants subjected to HLPC-MS/MS analysis as previously described (Walsky et al., 2012).
Chenodeoxycholic acid (UGT1A3) incubations were terminated after 15 minutes by removing a 100 µl-aliquot and quenched in 200 µl acetonitrile containing internal standard diclofenac (0.3 µM). Samples were vortexed and centrifuged at 2000 g for 10 minutes. Supernatants were analyzed via ultra-high-pressure liquid chromatography tandem mass spectrometry (UHPLC-MS/MS). Aliquots (11 µl) of sample extracts were injected directly onto a XB-C18 1.7 µm, 2.1 × 50 mm, 100 Å column (Phenomenex, Torrance, CA). Elution used a binary gradient at a flow rate of 0.6 ml min−1 with mobile phases H2O/0.1% formic acid (A) and acetonitrile/0.1% formic acid (B). Initial conditions 5% (B) were held for 0.4 minute, followed by a linear increase to 98% (B) over 1.2 minutes, which was held for 0.9 minute before returning to initial conditions at 2.1 minutes. CDCA-24-glucuronide eluted at approximately 1.3 minutes. Negative ion electrospray tandem mass spectra were recorded using an Applied Biosystems/Sciex (Framingham, MA) QTRAP 4000 mass spectrometer equipped with Analyst (version 1.6.2) operating software and Acquity UPLC System (Waters Corporation, Milford, MA). The ionspray voltage was set to −4 kV and the probe temperature was set at 500°C. Nitrogen was used as the collision gas and the collision activated dissociation, curtain gas, gas 1, and gas 2 values were set to −2, 15, 60, and 60, respectively. Multiple reaction monitoring (MRM) transition of m/z 567.4 → 391.3 was used during quantitative analysis of CDCA-24-glucuronide. Metabolite concentrations were determined against a CDCA-24-glucuronide standard curve (0.3–1000 nM) prepared in incubation matrix and treated identical to analytical samples.
R/S-oxazepam (UGT2B15) incubations were terminated after 60 minutes by removing a 50 µl-aliquot and quenched in 200 µl acetonitrile containing internal standard diclofenac (0.3 µM). Samples were vortexed and centrifuged at 2000 g for 5 minutes. Supernatant (170 µl) was evaporated under nitrogen (40–60 PSI at 37°C) and reconstituted in water (120 µl). Aliquots (10 µl) of sample extracts were injected directly onto a Kinetex Phenyl-Hexyl 1.7 µm, 2.1 × 100 mm column (Phenomenex) heated to 50°C. Elution used a binary gradient at a flow rate of 0.5 ml min−1 and consisting of mobile phases: 10 mM ammonium formate/1% isopropyl alcohol (A) and acetonitrile (B). Initial conditions 15% (B) increased to 32% (B) at 3.00 minutes, then to 63% (B) at 3.50 minutes, then to 96% (B) at 3.51 minutes and held for 0.49 minute before returning to initial conditions at 3.91 minutes. S-oxazepam glucuronide eluted at approximately 1.7 minutes and was separated from R-oxazepam glucuronide, which eluted at approximately 1.5 minutes. Positive ion electrospray tandem mass spectra were recorded using an Applied Biosystems/Sciex model API 6500 mass spectrometer equipped with Analyst (version 1.6.2) operating software and Acquity UPLC System (Waters Corporation). The ionspray voltage was set to 5 kV and the probe temperature was set at 500°C. Nitrogen was used as the collision gas and the collision activated dissociation, curtain gas, gas 1, and gas 2 values were set to 9, 25, 60, and 90, respectively. Multiple reaction monitoring (MRM) transition of m/z 463 → 287 was used during quantitative analysis of S-oxazepam glucuronide. The S-oxazepam glucuronide concentrations were determined against a standard curve (3.7–1000 nM) prepared in incubation matrix and treated identical to analytical samples.
Study Design and Statistical Data Analysis
Matched HLM samples were used in all proteomic and activity experiments. Measurement of the abundances of UGT enzymes (two datasets) and their catalytic activities (one dataset) was carried out in three different centers in a blinded manner between all analysts. Method validation and quality control steps were performed by the two laboratories independently as previously reported for the SIL-based (Fallon et al., 2013b) and QconCAT-based (Achour et al., 2014a) quantification. Independence of measurements was maintained to ensure that true representation of laboratory-specific bias could be assessed and to provide an objective measure of methodological variability in UGT quantification. Data analysis was carried out after all experimental work was completed.
Since a considerable proportion of the enzyme abundance and activity datasets did not follow Gaussian distribution (assessed using three normality tests: D’Agostino-Pearson, Shapiro-Wilk, and Kolmogorov-Smirnov tests), nonparametric statistical assessment was used. Absolute abundance data obtained using the two proteomic methodologies were compared by Mann-Whitney U-test and the distributions of the datasets were compared using Kolmogorov-Smirnov cumulative distribution test. Correlation analysis was performed using Spearman rank order correlation (Rs) test with t-distribution of the P value to assess correlations of data. Linear regression analysis was carried out to assess the linearity of relationships and scatter of the data. Assessment of fold differences was carried out using average fold error to assess bias between methods and absolute average fold error to assess scatter of one dataset in relation to the other. In all statistical analyses, a selected α-value of 0.05 was used for probability to indicate statistical significance of differences and correlations; however, when a dataset was assessed more than once for a particular test (multiple identical tests), for example in correlation matrices, a Bonferroni correction of the α-value was introduced based on the number of iterations in each dataset. Further details on statistical analysis are included in Supplemental Information. Statistical analysis was carried out using Microsoft Excel 2010 and GraphPad Prism version 7.01 (GraphPad Software, San Diego, CA). Graphs were generated using GraphPad Prism.
Results
In this study, we investigated the relationship between abundances of UGT enzymes and their catalytic activity. The abundance measurements of eight UGT enzymes were carried out using two established quantitative proteomic methods: the first using in-solution sample preparation with targeted proteomic analysis against stable isotope-labeled (SIL) standards, whereas the second used in-gel sample preparation in conjunction with targeted quantification using a quantitative concatemer (QconCAT) standard. Activity measurements were conducted using a set of developed selective high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) functional assays for glucuronides of seven UGT probe substrates: β-estradiol-3-glucuronide (for UGT1A1 activity), chenodeoxycholic acid (CDCA)-24-glucuronide (UGT1A3), trifluoperazine-N-glucuronide (UGT1A4), 5-hydroxytryptophol-O-glucuronide (UGT1A6), propofol-O-glucuronide (UGT1A9), zidovudine-5′-glucuronide (UGT2B7), and S-oxazepam glucuronide (UGT2B15). Although the datasets generated in this study are related to a total sample set of 60 livers, the activity assessment was carried out in 59 samples and the QconCAT-based proteomic analysis was carried out in only 24 samples. The SIL-based proteomic assessment was conducted on the whole sample set (n = 60) (see Supplemental Table 1).
Cross-methodology Comparison of Enzyme Abundance Measurements.
The analytical quality controls for the two methodologies used for measuring UGT enzyme abundances were established previously (Fallon et al., 2013b; Achour et al., 2014a). Supplemental Table 2 shows intra- and interday technical variability in the two methods, which was in overall agreement with the U.S. Food and Drug Administration bioanalytical method validation guidelines (www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM368107.pdf). Technical variability was consistent over several quality control runs. For the SIL-based technique, intraday variability was within 10% (expressed as CV) and interday variability was within 20% for the peptides used to report protein concentration, whereas these were within 15% and 20%, respectively, for QconCAT-based measurements. The exception was interday variability in measurements of UGTs 1A4 and 1A6 by both methods, which sometimes exceeded 20% but was consistently within 30%.
Assessment of matched data generated using SIL and QconCAT standards (n = 24) revealed significant differences between the abundance values for most of the datasets and their distributions (Fig. 1A; Supplemental Table 3). Only UGTs 1A1, 1A4, and 2B7 datasets showed no difference in abundances and dataset distribution. Fold error assessment (Fig. 1B) showed that the QconCAT methodology tended to consistently overestimate (average fold error = 2.03) abundance levels, and there was a significant spread of data across the two methodologies (absolute average fold error = 2.64). This bias and scatter are also reflected in individual data points shown in Fig. 1C.

Cross-methodology comparison of UGT abundance levels: box and whiskers plot of abundance measurements (n = 23 or 24 liver samples) of UGT enzymes quantified by SIL standards or QconCAT (A); fold difference (fold error) of matched values (i.e.,  for each enzyme i) (B); and correlation analysis of individual protein abundance measurements (n = 23–24) using the two methods (C). In (A) and (B), the boxes represent the 25th and 75th percentiles, the whiskers represent the minimum and maximum values and the bars represent the medians. In (A), the (+) sign represents the arithmetic mean. Differences were tested using Mann-Whitney rank order U-test: **P < 0.01; ***P < 0.001. In (B), the shaded area represents values within threefold, illustrating interchangeable abundance data. AFE is the average fold error and AAFE is the absolute average fold error. In (C), Rs is Spearman rank order correlation coefficient; green colored data points represent correlated enzyme levels between methods and gray data points reflect lack of correlation. Supplemental Information contains details of statistical analysis, and Supplemental Table 3 shows differences between generated data. Units of abundance measurements are picomoles per milligram HLM protein. QconCAT, quantitative concatemer standard; SIL, stable isotope-labeled peptide standards.
for each enzyme i) (B); and correlation analysis of individual protein abundance measurements (n = 23–24) using the two methods (C). In (A) and (B), the boxes represent the 25th and 75th percentiles, the whiskers represent the minimum and maximum values and the bars represent the medians. In (A), the (+) sign represents the arithmetic mean. Differences were tested using Mann-Whitney rank order U-test: **P < 0.01; ***P < 0.001. In (B), the shaded area represents values within threefold, illustrating interchangeable abundance data. AFE is the average fold error and AAFE is the absolute average fold error. In (C), Rs is Spearman rank order correlation coefficient; green colored data points represent correlated enzyme levels between methods and gray data points reflect lack of correlation. Supplemental Information contains details of statistical analysis, and Supplemental Table 3 shows differences between generated data. Units of abundance measurements are picomoles per milligram HLM protein. QconCAT, quantitative concatemer standard; SIL, stable isotope-labeled peptide standards.
Assessment of correlation between abundances measured using the two methods showed lack of correlation except for UGT1A1 (Fig. 1C), which showed significant correlation between the two datasets (Rs = 0.73, P < 0.0001), also corroborated by linear regression (R2 = 0.30), though indicating a level of scatter in the data. Although the UGT1A3 dataset showed strong correlation with less scatter (Rs = 0.72, P < 0.0001, R2 = 0.41), there was high discrepancy in the scale of data values indicated by the ranges and the fold differences (Fig. 1B; Supplemental Table 3).
With this level of difference between the abundance values of UGT enzymes assessed in the same set of samples using different methodological workflows, the most appropriate determinant of which values were more accurate was correlation with glucuronidation activity levels using previously optimized (Walsky et al., 2012) and newly developed activity assays.
Relationship between Activity and Abundance of UGT Enzymes.
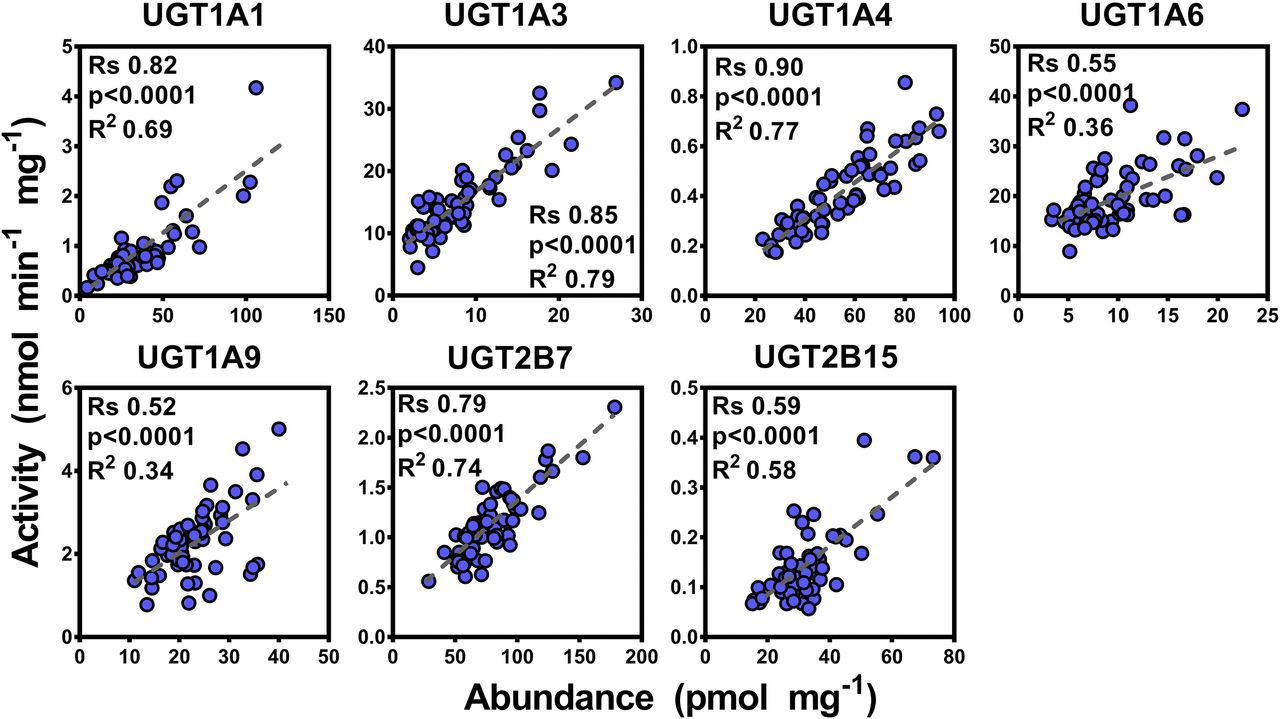
Seven activity assays were developed and optimized for UGT activity assessment as described in the Materials and Methods. Results of the activity characterization of microsomal samples are shown in Table 2. Correlation between activity and abundance levels was assessed using Spearman rank order test and linear regression with appropriate corrections applied to the significance α-value. Since there were differences in abundance values between the two methodologies, differences in correlation with glucuronidation activity were expected. The SIL-based abundance measurements correlated well with enzyme activities (n = 59), with correlations ranging from moderate for UGTs 1A6, 1A9, and 2B15 (Rs = 0.52–0.59, P < 0.0001, R2 = 0.34–0.58) to strong for UGTs 1A1, 1A3, 1A4, and 2B7 (Rs = 0.79–0.90, P < 0.0001, R2 = 0.69–0.79) (Fig. 2). This set of data presents a significant showcase of the linear relationship between catalytic activity and protein abundance for the main human hepatic UGT enzymes in a relatively large set of samples. On the other hand, most of the abundance data derived from the QconCAT methodology did not correlate well with the enzyme activity dataset (n = 24), except data related to UGTs 1A1, 1A3, and 2B7 exhibiting mostly moderate correlation (Rs = 0.40–0.79, P < 0.05, R2 = 0.30), as shown in Fig. 3.
Summary of enzyme activity data for seven UGT enzymes in human liver microsomal samples (n = 59)

Correlation between individual protein abundance and activity measurements for UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 in the stable isotope-labeled (SIL) quantification dataset (n = 59). Statistically significant, moderate-to-strong correlations are shown in blue color. Substrates: UGT1A1, β-estradiol; UGT1A3, chenodeoxycholic acid (CDCA); UGT1A4, trifluoperazine; UGT1A6, 5-hydroxytryptophol; UGT1A9, propofol; UGT2B7, zidovudine; and UGT2B15, S-oxazepam. Rs, Spearman rank order correlation coefficient; units of abundance measurements (x-axis), picomoles per milligram HLM protein; units of catalytic activity (y-axis), nanomoles (glucuronide) per minute per milligram HLM protein.
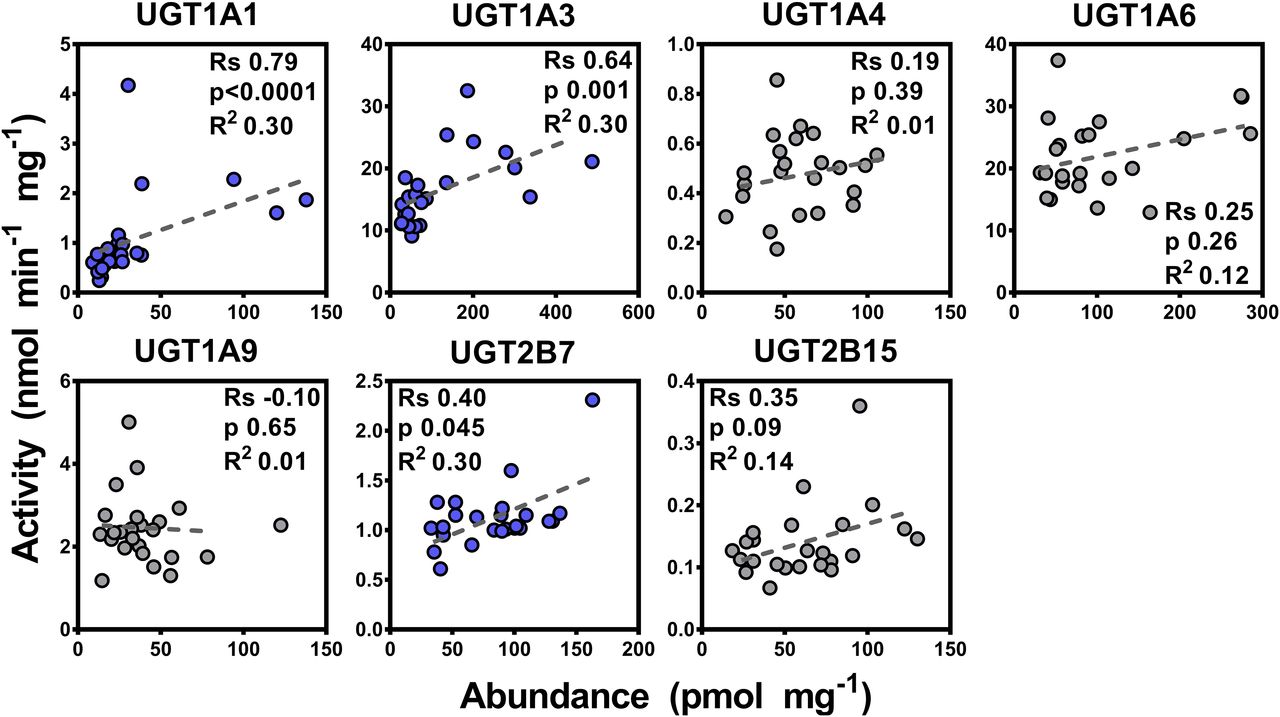
Correlation between individual protein abundance and activity measurements for UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 in the quantitative concatemer (QconCAT) quantification dataset (n = 23 or 24). Statistically significant, moderate correlations are shown in blue color, and nonsignificant, weak correlations are shown in gray. Substrates: UGT1A1, β-estradiol; UGT1A3, chenodeoxycholic acid (CDCA); UGT1A4, trifluoperazine; UGT1A6, 5-hydroxytryptophol; UGT1A9, propofol; UGT2B7, zidovudine; and UGT2B15, S-oxazepam. Rs, Spearman rank order correlation coefficient; units of abundance measurements (x-axis), picomoles per milligram HLM protein; units of catalytic activity (y-axis), nanomoles (glucuronide) per minute milligram HLM protein.
These findings confirmed that the extensive dataset (n = 60), obtained using SIL-based proteomic quantification, was the more reliable dataset, which prompted iterative testing of correlation between the activity measurements and the complete proteomic abundance dataset (10 enzymes; Fallon et al., 2013b) to establish the selectivity and specificity of the activity assays toward UGT enzymes. The abundance-activity correlation matrix is shown in Fig. 4 (data in Supplemental Table 4), with enzyme abundance on the x-axis and activity on the y-axis, with strong correlations shown in the diagonal line (in blue) representing the UGT enzyme activity toward its own developed probe in relation to its abundance levels measured using SIL-based quantification (the QconCAT matrix dataset is shown in Supplemental Table 5). The figure indicates specific activity of all UGT enzymes toward their respective substrates; however, some cases were complicated by the identification of apparent, possibly spurious, correlations with other UGT enzyme abundances (indicated in red), as shown in the case of UGT1A4 activity (with UGT2B4 abundance), UGT2B7 activity (with UGT2B15 abundance), and UGT2B15 activity (with UGT2B7 abundance). This finding necessitated a complete assessment of protein expression correlations in SIL-based and the QconCAT-based proteomic datasets (see below and Fig. 5).

Correlation matrix of individual protein abundance and activity measurements (abundance vs. activity) of UGT enzymes based on the extended dataset obtained using SIL standards (n = 59). Significant correlations are represented in blue (Rs > 0.5, Bonferroni corrected P < 0.01, R2 > 0.3). Spurious correlations are shown in red. Supplemental Table 4 shows the statistical analysis used to generate the activity-abundance correlation matrix. AZT, zidovudine; CDCA, chenodeoxycholic acid; EST, β-estradiol; 5HTOL, 5-hydroxytryptophol; OXAZ, S-oxazepam; PRO, propofol; Rs, Spearman rank order correlation coefficient; TFP, trifluoperazine.
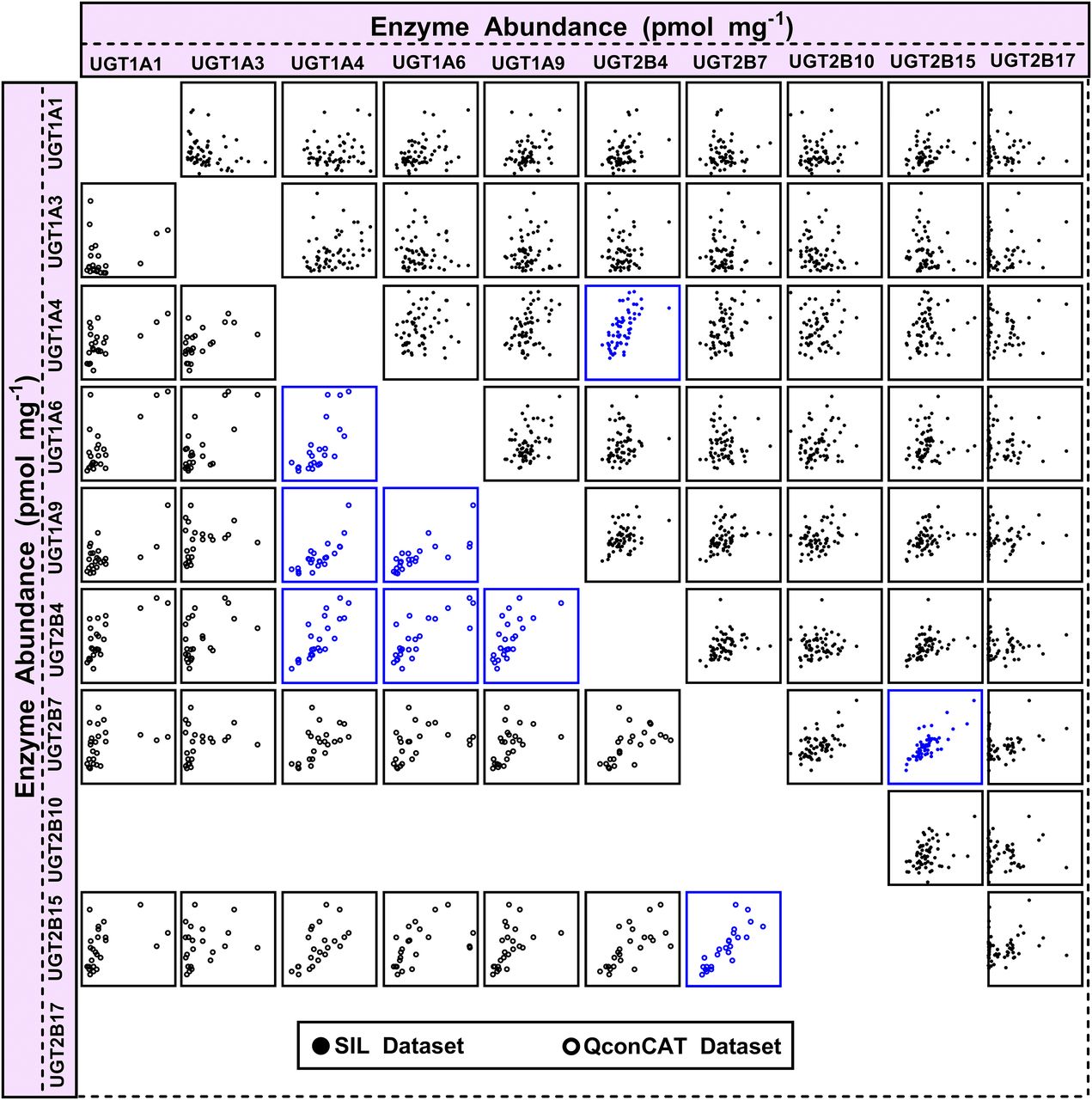
Correlation matrix of individual protein abundances of UGT enzymes (abundance vs. abundance) using the two proteomic methodologies: SIL standard-based quantification (closed circles, n = 60 livers) and QconCAT-based quantification (open circles, n = 23 or 24 livers). Significant correlations are represented in blue (Rs > 0.5, Bonferroni corrected P < 0.01, R2 > 0.3). x- and y-Axes represent abundance levels of UGT enzymes expressed in units of picomoles per milligram HLM protein; Rs, Spearman rank order correlation coefficient. Supplemental Table 6 shows the statistical analysis used to generate the abundance correlation matrix.
Correlations in Enzyme Expression Profiles.
Analysis of correlation between abundance measurements (correlation matrix in Fig. 5 with both axes representing abundance levels) consistently showed a relationship between the abundance of UGTs 1A4 and 2B4 (Rs = 0.66–0.82, P < 0.0001; R2 = 0.40–0.67) and between UGTs 2B7 and 2B15 (Rs = 0.73–0.91, P < 0.0001; R2 = 0.61–0.71) in both datasets generated using the two proteomic methods (Supplemental Table 6). These two correlations in the abundance levels of UGT1A4/2B4 and UGT2B7/2B15 enzymes are therefore expected to affect abundance-activity relationship assessment and consequently result in spurious correlations originating from correlation in natural protein expression, as reflected by the matrix in Fig. 4.
Other strong protein expression correlations were uncovered in the QconCAT-based dataset, including UGT1A4/1A9 (Rs = 0.75, P < 0.0001, R2 = 0.62) and 1A6/1A9 (Rs = 0.82, P < 0.0001, R2 = 0.59).
Consideration of Spurious Abundance-activity Correlations in Reaction Phenotyping.
The spurious correlations of enzyme abundance-activity measurements seen in this report in the cases of UGTs 1A4, 2B7, and 2B15 activity datasets raise a point of caution when conducting reaction phenotyping, e.g., using individual donor human liver microsomes and activity correlation analyses, where the results suggest that phenotyping may become complicated if the natural expression of enzymes at the protein level is strongly correlated. In these types of phenotyping experiments, the relative contributions of the different routes of metabolic elimination carried out by different enzymes are described, where correlations of expression and activity can be used to determine the contributing enzymes. This use of correlation analyses may confound the mapping of conjugative activity to the specific enzyme carrying out the biotransformation reaction when alternate strategies are not employed.
Discussion
Phase II drug metabolism is largely driven by the UGT family of enzymes, mainly expressed in the liver, which are responsible for approximately 35% of all conjugative metabolic pathways and are implicated in contributing to the metabolic clearance of nearly 60% of the 200 most prescribed therapeutic drugs in the United States in 2012 (Guillemette et al., 2014). The development of robust activity assays with selective UGT substrates and inhibitors has contributed to the progress of UGT-related phenotyping, where the relative contributions of different UGT enzymes to drug metabolism can be predicted, with potential applications in early characterization of drug candidates (Milne et al., 2011). In this line, Walsky et al. (2012) recently optimized reaction kinetic assays for five UGT substrates with a view to identify inhibitors for UGT enzymes, and the present report expands the list of these enzymes to also include UGT1A3 and UGT2B15.
Extrahepatic expression of certain UGT enzymes has also been highlighted in the literature (Sato et al., 2014; Margaillan et al., 2015a); however, since preparation methods for these tissues are less well-established, applying proteomic methods to assess abundances of these enzymes may be the preferred approach for IVIVE-PBPK clearance predictions. Particularly, with recent advances in proteomic methods, robust analyses of the protein expression patterns of UGT enzymes in different tissues and in vitro systems (Ohtsuki et al., 2012; Fallon et al., 2013b) made it possible to derive reliable relative expression factors (REFs) essential to these drug-related simulation exercises (Knights et al., 2016). More recent reports have, however, highlighted significant levels of interlaboratory discrepancy between abundance measurements of drug-metabolizing enzymes and drug transporters (Achour et al., 2014c; Badée et al., 2015), even when matched samples were analyzed (Harwood et al., 2016a), necessitating further investigations into the factors contributing to these discrepancies. To illustrate this point, the present study reports comparative analysis of a set of eight UGT enzymes quantified by two distinct proteomic methodologies (SIL and QconCAT-based approaches) in matching samples, with correlation of these abundances against rates of glucuronidation for seven UGT substrates, highlighting large interlaboratory discrepancies between the reported protein levels of these enzymes.
With SIL standard-based quantification, the results of this study demonstrated consistent and strong correlation between UGT abundances and their activity for all seven enzymes in a large set of samples, especially for UGTs 1A1, 1A3, 1A4, and 2B7. The cases of UGTs 1A6, 1A9, and 2B15 showed a significant but lower level of correlation, possibly due to less specific substrates or less efficient proteomic methods. The same substrates (propofol and zidovudine) were previously used to assess correlation of activity rates with UGTs 1A9 and 2B7 protein contents in human liver and kidney microsomal samples and demonstrated strong correlation in both cases (Margaillan et al., 2015b; Knights et al., 2016), pointing to the need for further optimization of the proteomic workflows in the cases where strong correlations were not demonstrated. In contrast, the QconCAT-based dataset showed overall poor correlation with catalytic activity, with moderate correlations observed only for UGTs 1A1, 1A3, and 2B7. Therefore, these intermethodology differences suggest that effective characterization of ADME protein expression levels in different systems should ideally be complemented with specific activity data, preferably generated within the same system or tissue samples, as previously advocated (Harwood et al., 2016b). The abundance data related to UGT1A3 is an interesting case, where the expression data from the two laboratories correlated and both datasets correlated with CDCA glucuronidation activity. However, large discrepancies were recorded in the scale of abundance levels, highlighting the relative nature of correlation analysis, where the relative distribution (rank order) of the data is the key factor, rather than the actual values.
Interindividual variability (n = 59) in glucuronidation activity ranged from 30 to 72% (expressed as CV), which is comparable to variability in UGT protein abundance for the same set of enzymes in the extended sample set (n = 60), with levels of variability ranging between 27 and 67%, in line with previous studies on UGT protein expression and activity that used similar methodologies (Sato et al., 2012; Margaillan et al., 2015b). In contrast, higher interindividual variability was recorded with the QconCAT abundance dataset (43%–101%, n = 23 or 24). Interlaboratory differences recorded in the abundance levels of the characterized UGT enzymes reached >30-fold in the cases of UGT1A3 and UGT1A6, with the QconCAT-based technique tending to consistently overestimate. A recent cross-laboratory study that looked into differences in measured abundances of two intestinal efflux transporters, P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2), reported up to three- and fivefold differences in abundance measured in matched human jejunal and Caco-2 samples, respectively (Harwood et al., 2016a). However, the laboratory-specific IVIVE relative expression factors (REFs) generated for these proteins were within twofold for both transporters and did not reflect significant differences in simulated pharmacokinetic outcomes (Harwood et al., 2016b), suggesting that systematic bias in methodology can lead to valid IVIVE conclusions if more suitable study designs are implemented, where specific REF values of ADME proteins are generated in the same or a similar setting. In contrast, another cross-laboratory study that compared quantitative proteomic data of several ADME proteins (P450s, UGTs, and transporters) from six independent laboratories (Wegler et al., 2017) reported large interlaboratory differences that led, in the case of organic anion transporting polypeptide transporters, to significant discrepancies in simulated uptake clearance of atorvastatin. The authors called for more effective standardization of proteomic methods across laboratories to generate more reliable proteomic data for drug assessment.
QconCAT methodology as a quantitative technique is not of itself flawed, as demonstrated not only by the consistency of cytochrome P450 measurements with activity (Achour et al., 2014a), but most notably, by experiments on the bacterial ribosome (Al-Majdoub et al., 2014). Although differences in accuracy between the two methods are difficult to assess due to lack of available purified UGT standards of known quantities, several factors may have led to the observed level of discrepancy. UGT enzymes are membrane-bound proteins and therefore are difficult to solubilize for reliable measurement, pointing to the importance of efficient sample preparation. In addition, digestion efficiency and quality (both purity and stability) of standards used for quantification can also contribute to differences in measurement. UGT enzymes introduce additional complexity at the level of peptide choice due to sequence homology, with some peptides being released more readily than others on proteolysis, most likely due to hydrophobicity and proximity to membrane embedded domains (Harwood et al., 2016a). These discrepancies seen especially with the QconCAT-based dataset will warrant further investigation into the sources of differences in reported end-point proteomic measurements of UGT enzymes, which will be the subject of a future publication.
The correlation matrix generated in this study for catalytic activity and SIL-based abundance data revealed the specificity of the substrates and activity assays used to measure glucuronidation rates, with activity rates strongly correlated with the protein content of their respective UGT enzymes. However, spurious correlations in relation to the activities of UGTs 1A4, 2B7, and 2B15 were also uncovered, which were not as strong as true correlations and could be explained by natural correlation of expression between UGT1A4/2B4 and UGT2B7/2B15, shown to be strong and significant in both SIL-based and QconCAT-based datasets and corroborated by recent literature (Achour et al., 2014c, Margaillan et al., 2015b). A possible ramification of such apparent relationships can be compromised reaction phenotyping of substrates of such enzymes if phenotyping is carried out using correlation of activity with enzyme content in an in vitro system, such as human liver microsomes. Although correlation of UGT-isoform selective activity with metabolic turnover of a new chemical entity is useful in identifying the UGT isoforms responsible for metabolism, confirmatory approaches utilizing isoform-selective chemical inhibitors, recombinantly expressed UGT enzyme analyses, and genotyped tissue fractions should also be employed (Zientek and Youdim, 2015).
In conclusion, the current study investigated intermethodology differences in abundance measurements of eight UGT enzymes and correlations of such expression levels to glucuronidation activity using a panel of seven specific substrates. With strong and specific correlations between UGT abundances and activity rates, this study highlights the importance of using activity data to establish confidence in UGT abundance levels used in metabolic reaction phenotyping and IVIVE prediction of drug clearance. The main limitation of this approach is that concomitant abundance and activity measurement of ADME proteins is not always possible in all laboratories.
Acknowledgments
We thank Dr. Larry Tremaine from Pfizer for facilitating the interlaboratory collaboration and for thoughtful review of the manuscript. B.A., J.B. and A.R.-H. thank the Michael Barber Centre for Collaborative Mass Spectrometry (MBCCMS), University of Manchester, for allowing access to LC-MS/MS instrumentation.
Authorship Contributions:
Participated in research design: Achour, Dantonio, Niosi, Novak, Fallon, Barber, Smith, Rostami-Hodjegan, Goosen.
Conducted experiments: Achour, Dantonio, Niosi, Novak, Fallon.
Contributed new reagents or analytical tools: Niosi, Novak.
Performed data analysis: Achour, Dantonio, Niosi, Novak, Rostami-Hodjegan, Goosen.
Wrote or contributed to the writing of the manuscript: Achour, Dantonio, Niosi, Novak, Fallon, Barber, Smith, Rostami-Hodjegan, Goosen.
Footnotes
- Received May 10, 2017.
- Accepted July 31, 2017.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- AZT
- zidovudine
- CDCA
- chenodeoxycholic acid
- HLM
- human liver microsomes
- HPLC
- high-pressure liquid chromatography
- IVIVE
- in vitro-in vivo extrapolation
- LC
- liquid chromatography
- MRM
- multiple reaction monitoring
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetics
- QconCAT
- quantification concatamer
- REF
- relative expression factor
- SIL
- stable isotope labeled
- UGT
- uridine-5′-diphospho-glucuronosyltransferase
- UHPLC
- ultra-high-pressure liquid chromatography
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}