


Abstract
During the process of drug discovery, the pharmaceutical industry is faced with numerous challenges. One challenge is the successful prediction of the major routes of human clearance of new medications. For compounds cleared by metabolism, accurate predictions help provide an early risk assessment of their potential to exhibit significant interpatient differences in pharmacokinetics via routes of metabolism catalyzed by functionally polymorphic enzymes and/or clinically significant metabolic drug-drug interactions. This review details the most recent and emerging in vitro strategies used by drug metabolism and pharmacokinetic scientists to better determine rates and routes of metabolic clearance and how to translate these parameters to estimate the amount these routes contribute to overall clearance, commonly referred to as fraction metabolized. The enzymes covered in this review include cytochrome P450s together with other enzymatic pathways whose involvement in metabolic clearance has become increasingly important as efforts to mitigate cytochrome P450 clearance are successful. Advances in the prediction of the fraction metabolized include newly developed methods to differentiate CYP3A4 from the polymorphic enzyme CYP3A5, scaling tools for UDP-glucuronosyltranferase, and estimation of fraction metabolized for substrates of aldehyde oxidase.
Introduction
In an era where combination drug therapy to treat several conditions simultaneously is common, drug companies emphasize the need for optimal absorption, distribution, metabolism, and excretion (ADME) properties, with the purpose of optimization of efficacy and minimization of the risk of adverse events. This includes a proper assignment and an extensive understanding of the routes of metabolism to aid in the prediction of human pharmacokinetics, and to help avoid a potential “object drug” scenario when coadministration is likely. The attributes of the coadministered drug and/or the patient has the potential to increase the probability of a drug-drug interaction (DDI), i.e., enzyme inhibitor, inducer, polymorphic genotype. Hence, the greater the percentage attributed to a single metabolic route, the greater the potential for a DDI and possible “black box warning” being issued as part of the drug package insert. Furthermore, the pharmacokinetic implications of a single polymorphic enzyme being responsible for a majority of the metabolism of a drug may have either an efficacy (extensive metabolizers) or toxicological (poor metabolizers) impact on exposure. To address these concerns, early drug discovery teams strive for balanced metabolism across multiple enzymes and clearance mechanisms (hepatic, renal, biliary). These discovery efforts center on in vitro reaction phenotyping to support enhanced chemical design.
There is a general agreement among the major regulatory agencies that pharmaceutical companies should provide a characterization of the metabolic profile of a new chemical entity (NCE) and understand the enzymology of the major clearance mechanisms (reaction phenotyping) using various in vitro study tools (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf). Reaction phenotyping is the semiquantitative in vitro estimation of the relative contributions of specific drug-metabolizing enzymes to the metabolism of a test compound. The relative enzyme contributions quantified in these studies guide the need for appropriate clinical DDI studies, impact polymorphic genomic status for a drug-metabolizing enzyme, and in some cases may be affected by organ impairment. Regulatory guidance has recommended that if a specific clearance mechanism is responsible for 25% or more of a drug’s elimination, in vivo assessment is required to understand the clinical impact, either through DDI, pharmacogenomics, or disease state [U.S. Food and Drug Administration (USFDA), 2012]. However, the current thinking of most preclinical ADME scientists is that when in vitro phenotyping studies estimate a single route of clearance contributing to >10% of the total metabolism, a complete kinetic study should be used to aid in predicting the clinical impact. However, definitive values can only be determined following completion of the human in vivo 14C-radiolabeled study in which the fraction of dose excreted as metabolites can be determined and assigned to a primary clearance pathway.
Given that in vivo 14C-labeled human ADME studies often occur late in the development life cycle of a drug due to cost and required resources, companies generally embark on a more pragmatic approach to estimate the fraction metabolized (fm) for major pathways, utilizing reaction phenotyping methodologies. As such, there is an emphasis on having a semiquantitative and qualitative understanding of the major human clearance mechanism(s) involved. In the absence of in vivo human metabolism information, the use of human liver–derived in vitro systems (primary hepatocytes or liver homogenates) is recommended, as the majority of drug metabolism occurs in this organ.
There is a hierarchical strategy for investigating object DDI potential; one strategy is to control overall cost while providing data sufficient to protect patient safety in clinical trials. Therefore, before embarking on studies to identify enzymes involved in the metabolic fate of a NCE, one must first determine which metabolic reactions comprise the major clearance pathways (Table 1).
Major metabolic clearance mechanisms involved in the elimination of drugs
The criteria presented in Table 1 serve as a guide to whether the enzymes involved are oxidative, conjugative, hydrolytic, or reductive. For conjugation reactions, the identity of the conjugate (e.g., glucuronide, sulfate, etc.) will give direction as to which family of enzymes should be examined. For oxidative metabolic pathways, a number of approaches can be used to help identify the enzyme family involved (Table 2).
Recommended conditions and inhibitors useful in identifying the role of drug-metabolizing enzymes
In 2007, Zhang and coworkers presented an industrial perspective on understanding NCEs as victims of an interaction through the use of in vitro cytochrome P450 reaction phenotyping studies, which laid out the then-current strategies used to enable the design and priority of follow-up clinical studies to support regulatory filing (Zhang et al., 2007). The current article aims to present the most recent advances in the field of reaction phenotyping, with emphasis not only on cytochrome P450 (P450) but also on aldehyde oxidase (AO) and UDP-glucuronosyltransferases (UGTs).
Discussion
Cytochrome P450s
Most of the experience in the discovery and development setting has been focused on cytochrome P450, a superfamily of heme-containing proteins. In short, the focus on cytochrome P450s is due to their predominant role in drug metabolism. This attention has been rightfully positioned due to the significant number of drugs on the market being metabolized by this enzyme family (Williams et al., 2004; Wienkers and Heath, 2005; Lamb et al., 2007).
The human P450 enzymes can be classified into three categories based on the importance played in the metabolism of drugs:
Major P450 enzymes: CYP1A2, 2C8, 2C9, 2C19, 2D6, and 3A4. Of all the P450s, these six presently are involved in the majority of P450 catalyzed drug biotransformation reactions (Danielson, 2002; Niwa et al., 2009).
Cytochrome P450 enzymes of emerging importance: 3A5. This P450 enzyme has recently received more attention in the scientific literature, as its involvement in the metabolism of CYP3A substrates in vivo is more than previously thought (Hesselink et al., 2003; Patki et al., 2003; Huang et al., 2004; Li et al., 2012). Recent advances in tools to help delineate between CYP3A4- and 3A5-mediated metabolism have emerged and will be discussed in more detail.
Cytochrome P450 enzymes that have not been shown to have a large fm in drug clearance and/or metabolize a limited range of drug substrates: CYP1A1, 1B1, 2B6, 2A6, 2E1, 4A11, 2J2, etc. A number of these P450s are extrahepatic, and usually involved in the metabolism of endogenous substrates (Raccor and Kaspera, 2013).
A number of different approaches are well established to characterize the P450-mediated metabolic clearance of compounds, namely:
Examination of the metabolic reaction of interest in the absence and presence of cytochrome P450 isoform–specific chemical inhibitors and/or inhibitory antibodies.
Determination of whether heterologously expressed recombinant human cytochrome P450s (rhP450s) are able to catalyze the reaction of interest, and rate of metabolism (nanomole per minute per picomole of P450).
Correlation of the rate of a metabolite formed in the reaction of interest and comparing it to a particular cytochrome P450–specific marker activity across a panel of liver microsomal samples characterized for their activity levels of the various cytochrome P450 isoforms from individual donors.
The science underlying these tools has developed over the last two decades, reducing the need to use several approaches simultaneously to make unequivocal conclusions regarding the cytochrome P450 isoform responsible for the metabolism of a drug of interest. It should be noted that any system in isolation is inadequate for assigning a biotransformation reaction to a specific cytochrome P450. One should consider at least two approaches, with one of these being the use of selective inhibitors/inhibitory antibodies. If the interpretation of these two approaches is consistent, no further experimentation is required. If not, a third approach is necessary, with interpretation made based on agreement between two of the three approaches. However, a caveat with this strategy is the potential metabolic stability of the test compound. As chemists strive to synthesize compounds with reduced susceptibility to metabolism, standard phenotyping approaches become more qualitative and less quantitative, since turnover is too low for slowly metabolized compounds to delineate with any confidence the relative contributions from multiple enzymes.
Recent Advances in Strategies to Assess Oxidative Metabolizing Enzymes
High-quality chemical tools are available to determine the activity and inhibition of most of the major cytochrome P450s involved in xenobiotic metabolism. A number of reviews describe cytochrome P450 reaction phenotyping and substrate/inhibitor specificities of various P450 enzymes (Newton et al., 1995; Zhang et al., 2007; Harper and Brassil, 2008; Emoto et al., 2010). However, this review is not intended to restate this topic, but rather focus on emerging data. Of interest is a recent publication by Khojasteh and coworkers (2011), who provided a thorough review of the published data by examining the potency and selectivity of cytochrome P450 inhibitors (Table 3). Readers are directed to this review to obtain a more detailed analysis of the literature. In summary, their analysis showed that the most selective inhibitors available are furafylline for CYP1A2 (KI [inhibitor concentration at the 1/2 maximal inactivation rate] 0.6–3µM) and 2-phenyl-2-(1-piperidinyl)propane (PPP) for CYP2B6 (KI ∼ 6 µM), both of which are irreversible inhibitors requiring preincubation, whereas selective reversible inhibitors include montelukast for CYP2C8 (Ki 0.01–0.15 µM), sulfaphenazole for CYP2C9 (Ki [inhibitory constant] 0.12–0.7µM), (–)-N-3-benzyl-phenobarbital for CYP2C19 (Ki ∼ 0.08 µM), and quinidine for CYP2D6 (Ki 0.03–0.4 µM). For CYP2A6, tranylcypromine was shown to be the most widely used inhibitor (Ki 0.04–0.2 µM), but Khojasteh and coworkers (2011) reported that either 3-(pyridin-3-yl)-1H-(pyrazol-5-yl)methanamine (PPM) (Ki 0.3 µM) or 3-(2-methyl-1H-imidazol-1-yl)pyridine (MIP) (Ki 0.25 µM) could replace tranylcypromine as a more selective CYP2A6 inhibitor. Given that few drugs are metabolized by CYP2E1, quantification of its contribution to metabolism is not routine in phenotyping studies. However, among the inhibitors for this enzyme, 4-methylpyrazole appears to be selective. For CYP3A4, ketoconazole is widely used in phenotyping studies, although at best, any inhibition of metabolism is likely a composite of contributions from CYP3A4 and 3A5.
List of selective inhibitors of cytochrome P450 isoforms
Cytochrome P450s 3A4/3A5.
In humans, four closely related members of the 3A family have been reported (CYP3A4, CYP3A5, CYP3A7, and CYP3A43) (Daly, 2006). Of these, CYP3A43 is not considered important in drug metabolism, and as such will not be discussed in this review. In contrast, CYP3A4 is generally thought to be the predominant form expressed in the liver and intestine (Kawakami et al., 2011), and is the most important enzyme in drug metabolism. CYP3A5 is a polymorphically expressed enzyme with the frequency of the wild type, CYP3A5*1, being only 5–15% in the Caucasian population, 25–40% in various Asian ethnic groups, and approximately 40–60% in Africans and African Americans (Kuehl et al., 2001; Daly, 2006). CYP3A7 has been found to be predominantly a fetal-expressed enzyme, with expression levels shown to fall by 50% from week 13 to full-term pregnancy (Stevens et al., 2003), but which then remain constant during the first 6 months of infancy. The fluctuating CYP3A7 levels are an important consideration due to the emphasis as of late on pediatric investigation plans for drugs coming to the market. Expression of CYP3A7 in adult human liver has also been measured (Leeder et al., 2005). In most cases, adult levels are extremely low; however, there are exceptions. It has been found that for about 10% of the adult population, CYP3A7 abundance remains high in the intestine and liver due to multiple high-expression polymorphisms (CYP3A7*1B and *1C) (Burk et al., 2002).
There is some uncertainty with respect to the clinical significance of the CYP3A5 polymorphism. Some CYP3A substrates are reportedly affected by the CYP3A5 genotype, such as tacrolimus (Barry and Levine, 2010), verapamil (Jin et al., 2007), vincristine (Dennison et al., 2006, 2007, Santoro et al., 2013), and cyclosporine (Zhu et al., 2011). Tacrolimus is one example which shows clinical relevance of the CYP3A5 genotype worthy of further discussion. Tacrolimus is an immunosuppressant that is metabolized three times more efficiently by CYP3A5 compared with CYP3A4 (Barry and Levine, 2010). Hence, maintaining the required minimal tacrolimus trough concentrations in patients who express CYP3A5 variants *1/*1 and *1/*3 requires approximately twice the dose compared with patients with the *3/*3 (inactive) genotype (Iwasaki, 2007). Given the significant overlap in substrate specificity between CYP3A5 and CYP3A4, which share 84% similarity of their amino acid sequences (Liu et al., 2007), identification of a suitable in vitro tool to distinguish the enzymatic contributions of each CYP3A isoform has been problematic. As such, the ability to predict the clinical impact of the CYP3A5 genotype on the pharmacokinetics of a CYP3A substrate has suffered too. Given that many drugs are metabolized by CYP3A, having the appropriate in vitro tools to understand the relative roles of CYP3A4 and CYP3A5 is therefore highly desirable.
Mifepristone, Raloxifene, OSI-930, Azamulin, SR-9186, and CYP3cide.
The inability to distinguish between the contributions of CYP3A4 and CYP3A5 in vitro has led to treating both enzymes as a single entity, CYP3A. Hence, there has been uncertainty in the quantitative assessment of each isoform’s contribution to metabolism, which is a significant issue since CYP3A is a primary enzyme involved in the clearance of more than half of all drugs. A number of compounds have previously been reported to show a preference for inhibiting CYP3A4 relative to CYP3A5 (Fig. 1). In this regard, mifepristone has been reported as a selective CYP3A4 inactivator using a recombinant system (Soars et al., 2006). Recently, mifepristone was examined using both human liver microsomes (HLMs) and recombinant enzymes. It was found that mifepristone did not have suitable properties to use in reaction phenotyping of CYP3A4 versus CYP3A5 (M. A. Zientek et al., manuscript in preparation). In short, Zientek and colleagues found the inhibitory specificity of mifepristone was not universal among CYP3A substrates, and thus the usefulness of mifepristone as a tool in CYP3A phenotyping is limited. Raloxifene has also been described as inactivating CYP3A4 and not CYP3A5 (Pearson et al., 2007). It was hypothesized that CYP3A5 may exhibit reduced or minimal time dependent inactivation at comparable rates of raloxifene metabolism, leading to an enhanced potential for systemic exposure to bioactivated intermediates. Unfortunately, raloxifene is also known to inhibit other major cytochrome P450 enzymes, with IC50 values ranging from 0.39 to 4.21 μM (VandenBrink et al., 2012), limiting its application as an inhibitor to differentiate between CYP3A4 and CYP3A5.

Structures of selective CYP3A4 inhibitors from the literature.
Other compounds have been further characterized, but only a few have been fully assessed to provide confidence in their ability to differentiate CYP3A5 from CYP3A4 metabolism. Of the compounds that show promise but lack a full assessment are OSI-930 and azamulin. OSI-930 [3-((quinolin-4-ylmethyl)amino)-N-(4-(trifluoromethoxy)phenyl) thiophene-2-carboxamide] is an experimental anticancer agent which has been shown to inactivate CYP3A4 and not CYP3A5 (Lin et al., 2011). Studies therein demonstrated that OSI-930 was able to act as a time-dependent inhibitor of human CYP3A4 with a KI of 24 μM, a maximum inactivation rate (kinact) of 0.04 minute−1, and a partition ratio of approximately 23. The kinact/KI, a measure of the efficiency of a compound to act as a time-dependent inhibitor, was 0.0017 μM−1 ⋅ min−1. The inactivation was reported to be primarily due to heme modification rather than protein modification, and the inactivation required cytochrome b5. The authors also showed that this CYP3A4 inactivator exhibited no effect on CYP3A5 activity under the same conditions, even in the presence of cytochrome b5. Azamulin [14-O-(5-(2-amino-1,3,4-triazolyl)thioacetyl)-dihydromutilin] is an azole derivative of the pleuromutilin class of anti-infectives. The inhibitory potency of this compound toward 18 cytochrome P450s using HLMs or microsomes from insect cells expressing single isoforms has previously been reported (Stresser et al., 2004). In a competitive inhibition model, IC50 values for CYP3A (0.03–0.24 μM) were at least 50-fold lower than all other non-CYP3A enzymes. The IC50 value with rhCYP3A4 was 15-fold and 13-fold more potent than those of rhCYP3A5 and rhCYP3A7, respectively. Although both of these agents show potential promise, the experimental strategy followed in assessing their selectivity toward CYP3A4 or 3A5 had a significant shortcoming. In this regard, testing the putative selective inactivator was not performed using HLM from CYP3A5 expressers and nonexpressers. This lack of full characterization, we believe, makes it difficult to recommend any of these compounds to be used as a selective tool for phenotyping purposes.
SR-9186
Li and coworkers (2012) have recently reported the selectivity of 1-(4-imidazopyridinyl-7 phenyl)-3-(4′-cyanobiphenyl) urea (SR-9186) (Fig. 1). SR-9186 was optimized via structural refinement following initial screening, targeted to obtain >1000-fold selectivity for the inhibition of rhCYP3A4 versus rhCYP3A5. In vitro studies confirm selectivity with inhibitory IC50 values against three probes: midazolam to 1′ hydroxymidazolam, testosterone to 6β-hydroxytestosterone, and vincristine to vincristine M1 metabolite. IC50 values ranged from 4 to 38 nM for rhCYP3A4, and 0.36 to 1.5 μM for rhCYP3A5. The data represented a large concentration window in which CYP3A4 was inhibited >90%, but CYP3A5 less so (<20%). IC50 values for SR-9186 at low HLM concentration (<0.05 mg/ml) were equipotent to that found using recombinant enzymes. Increases in microsomal protein were found to impart a large reduction in potency owing to the effect of nonspecific microsomal binding.
Selectivity of SR-9186 was compared with that of ketoconazole toward other hepatic incubations containing 1 mg/ml HLM (chosen to mimic conditions common in phenotyping experiments). SR-9186 was studied at 2.5 μM, approximately 10-fold greater than the CYP3A4 IC50 in the presence of 1 mg/ml microsomal protein, and 1 μM ketoconazole on the basis of historical precedence. SR-9186 demonstrated selectivity over CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A5 greater than or equal to that of ketoconazole. Preincubation of SR-9186 with or without NADPH in HLMs did not alter the selectivity of SR-9186, thus indicating a reversible interaction (Li et al., 2012).
Since the use of multiple-donor (nonbiased) pools of HLMs is common for phenotyping purposes, and CYP3A5 expressers are found in a minority of the population, the effect of CYP3A5 is diluted in a pooled microsomal sample. This dilution translates to an artificial lack of the influence of CYP3A5 and a misrepresentation of this polymorphic enzyme. In testing the abilities of SR-9186, Li and coworkers (2012) examined whether the large differences between CYP3A4 and CYP3A5 inhibition observed using recombinant enzyme would translate to physiologic samples, through evaluations using individually genotyped donors, with *1/*1, *1/*3 and *3/*3 CYP3A5 polymorphisms. The maximal percent inhibition was highly significant, with individual donors lacking at least one CYP3A5*1 gene exhibiting greater maximum inhibition.
It was concluded that, for phenotyping experiments, 2.5 μM SR-9186 preferentially inhibited CYP3A4 in microsomal incubations. Moreover, depending on the concentration of microsomal protein used in the incubation, SR-9186 concentration may need to be adjusted. Of practical importance, the authors also stressed that, when dissolved in aqueous solutions, SR-9186 extensively binds to plastic, leading to large errors at low concentrations. This problem is circumvented using organic solvents or in the presence of proteins such as hepatic microsomes, and is of particular importance when dilutions are made. Dilutions should be made in organic solvents and directly transferred to the incubation.
CYP3cide
Walsky and colleagues (2012b) have shown that 1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine, or CYP3cide, is a potent, efficient, and specific time-dependent inactivator of human CYP3A4. CYP3cide demonstrated an inactivation efficiency (kinact/KI) of 3300–3800 ml·min−1·μmol−1 in HLM from donors of nonfunctioning CYP3A5 (*3/*3). This efficiency was shown to equate to an apparent KI between 420 and 480 nM with a maximal inactivation rate (kinact) equal to 1.6 minutes−1. A similar degree of potency was also seen using another CYP3A substrate: testosterone. The inactivation activity of CYP3cide was further illustrated with the determination of the partition ratio, which approached unity. This very low partition ratio indicates that, on average, CYP3cide inactivates the CYP3A4 enzyme every other time CYP3cide is metabolized.
It is recommended that, for incubation of HLM, CYP3cide be used at between 0.25 and 2.5 μM in the presence of NADPH (for 3–5 minutes), followed by dilution into an incubation mixture containing the test compound. Data should then be compared with those of a control (no CYP3cide) and an incubation containing ketoconazole at a standard concentration (usually ∼1 μM). A method without the dilution step was also proposed to investigate compounds that have extremely low microsomal clearance where higher concentrations of enzyme (e.g., up to 10 mg/ml) might be required to discern the CYP3A4 contribution to metabolism. Under such conditions CYP3cide would need to be incubated at a higher concentration, taking into account the microsomal binding and thus ensuring a free concentration of approximately 0.5 μM (experimental unbound fraction in microsomes (fu,mic) at 0.8 mg/ml = 0.56).
CYP3cide and SR-9186 highlight a significant advancement in being able to differentiate between CYP3A4 and CYP3A5 activities, potentially helping elucidate the individual contributions of the two enzymes in vivo. We believe that the strategic use of SR-9186 or CYP3cide in reaction phenotyping would follow a standard cytochrome P450 reaction phenotyping study investigating metabolism by rhP450 enzymes in combination with HLM in the presence of P450-selective inhibitors (Fig. 2). If after these two studies it is shown that the NCE can be metabolized by rhCYP3A5, and ketoconazole inhibits metabolism in HLM, then a follow-up experiment focused on the CYP3A4 versus CYP3A5 contribution using CYP3cide should be undertaken comparing polymorphic CYP3A5 HLM pools (CYP3A5 *1/*1 versus *3/*3). If one or the other of the metabolic routes is subsequently found to comprise greater than 25% of the total clearance, an appropriate clinical strategy would need to be established (Tseng et al., 2014). Factors to be considered would be inclusion/exclusion of patients with specific genotypes, comedications, and target populations in which expression of enzymes may differ.

Decision tree for the estimation of CYP3A contribution to the metabolism of a compound. kdeg, degradation constant.
Further Applications of Novel Cytochrome P450 3A Inhibitors.
The intrinsic stability of a compound will often dictate, especially during early drug discovery, whether P450 fm values are estimated using chemical inhibitors or rhP450s. In this context, rhP450s are often used when investigating more metabolically stable compounds. With the identification of more specific CYP3A inhibitors, the opportunity exists to optimize phenotyping approaches using rhP450s.
A prerequisite in being able to estimate an appropriate fm using rhP450s is the confidence in their scaling to the intrinsic hepatic clearance (CLint) in HLMs. The CLint per unit amount of cytochrome P450 (intrinsic activity or turnover number) varies between HLM and rhP450s (Dickins et al., 2007), which is attributed to differences in the concentrations of accessory proteins, the lipid microenvironment, and relative abundance of P450 isoform in each system (Tang and Stearns, 2001; Nakajima et al., 2002; Venkatakrishnan and Obach, 2005). In this regard, two approaches have been adopted to help bridge the gap between drug catalytic rates observed in rhP450s and HLMs. The first approach uses a relative activity factor (RAF), which defines the amount of rhP450 required to give an equivalent reaction velocity to that of the particular HLM sample (Crespi, 1995; Proctor et al., 2004). However, this approach does not address interindividual variability in P450 expression, nor the apparent substrate specificity of RAFs. This may be overcome through another approach, the use of intersystem extrapolation factors (ISEFs), which compares the intrinsic activities of rhP450 versus liver microsomes and provides P450 abundance scaling by mathematical means. This latter approach uses the RAF and adjusts for the actual amount of liver microsomes cytochrome P450 isoform presents (Proctor et al., 2004).
Numerous publications have presented the utility of each approach, and consequently, a detailed overview falls outside the scope of the current review. Nonetheless, it should be noted that observations from different laboratories have demonstrated the benefit of RAFs (Galetin et al., 2004; Stringer et al., 2009), whereas others have shown an improved approach through the use of ISEFs (Youdim et al., 2008; Chen et al., 2011; Crewe et al., 2011). The advantages of one or the other of the approaches seem very much dependent upon the laboratory conditions: HLM pools and expression systems used, experimental approach (e.g., metabolite formation or substrate depletion), and the probe substrate(s) used.
Given the major role played by CYP3A in metabolic clearance of many drugs, an opportunity exists through the use of the newly reported, more specific CYP3A inhibitors to establish more reliable RAFs and ISEFs. This could also provide an opportunity to better delineate between the two mathematical approaches. Currently, CYP3A probes such as midazolam and testosterone are routinely used to estimate an RAF or ISEF, enabling scaling of rhCYP3A4 CLint to that in HLM, from which an estimation of CYP3A4 contribution can be determined. However, given that the metabolism of CYP3A substrates is mediated by both CYP3A4 and 3A5, it is unlikely that such values are totally accurate. In this regard, the aforementioned specific CYP3A4 inhibitors could be used to establish the appropriate Michaelis constant (Km) and maximum reaction rate (Vmax) kinetic parameters in HLM for both the CYP3A4 and 3A5 reactions. Here, the CYP3A5 parameters would be those estimated in HLM in the presence of the inhibitors and those for CYP3A4 determined from the difference between the HLM with no inhibitor and that with inhibitors. This is clearly an area where further research is required.
Cytochrome P450 2C9.
CYP2C9 is one of the most abundant P450 enzymes in the human liver, accounting for up to 20% of the total hepatic P450 protein content and 15–20% of P450-mediated xenobiotic metabolism (Ahlstrom et al., 2007). CYP2C9 is highly polymorphic, the most common variant being wild-type allele CYP2C9*1. The most well studied and clinically significant allelic variant is CYP2C9*3, with 0.4% of white individuals being homozygous carriers of CYP2C9*3 and 15% being heterozygous (Wang et al., 2009). Consequently, individuals expressing CYP2C9*1/*3 and CYP2C9*3/*3 demonstrate significantly reduced oral clearances of commonly used drugs of between 40 and 50% and 70 and 80%, respectively (Kumar et al., 2008). These polymorphisms also influence the extent of a DDI (Kumar et al., 2008). In vitro studies to date have had to rely heavily on rhCYP2C9 for evaluation of CYP2C9 polymorphisms, due to the limited availability of human liver preparations from individuals with CYP2C9*1/*3 and CYP2C9*3/*3. Although such systems allow characterization of the allelic variants, they lack a true physiologic environment and do not allow study of the contributions of other enzymes to total metabolism. As such, pooled HLMs which contain oxidative P450 enzymes remain an important in vitro tool for simultaneous examination of drug effects on multiple P450 pathways. However, only a limited supply of HLMs that express CYP2C9 allelic variants exist for genotype-dependent studies. Therefore, having a reliable in vitro model system to help understand CYP2C9 polymorphisms would be valuable.
To achieve a tool to investigate CYP2C9 polymorphisms, Flora and Tracy (2012) exploited the mechanism-based inactivation property of the thiophene derivative tienilic acid (Hutzler et al., 2009). They suggested that the total reduction in catalytic activity would be the same regardless of whether this reduction derived from the presence of a CYP2C9*3 allele or tienilic acid–exposed CYP2C9 enzymes; the catalytic activity of CYP2C9 polymorphisms could be mimicked using tienilic acid in pooled HLMs. CYP2C9-mediated activity was initially measured following flurbiprofen to 4′-hydroxyflurbiprofen formation, in commercially available CYP2C9*3/*3 HLMs. In each case, 4′-hydroxyflurbiprofen formation was substantially lower than that observed in pooled HLMs. Using a standard experimental approach to assess time-based inhibition, tienilic acid was preincubated with HLM in the presence of NADPH for 30 minutes, after which the primary incubation was diluted 20-fold (2 mg/ml to 0.1 mg/ml) into an activity mix containing a relevant CYP2C9 probe substrate. This dilution step ensured the removal of any residual tienilic acid. Under these experimental conditions, CYP2C9 activity was reduced in a dose- and microsomal protein concentration–manner. Increasing the tienilic acid concentration increased the inactivation of CYP2C9, whereas increasing the protein concentration tended to decrease CYP2C9 inactivation at a given tienilic acid concentration. Of the three total protein concentrations, 0.1 mg/ml resulted in the most rapid loss of CYP2C9-mediated activity. A 5 μM tienilic acid concentration resulted in a loss of approximately half of the activity in the 0.1 mg/ml HLM group; 15 or 40 μM tienilic acid was necessary for the 0.2 and 0.5 mg/ml HLM groups, respectively. The tienilic acid titration curve determined at 0.1 mg/ml was used to estimate that 4 and 14 μM tienilic acid concentrations would be required to generate 40–50% and 70–80% losses of CYP2C9 activity and, therefore, to mimic the CYP2C9*1/*3 and CYP2C9*3/*3 genotypes, respectively. The authors also ensured that following an approach of this type would not affect other P450 enzymes. Metabolism of probe substrates for P450s 1A2, 2B6, 2C8, 2C19, 2D6, 2E1, and 3A4, respectively, were shown to be unaffected by tienilic acid in pooled HLMs, suggesting that the tienilic acid–based model is selective for CYP2C9. Although it has been shown that tienilic acid can be used to selectively decrease total CYP2C9 catalytic activity such that it resembles the net CYP2C9 catalytic activity observed in CYP2C9 polymorphic variants, this would rely on the test compound itself demonstrating a degree of metabolic turnover at the recommended 0.1 mg/ml microsomal protein concentration, which is lower than the 1 mg/ml protein concentration often used in most discovery phenotyping studies. One could envision applying this approach later in the development phase, where perhaps metabolite standards may be available permitting kinetic assessments to help delineate the impact of CYP2C9 polymorphic variants.
The previous section described experimental approaches performed under nonfortified protein conditions. However, several publications have previously reported the rate of metabolism of CYP2C9 substrates by HLMs increased following the addition of fatty acid–free serum albumin to incubations (Wang et al., 2002; Zhou et al., 2004). After accounting for binding to albumin, lower Km values were estimated for a number of prototypical substrates, such as phenytoin p-hydroxylation and tolbutamide tolymethyl-hydroxylation, in HLM incubations supplemented with serum albumin. This observation, subsequently referred to as the “albumin effect” by Rowland et al. (2008a), was due to the sequestration of long-chain unsaturated fatty acids (e.g., C18:1n-9, C18:2n-6, and C20:4n-6) by serum albumin. These fatty acids are released from the microsomal membrane during the course of an incubation or microsomal preparation, and were shown to act as potent competitive inhibitors of CYP2C9. The studies showed that bovine serum albumin (BSA) and essentially fatty acid–free human serum albumin reduced the Km values for phenytoin hydroxylation (based on unbound substrate concentration) HLMs and rhCYP2C9 by approximately 75%, with only a minor effect on Vmax. A mixture of arachidonic, linoleic, and oleic acids, at a concentration corresponding to 1/20 of the content of HLMs, doubled the Km for phenytoin hydroxylation by CYP2C9, without affecting Vmax. The overall impact was an increase in the in vitro intrinsic clearance, which resulted in more predictive in vitro–in vivo extrapolation (Rowland et al., 2008a).
In light of the albumin effect, when a contribution by rhCYP2C9 is observed during standard phenotyping studies, follow-up incubations should be considered in the presence of 2% fatty acid–free BSA. However, one must adopt a pragmatic approach when looking to conduct these studies, and to put the metabolic clearance into context with the overall clearance. If reaction phenotyping studies suggest a significant involvement of CYP2C9 (>60%) and that metabolism is a major clearance route, then there is potential for a clinical interaction. This is pertinent for compounds with a narrow safety margin owing to the likely greater exposure in 2C9 poor metabolizers; hence, additional in vitro studies would serve to support the clinical risk assessment. As such, studies in the presence of BSA are best considered where preliminary studies estimate contributions by CYP2C9 to be >25%, to better verify if CYP2C9 contributions are accurately estimated. Given that test compounds will have a decreased rate of apparent intrinsic clearance in the presence of BSA (due to the increased protein binding), the most appropriate phenotyping studies utilizing BSA are those with recombinantly expressed human cytochrome P450s. The impact of BSA’s scavenging of the inhibitory free fatty acids on overall clearance can only be estimated once corrected for the protein binding. Consequently, to best estimate the fm, HLM and rhP450 incubations would all need to include BSA. Moreover, if protein content differs between incubations, then a suitable correction for protein binding will need to be applied before estimating the fm. Last, as stated, CYP2C9/BSA experiments do not come without added complexities; therefore, it is suggested these experiments be reserved for more mechanistic understanding of the potential clinical risk of a particular compound or compound series.
Key Emerging Routes of Metabolism and Associated Phenotyping Tools
Oftentimes in early drug discovery, an initial assessment of hepatic clearance is conducted with NADPH-fortified human liver microsomes. When scaled to in vivo hepatic clearance, this method provides an early prediction of the fraction of drug extracted by the liver during first pass in humans. If the clearance is noted to be high in humans, this candidate has a high potential to be eliminated from further development. A known limitation of this early screening strategy is that this method only provides an understanding of P450-mediated, and perhaps flavin monooxygensase, clearance (depending on assay procedure). Therefore, these efforts to reduce P450-mediated clearance help to channel chemical synthesis into less-understood routes of human metabolism. In this regard, the drug metabolism and pharmacokinetic scientist has seen a marked increase in alternative routes of clearance for new chemical entities, especially in the discovery phase. These alternative routes include both the UDP-glucuronosyltransferases and, more often in recent times, aldehyde oxidase.
UDP-glucuronosyltransferases
UGTs are a superfamily of enzymes that catalyze the transfer of glucuronic acid to a drug molecule in the presence of the cofactor uridine diphosphate glucuronic acid (UDPGA), yielding a drug glucuronide conjugate. UGTs have been cited as a critical route of drug metabolism, second only to cytochrome P450 as the initial mechanism of metabolism (Williams et al., 2004; Wienkers and Heath, 2005). In 2004, Williams and coworkers identified UGTs as responsible for the metabolism of approximately 10% of the top 200 prescribed drugs (Williams et al., 2004). This is probably an underestimate due to limitations in in vitro methodology used in the past and the instability of glucuronide conjugates excreted in bile and eliminated in feces.
The site of glucuronidation is generally an electron-rich oxygen, sulfur, or nitrogen heteroatom. This superfamily of enzymes has isoform nomenclature similar to those found in the cytochrome P450 family. The subfamilies which are known to metabolize xenobiotics are mainly found in the 1A and the 2B groups. The isoforms deemed of most interest to drug metabolism due to polymorphic nature and broad substrate specificity by the USFDA and EMA are UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf). Enzyme expression of the majority of these enzymes is found on the luminal side of the endoplasmic reticulum. This location of expression is thought to impede the access of the UDPGA cofactor to the enzyme in vitro, and hence pore-forming reagents have been used to increase accessibility. Since the UGT-catalyzed reactions are becoming more recognized in drug metabolism, a noticeable increased effort has been observed in the literature to mimic the tools established for cytochrome P450s.
The speed at which one can establish an in vitro to in vivo extrapolation for UGTs is affected by hurdles which have impeded development of a universal phenotyping paradigm for the UGT family of enzymes. Of particular note is the well documented underprediction of in vivo hepatic clearance from in vitro human liver homogenates, and to a lesser extent human hepatocytes (Mistry and Houston, 1987; Boase and Miners, 2002; Soars et al., 2004; Ito and Houston, 2005; Riley et al., 2005; Miners et al., 2006; Kilford et al., 2009). The underpredictions may be attributed to any number of factors, including the influence of incubation conditions, dependence on a consistent purified enzyme source, a lack of an easy quantitation method to assess the abundance of the enzyme, a lack of specific substrates and/or identification of specific metabolites to establish RAF or ISEF calculations from human liver microsomes to recombinant human UDP-glucuronosyltransferases (rhUGTs), release of free fatty acids from the microsomal membrane which are potent inhibitors of certain UGT enzymes, and the observation of atypical glucuronidation kinetics. However, great strides have been made to lay the groundwork for a UGT phenotyping strategy similar to that for P450 enzymes, and this will be the focus of the subsequent UGT sections. In this context, a summary of some of the most appropriate specific substrates and inhibitors will be discussed for both the correlation analysis and chemical inhibition phenotyping. Moreover, efforts to quantify the abundance of UGTs in human tissues will be explored and a summary strategy will be presented for interpreting the fraction metabolized by the UGT family.
In Vitro UGT Assay Optimization
UGT in vitro assay conditions have been shown to impact the efficiency of many UGTs. The addition of albumin, saccharolactone, alamethicin, MgCl2, uridine-diphosphate-glucuronic acid trisodium salt (UDPGA), and choice of buffer has been investigated (Fisher et al., 2001; Boase and Miners, 2002; Court, 2004; Soars et al., 2004; Engtrakul et al., 2005; Mutlib et al., 2006; Rowland et al., 2008b, 2009; Manevski et al., 2011; Walsky et al., 2012a). The influence of resulting optimized conditions and reagents can be narrowed to a few key guidances. There is universal agreement that the key source of the transferred glucuronide (UDPGA) to the substrate should be included in excess at a concentration of 5 mM to initiate the in vitro reaction (Kilford et al., 2009; Walsky et al., 2012a). The literature also shows that Tris buffer is preferred to sodium phosphate buffer for a majority of UGTs to achieve the highest enzyme activity, as phosphorylation of UGTs will adversely affect the selectivity and enzyme activity (Basu et al., 2005), thus it is suggested that 100 mM Tris buffer pH 7.5 at 37°C be used for in vitro experiments.
Saccharolactone has been incorporated into the UGT reaction mixture to inhibit the ability of endogenous β-glucuronidases to remove the glucuronide from the substrate conjugate during the incubation. In some instances saccharolactone has been shown to preserve the glucuronide (Bauman et al., 2005). However, in other instances, saccharolactone did not increase glucuronide formation rates, and in fact, even lowered the glucuronide formation levels (Kaivosaari et al., 2008; Oleson and Court, 2008; Walsky et al., 2012a). It is the authors’ opinion and experience that saccharolactone is not necessary due to its potential detrimental effects; hence, it can be excluded from in vitro incubations. Nevertheless, a comparison of the cumulative metabolite formation rates to the parent depletion in a UGT cofactor–fortified reaction is warranted, and if the rates are not complimentary, the incubation should be repeated with the inclusion of saccharolactone.
Divalent metal ions such as MgCl2 are also commonly used (Boase and Miners, 2002; Court, 2004; Walsky et al., 2012a), with final concentrations ranging from 4 to 5 mM. These concentrations have been shown to increase UGT activity in both recombinant UGTs and human liver microsomes by up to 4-fold.
Another critical in vitro UGT reagent is alamethicin. Alamethicin is a pore-forming peptide which is thought to increase accessibility of the UDPGA cofactor and substrate to the luminal side of the endoplasmic reticulum. It has been shown to increase the activity of the UGT enzyme significantly (Fisher et al., 2001), and its inclusion is considered critical to produce enzyme activity for adequate sensitivity. When optimizing the concentration of alamethicin, Walsky and colleagues (2012a) noted that, at lower microsomal concentrations, a greater amount of alamethicin was needed to activate the enzyme than at higher microsomal concentrations. A concentration of 10 µg/ml alamethicin was suggested to provide adequate levels of pore formation for microsomal protein levels from 0.01 to 0.5 mg/ml. Interestingly, no increase in activity has been observed with the addition of alamethicin to recombinant UGTs expressed in insect-cell baculovirus (Kaivosaari et al., 2008; Walsky et al., 2012a).
One of the more recent discoveries influencing UGT activity is the inclusion of fatty acid–free BSA to the reaction buffer. Rowland and colleagues (2008b) were the first to indicate that the inclusion of BSA in the incubations of UGT1A9 and UGT2B7 enhances the clearance rates in vitro. More specifically, the addition of BSA to propofol and zidovudine as specific substrates decreased the observed Km without affecting Vmax (Rowland et al., 2008b, 2009). The reasoning was akin to that described for CYP2C9, whereby BSA was acting as a sink to bind free fatty acids released from the microsomal membranes during preparation, thus avoiding the competitive inhibition imparted by the fatty acids. Additional UGT/BSA studies conducted in a separate laboratory confirmed the effects of BSA on UGT2B7, and provided evidence that not only was the Km decreased, but the Vmax of UGT1A9 was also increased (Manevski et al., 2011, 2012).
More recently, the influence of BSA on additional UGTs has been tested. In these experiments, rhUGT1A7, 1A8, 1A10, 2A1, 2B15, and 2B17 expressed from baculovirus-infected insect cells were assessed with both selective and nonselective substrates (Manevski et al., 2013). Due to the use of two estradiol isomers as substrates and the observed high concentration-dependent binding of these isomers to BSA, the BSA concentration was reduced from the published 2% to 0.1%. The lower concentration of BSA had been shown to be sufficient to reduce the inhibitory effect of the free fatty acids (Manevski et al., 2011). The study concluded that each of the isoforms tested, with the exception of 2B17, showed a reduced Km and an increased Vmax. The increased Vmax effects observed with certain substrates indicate more complex interactions than just the removal of competing free fatty acids. Interestingly, UGT1A8 had a sharp drop in Km value with the rather large substrate 17β-estradiol, but no effect on Km was observed with much smaller substrates. In a similar experiment, Shiraga and colleagues showed kinetic benefit from low levels of BSA with UGT1A9 when evaluating darexaban. At 0.1% BSA, kinetic parameters similar to those obtained at 2% BSA were observed when corrected for the free fraction (Shiraga et al., 2012); however, the effects were most noticeable on the unbound Km and not the Vmax.
Certain UGTs have been found to be highly expressed in the kidneys and intestine, and consequently have been implicated as a major contributor toward the total clearance of UGT-cleared substrates (Takizawa et al., 2005; Dalvie et al., 2008). As such, there has been particular interest in the effects of BSA on these enzymes and whether their expression and the sequestration of free fatty acids liberated during microsomal preparation are affected similarly to that observed in the liver. One UGT exemplar is UGT1A9, which is highly expressed in the kidney. It has been reported that the expression levels of UGT1A9, on a picomole per milligram basis, are 3- to 4-fold higher than in the liver (Harbourt et al., 2012). Hence, it is not surprising that Gill and colleagues (2012) reported in vitro renal intrinsic clearance rates two times higher on a per gram of tissue level compared with liver for UGT1A9 when BSA was added to the incubation. Furthermore, when the scaled renal UGT clearance from kidney microsomes was added to hepatic UGT clearance, an improvement of the in vitro to in vivo extrapolation of glucuronidation clearance was observed. The inclusion of BSA brought 50% of the drugs tested to within 2-fold of the clinically derived values, when the in vitro data were scaled using tissue-specific scaling factors. The authors also state that the magnitude of effect on the clearance rates in each tissue may be influenced by the overall free fatty acid content of that tissue.
When the literature is summarized, the UGTs which kinetically benefit from the inclusion of BSA in the incubation reaction include 1A7, 1A8, 1A9 1A10, 2A1, 2B7, and 2B15, whereas UGT2B17, 1A1, 1A4, and 1A6 are affected to a much lesser extent, if at all (Manevski et al., 2011; Gill et al., 2012; Walsky et al., 2012a). On the practical side, when incorporating BSA into reactions to enhance UGT activity as well as the clinical predictability of clearance, it has been recommended that the free fraction in the incubation reaction be assessed in the presence of BSA via equilibrium dialysis, regardless of the tissue investigated (i.e., kidney, liver, or intestine) (Waters et al., 2008; Gill et al., 2012; Walsky et al., 2012a). Once this measurement is obtained, the unbound fraction from the incubation should be incorporated into the mathematical method to calculate clearance associated with UGTs in the liver, kidney, and gastrointestinal tract (Walsky et al., 2012a; Manevski et al., 2013). This mathematical correction of the free unbound clearance will then provide a better overall assessment of in vivo clearance prediction, and hence a more thorough assessment of the fraction metabolized via the UGT route(s) in relation to other routes of metabolism. Regardless, utilizing the minimum amount of BSA to decrease the free fatty acids produced during microsomal preparation may be the best method to follow. Therefore, the work which Manevski and colleagues have presented provides a good compromise in keeping the concentration of BSA down and achieving the goal of limiting the concentration of detrimental free fatty acid affecting the UGT, while still limiting the impact on the free concentration of drug. However, every UGT may not be at optimal activity at such low concentrations of BSA (personal communication with Kimberly Lapham, Pfizer Worldwide Research and Development), and therefore the BSA concentration may need to be increased depending on free fatty acid concentration in the source of the enzyme (HLM > sf9 cells > Escherichia coli) and the enzyme’s sensitivity to the free fatty acid exposure (Marheineke et al., 1998; Rowland et al., 2007, 2008a).
Basic UGT In Vitro Phenotyping Assays
As with P450, common UGT phenotyping assays consist of chemical inhibition for a specific UGT, the use of recombinant UGTs, and correlation analysis (Bauman et al., 2005; Court, 2005; Walsky et al., 2012a). The in vitro correlation analysis utilizes verified specific substrates for each isoform of UGT with a characterized bank of donor human liver microsome samples. In the presence of UGT cofactors, the rate of formation of a specific substrate’s glucuronide metabolite is compared linearly with the rate of formation of the compound of interest’s glucuronide conjugate. A nice example of the correlation method for UGTs has been published by Court (2005) (Fig. 3), who reported a significant (P < 0.0001, R2 = 0.97) correlation between formation of the S-oxazepam glucuronide, a specific UGT2B15 probe substrate, and S-lorazepam glucuronide. The correlation assay is most useful as a companion to the other methods mentioned rather than a stand-alone phenotyping tool due to the need for previous metabolite identification and analytical method development for each glucuronide. Instead of monitoring glucuronide formation, parent drug disappearance is followed using this approach when the compound has been found to be exclusively metabolized by a single UGT isoform and the rate of disappearance along with the UGT contribution can be compared with other metabolizing enzymes.
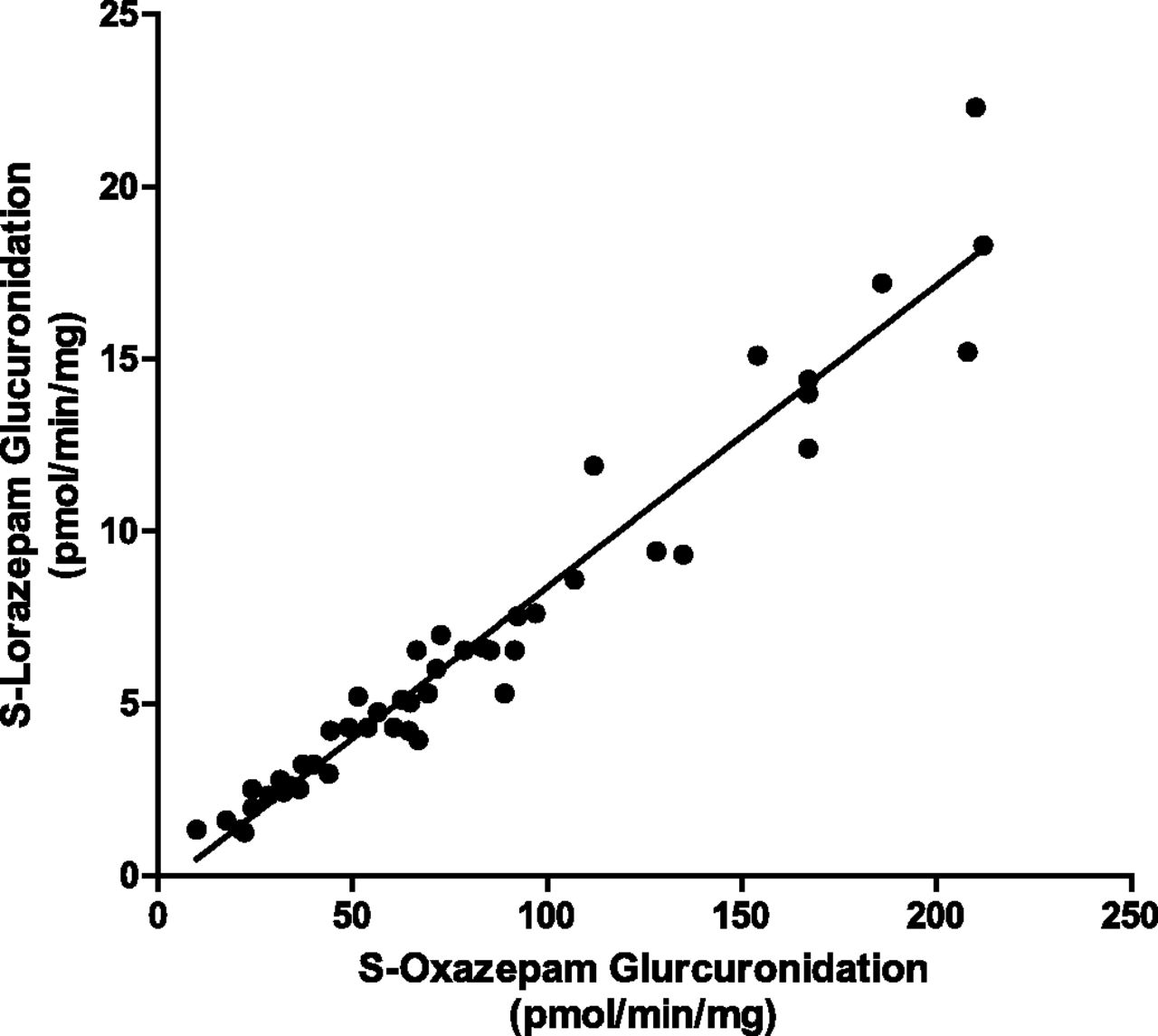
Example of a UGT2B15 glucuronidation correlation analysis of S-oxazepam versus S-lorazepam (P < 0.0001, Rs = 0.97), conducted by and with permission of M. H. Court and previously published in the Methods of Enzymology (Court, 2005).
To aid in the utilization of the correlation analysis, a list of some of the most well understood specific substrates and metabolites for each isoform can be found in Table 4. Not all of the substrates listed in the table are completely specific for the isoform noted, nor have they been completely vetted for all assay conditions (i.e., inclusion of BSA, buffer, optimal pH, etc.); therefore, some caution or preparatory validation work is recommended.
List of inhibitors and substrates of UGT isoforms
In 2012, Walsky and colleagues optimized kinetic assay conditions for multiple UGT-specific substrates in an effort to identify inhibitors of each UGT isoform (Walsky et al., 2012a). The assay conditions optimized included the use of albumin, saccharolactone, alamethicin, MgCl2, and the type of buffer used. Those UGTs and substrates which were optimized are noted in Table 4, have been shown elsewhere to be quite specific for each isoform, and are the preferred tool compounds for UGT phenotyping (Court et al., 2003; Krishnaswamy et al., 2004; Lepine et al., 2004; Soars et al., 2004; Court, 2005; Uchaipichat et al., 2006a; Itäaho et al., 2008; Miners et al., 2010).
UGT Phenotyping Using Chemical Inhibitors
As stated previously, chemical inhibition phenotyping has provided sufficient understanding of the fraction metabolized for cytochrome P450, and is widely accepted as one of the gold standards for reaction phenotyping. This method has also been acknowledged and accepted by both the FDA and EMA’s guidance (http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf). A next logical step for this method was, and still is, to expand it to UGTs.
The selectivity of a compound as an inhibitor is critical to its utility as a phenotyping tool. To be effective, the 90% inhibition concentration (IC90) of a tool inhibitor should be selective enough to show very little, if any, effect on other enzymes responsible for metabolism. Phenotypic testing at the IC90 allows for almost complete inhibition of the enzyme while providing for an understanding of the fraction metabolized through the decrease in the rate of the loss of parent versus time. Unfortunately, identification of specific UGT inhibitors is only slowly evolving. However, multiple publications in the literature have identified specific inhibitors (Table 4). The most specific UGT inhibitors have been reported for four of the UGT isoforms—UGT1A1, 1A4, 1A9, and 2B7—and the evidence for the inhibitors of these enzymes is discussed below. Although not always shown in the published result, a theoretical IC90 can be achieved by applying a three-parameter fit to the reported IC50 data. A few examples of this method have been provided as a reference value.
UGT1A1, a highly polymorphic enzyme, accommodates a wide array of chemical substrates. A number of chemical inhibitors have been identified that are useful for UGT phenotyping purposes, and among those, two stand out: erlotinib and atazanavir. Erlotinib has been reported to be a specific inhibitor of UGT1A1 with a Ki equal to 64 nM, and when tested at 100 µM, inhibited 4-methyl umbelliferone glucuronidation by almost 90% and was 5-fold selective compared with the other isoforms (Liu et al., 2010). Atazanavir, also identified as a UGT1A1 inhibitor, is both potent and specific for UGT1A1 with an IC50 equal to 1.9 µM. The reported mode of inhibition is thought to be through a linear mixed-model mechanism (Zhang et al., 2005). UGT1A4’s tendency to catalyze quaternary ammonium glucuronides helps to identify the utility and possible contribution of this isoform to metabolism of compounds. With this type of structural information, one can identify potential UGT1A4 substrates, and through the use of hecogenin in a substrate depletion reaction as a specific inhibitor of UGT1A4, one can confirm UGT1A4 contribution. It has been reported that the IC50 of hecogenin is equal to 1.5 µM (Uchaipichat et al., 2006a; Miners et al., 2010), and therefore, from this a theoretical IC90 of 15 µM is derived. UGT1A9 is mainly expressed in the kidney, but is also found in other tissues such as the colon, liver, ovaries, and testis (Green et al., 1995; Strassburg et al., 1998; Wiener et al., 2004). It is widely accepted that niflumic acid is a specific inhibitor of UGT1A9 with a potent Ki equal to 0.0275 µM (Mano et al., 2006; Gaganis et al., 2007), and theoretically an IC90 value of 0.2 µM.
UGT2B7 is thought to be perhaps the most important UGT in drug metabolism due to its diverse substrate specificity, which includes nonsteroidal anti-inflammatory drugs, opioids, and many other drug agents from many different therapeutic classes. Fluconazole is considered a competitive and specific inhibitor of UGT2B7; however, it is also a very good CYP3A4 inhibitor. To that end, great care needs to be taken to isolate each route of metabolism by using the appropriate cofactors (UDPGA versus NADPH) in human liver microsomes or human liver S9 fractions. The competitive Ki of fluconazole is equal to 143 µM in HLM and 73 µM in rhUGT2B7. A concentration of fluconazole above 1.3 mM in a UGT chemical inhibition study should be enough to provide a good estimate of the fraction metabolized for UGT2B7 in HLM with the appropriate cofactor. For the other UGT isoforms, a more in-depth assessment is needed for generalized use. With the establishment of more optimized assay conditions across a number of laboratories, more specific inhibitors will inevitably be published in the literature in the near future (Miners et al., 2010; Walsky et al., 2012a).
UGT Tissue Abundance and Estimating Fraction Metabolized from Recombinant UGTs
An accurate measure of tissue abundance provides the ability to scale measured activity in vitro to organs responsible for metabolizing a drug agent of interest. Without this abundance of information in tissues, it is impossible to quantitatively extrapolate relative contributions of rhUGTs to human tissue, similar to what has been demonstrated with the P450 enzymes. The abundance is also critical to scale beyond just a single organ, to whole-body clearance via physiologically based pharmacokinetic prediction, and thus providing an avenue for population variability assessment.
Historically, quantification of UGTs in tissue homogenates was assessed utilizing immunometric methods, such as enzyme-linked immunosorbent assay or Western blotting. Unfortunately, the results are disparate and nonquantitative due to the high sequence homology among UGTs leading to the lack of specificity of the antibodies used for immunoquantification. As of late, multiple laboratories have undertaken the initiative to use absolute quantitative proteomics using either heavy atom standards in conjunction with liquid chromatography–mass spectrometry (LC-MS) or S-tag fusion proteins as recombinant standards (Fallon et al., 2008, 2013; Milne et al., 2011; Harbourt et al., 2012; Sato et al., 2012; Schaefer et al., 2012; Sridar et al., 2013). Both methods seem to have specific requirements to ensure success. The heavy atom standard method is in need of multiple items to be successful: 1) purified protein source from an expression system, 2) a complete trypsin digestion of the enzyme, and 3) a highly sensitive instrument. In contrast, the S-tag fusion protein method needs very specific antibodies for accurate quantification, and the method is also reliant on sensitivity similar to Western analysis. Obtaining accurate quantification with either Western analysis or the S-tag fusion protein method suffers from a similar issue: incomplete protein solubilization. This incomplete solubilization is especially common with any hydrophobic membrane proteins, such as UGTs. The issue results in a wide range of protein quantification results from one laboratory to another, with only minor modifications in the method used to solubilize the protein. Also noteworthy are the laboratory differences in the subcellular fractionation approaches that affect the protein recovery parameters used to scale up to organ level. Fortunately, great strides have been made in the use of a heavy atom in quantification due to procedure modifications. Keys to this success include decreased peptide loss during sample preparation and efforts to ensure complete trypsin digestion. Other fixes include the use of the stable isotope–labeled peptides as internal standards for the assay during the digestion of proteins and the use of multiple specific heavy atom internal standards to verify specificity among abundance assessments.
The majority of the published UGT abundance data in the literature have been attributed to two laboratories. Those laboratories are based at The University of North Carolina at Chapel Hill and Tohoku University in Japan, both of which have used a heavy isotope internal standard to achieve results. For many of the isoforms tested at both of these two laboratories, some of the same stable isotope–labeled peptides were used as internal standards, and thus it is not surprising that similar abundances were observed. A summary of the quantification data available in the literature is presented in Table 5.
Summary of literature-derived UGT abundances in the liver, intestine, and kidney
When one scrutinizes the data, there are noticeable differences in the magnitude of the abundance for which enzymes have been tested using both the heavy atom standards in conjunction with LC-MS and S-tag fusion proteins as recombinant standards. Both UGT1A1 and UGT1A6 have been tested in both methods, where the S-tag fusion protein method when scaled places the abundance at the 0.1–0.5 pmol/mg of microsomal protein, whereas the heavy isotope method quantifies a 3–120 pmol/mg of microsomal protein. The magnitude difference in abundance is not all that surprising given the numerous sources of complicating factors, as stated previously. However, the overwhelming weight of data support the LC-MS method utilizing the heavy isotope as the method of choice to quantify UGTs at this time.
More recently, Achour and colleagues (2014) investigated another method to quantify UGT abundance. Their studies detail the results of a QconCAT (quantification concatamer) method to ascertain abundance quantification of both UGTs and P450s. This technology has the benefit and ability to quantify multiple enzymes at once via an internal standard of concatenated peptide sequences, which are trypsinized to match the cleavage site of the native protein. It was reported that, for the most part, abundance values were similar using the QconCAT method compared with that reported via liquid chromatography–tandem mass spectrometry (Achour et al., 2014).
One reassuring piece of evidence that consensus abundance values will be ascertained is that there are a number of assessments that have to be conducted with different internal standards, leading to similar abundance results. Although the median levels are only determined from a small number of studies, UGT1A1, 1A3, 1A6, 1A9, and 2B7 appear to have a reasonable overlap with a number of different heavy atom internal standards. The median abundances estimated are approximately 24, 10, 5, 26, and 58 pmol/mg of microsomal protein, respectively. However, due to the consensus values coming from these few assessments, caution should still be used when utilizing these data. Perhaps a more reasonable use of the data is to allow for a range of values obtained from Table 5.
As the availability of UGT abundance data from numerous laboratories increases, a putative methodology can be applied to understand the relationship between recombinant enzymes and human liver, intestinal, and kidney microsomes on a per-picomole basis. This relationship can be used for scaling purposes (for example, with use of ISEFs) and more importantly, for further understanding of the impact of population variability of a drug’s exposure in humans. The proteomics approaches have had particular utility with compounds which display extremely low clearance in vitro due to the now-understood low abundance of the responsible enzyme in human liver homogenate and the inability to push the protein level high enough without encountering detrimental effects from protein and lipid binding. This realization has provided the rhUGTs with a unique opportunity to lower overall protein levels, with greater abundance of UGT enzyme per milligram of protein, and potentially greater activity on a picomole basis to push the reaction to measurable clearance rates. These results can then, in turn, be scaled to in vivo clearance using the appropriate scaling factors and applied to the variability in the population. Of course, this in vitro ISEF relationship is enzyme source–specific for both the lot of recombinant enzyme and the corresponding pooled or single-donor human liver microsomes. As such, caution should also be used when using an ISEF among multiple vendors of recombinant enzymes and liver microsomes.
With the current methods and reagents available, a preliminary ISEF can be established for UGT1A1, 1A6, 1A9, and 2B7 due to the combination of the abundance and the availability of both rhUGTs and hepatic liver fractions matched with substrates and metabolite kinetics. In lieu of certainty of a UGT’s enzyme abundance, relative activity factors are still an option to connect the recombinant enzyme to those found in liver tissue fractions. Although these types of scaling factors do not allow for relating understanding of activity differences on the enzyme subunit level and the understanding of population variability, they do, however, allow for scaling recombinant enzymes to whole-organ clearance and provide a general prediction of clearance dictated by a particular lot of tissue fraction. Two nice examples of the use of RAF to extrapolate to whole-body clearance for UGTs were published by Gibson et al. (2013) and by Kato et al. (2012). Gibson and colleagues used RAFs to scale rhUGT1A1, 1A9, and 2B7 to whole-organ clearance of the liver and UGT1A9 and 2B7 in the kidney, whereas Kato et al. described RAFs for UGT1A4, 1A9, and 2B7 in the liver. Both groups describe their application of these derived RAFs to the quantitative involvement of each isoform to their respective compounds. Their work could be expanded to derive ISEFs, if the enzyme abundance data presented in Table 5 represent that of the pooled microsomal lot, and if the corresponding recombinant lot of enzyme abundance has been measured.
On a broader scale, obtaining the abundances and the methods used to generate these data provides the ability to perform physiologic-based pharmacokinetic predictions, which can be expanded to polymorphic populations and even the effects that disease states have on enzyme expression. Moreover, abundance evaluations can be extended to extrahepatic tissue to provide a baseline for all enzyme-expressing tissues, which currently are not well defined (i.e., individual section of the intestine and colon, etc). These data together will also help provide the ability to remedy the noted underpredictions observed from scaling of human liver fractions and hepatocytes alone. Additional work will have to be completed in the field to assess the most perfused tissues and the appropriate in vitro assay conditions to facilitate greater quality predictions of human UGT clearance.
When the three UGT phenotyping methods (chemical inhibition, recombinant enzymes, and the correlation analysis) are used together, a strategy is apparent based upon each method’s strengths and weaknesses, and is shown in Fig. 4. The strategy to identify UGT contribution starts with a simple HLM substrate depletion assay incorporating all of the necessary UGT reagents mentioned earlier in the In Vitro UGT Assay Optimization section. At this early stage, and due to the lack of understanding of which UGT is responsible for clearance, it is advisable to leave BSA out of the reaction due to the observed detrimental effect on affinity and CLint of certain UGTs (Walsky et al., 2012a). Once the UGT contribution is confirmed, a panel of recombinant enzymes is assayed to inform subsequent assessments and appropriate reagents to include. Both the chemical inhibition and recombinant UGT assays are then initiated to provide two separate assessments of the fraction metabolized via UDP-gucuronysltransferase (fm,(UGT)), and if the two methods are in agreement, no further assessment is needed. However, if a discrepancy is observed, the more labor-intensive correlation analysis utilizing the glucuronide metabolite is necessary to provide clarity.

Strategy to assess individual UGT isoforms contributing to total UGT clearance.
Aldehyde Oxidase
AO is a molybdenum-containing enzyme and is part of a large class of enzymes which incorporate molybdenum, a transition metal, and a flavin cofactor to enable catalytic activity. Interest in AO as a drug-metabolizing enzyme has elevated in the past few years. This interest is due mainly to two reasons. The first reason is attributed to the introduction of substituents, such as polar heterocycles or azaaromatics (e.g., pyridyl, pyrimidyl, quinolinyl), to reduce lipophilicity during early drug design in mitigating cytochrome P450 metabolism and to achieve the required metabolic stability in humans. Unfortunately, in some cases, these modifications render the new molecule vulnerable to AO metabolism (Rettie and Fisher, 1999; Pryde et al., 2010). The second and perhaps more important reason can be attributed to the necessary use in drug design of azaaromatic chemical scaffolds, which are required to achieve specific binding interactions at the site of a therapeutic target (i.e., kinases) (Knight and Shokat, 2005; Liu and Gray, 2006; Knight, 2010). This switch in metabolic routes from P450 to AO moves drug researchers from a well understood drug-metabolizing enzyme family to one where a testing strategy is much less established.
Aldehyde oxidase is different from P450 and UGTs, as it has only one recognized active isoform in humans. Therefore, the major emphasis is the recognition of aldehyde oxidase activity in the metabolism of a compound and placing it in perspective with the other recognized routes of metabolism. However, this does not change the major strategic methods one utilizes to establish the fraction metabolized via aldehyde oxidase [fm(AO)], chemical inhibition, and recombinant enzyme phenotyping. When assessing the fm(AO) of an NCE known to have multiple enzymes contributing to its hepatic metabolism, one needs to assess the AO contribution using a complete complement of human hepatic enzymes such as freshly isolated hepatocytes, cryopreserved hepatocytes, and liver S9 fractions.
There are certain challenges when assessing the fraction metabolized by AO in humans. These include greater than human hepatic blood flow clearances for AO substrates (indicating an extrahepatic component to AO clearance), the documented AO polymorphisms in the human populations (Hartmann et al., 2012) that affect the fm among subpopulations, and in vitro enzyme stability to extrapolate the impact and contribution to human clearance. Some of these challenges, where supporting data exist, are addressed in the following sections.
In Vitro Sources of AO Activity and Their Utility in Assessing Fraction Metabolized
Liver homogenates have been shown to have significant differences in activity between laboratories and manufacturers, and thus have fueled debates as to the underlying cause of the varying activity. The cause has, to a large extent, been attributed to the treatment of these sources during both preparation and storage. In a review, Garattini and Terao (2013) suggested that aldehyde oxidase 1 (AOX1) is a relatively unstable protein that after repeated freeze/thaw cycles rapidly becomes inactivated. However, contrary to this. Otwell and colleagues (2013) reported only a 12% change in AO activity following 10 freeze/thaw cycles of a cytosolic fraction. Otwell and colleagues also investigated the stability of AO activity over long-term storage at −70°C of different lots of pooled S9 liver fractions prepared from 2001 through 2011. Although different pooled donor lots were tested annually, activity was found to be within 2-fold across all lots tested, suggesting that these factors do not impact AO activity. If storage and freeze/thaw cycles are of little impact on activity of AO, one could attribute reported differences to preparation of the cellular fraction. Since no definitive data have been published on the impact of liver homogenate preparation on AO activity, the authors conducted a comparison of three different manufacturers of pooled donor liver S9 and cytosol (Fig. 5).

Comparison of aldehyde oxidase enzyme activity from donor-matched cytosol and S9 fractions obtained from three different manufacturers. The cross-hatched bars are from manufacturer A, the black and white diagonal lined bars are from manufacturer B, and the black and white horizontal bars are from manufacturer C.
Both human pooled liver S9 and cytosol were tested using three AO-specific probe substrates, as previously described (Zientek et al., 2010). The number of donors included in each manufacturer’s pooled liver fraction was 10, 50, and 150. Each source comprised a relatively equal mixture of both male and female donors. Large differences in activity were observed from manufacturer to manufacturer, and were assumed to be attributed to differences in the preparation of the liver fraction, enzyme denaturation, or attributes of the donors associated with each pool. The 50-donor pooled lot had the lowest activity (Fig. 5, manufacturer B), whereas activities of the 10-donor pool (Fig. 5, manufacturer A) were comparable to those of the largest pool with 150 donors (Fig. 5, manufacturer C). As the two largest pooled lots had quite disparate activities, yet contained enough donors to account for population variability (Hutzler et al., 2014), the observed difference likely originates during the preparation process. Therefore, when choosing a pooled source of enzyme, it is suggested that the overall enzyme activity should be assessed and considered to maximize sensitivity and dynamic range.
Purified recombinantly expressed AOX1 enzyme has also been used to understand AO metabolism. To date, commercial sources providing active recombinant expressed AO enzyme are limited. However, those laboratories which have expressed the enzyme, via either insect cells (Zhang et al., 2011) or bacterial systems (Alfaro et al., 2009; Hartmann et al., 2012), have observed active enzyme. Barr and coworkers (2013) compared a recombinant source of the enzyme prepared in their laboratory to that of a commercially available lot of human liver cytosol. Through the measurement of catalytic rate constant using absolute abundance of the enzyme, it was determined that the recombinant enzyme was far less active on a per-picomole basis compared with isolated human liver homogenates. It was hypothesized that the overexpression of the recombinant enzyme is compromised in the absence of corresponding cofactors, such as native sulfurase, which catalyzes the conversion of the oxo to the sulfido form of the molybdenum cofactor required for the activity of AO. Attempts have been made in other laboratories to coexpress the human sulferase with AO; unfortunately, these attempts have been unsuccessful at increasing active enzyme (Hartmann et al., 2012). Despite a greater amount of total protein needed to achieve realistic levels of activity compared with the native cytosolic enzyme, the recombinant enzyme can still provide a viable pure source for AO metabolism experiments. In this regard, the utility of aldehyde oxidase recombinant enzyme for fm(AO) assessments has progressed over recent years.
In two separate publications, the absolute quantification of liver AO was achieved using liquid chromatography–tandem mass spectrometry and stable labeled peptide standards. Unfortunately, the levels between the two methods vary greatly from 0.74–2.3 pmol/mg cytosolic protein (Fu et al., 2013) to 21–40 pmol/mg cytosolic protein (Barr et al., 2013). The discrepancy, whether due to enzyme source, preparation, or differences in the quantification method, warrants a number of further assessments to reach a consensus and provide a best practice for quantifying AO. Once completed, these best practices can be applied to both recombinant enzymes and cytosolic fractions to build intersystem extrapolation factors to scale clearance to human liver, similar to what has been achieved for P450 and UGTs. These absolute abundance methods also have utility to assess other tissues which are thought to significantly contribute to overall AO metabolism and systemic clearance. Hence, studies have reported AO expression in the kidney, pancreas, prostate, testis, ovary, and especially the respiratory system (Wright et al., 1995; Moriwaki et al., 2001; Yasuhara et al., 2005). AO has also been observed via immunostaining in the adrenal gland, cortex, and zona reticularis. Moreover, it has been reported that both the liver and adrenal glands have a similar abundance of AO and are the richest sources of AO mRNA transcript (Zientek et al., 2010). With the inclusion of the additional tissues and their impact on clearance rates well beyond human hepatic blood flow (∼20 ml/min per kg), compounds which have underpredicted clearance due to in vitro liver assessments alone could be accommodated in physiologically-based pharmacokinetic modeling.
In Vitro Considerations to Assess Aldehyde Oxidase Metabolism
The deviation of experimental conditions reported in the literature for AO investigations has been quite limited. This may be because, when fully assembled, AO is catalytically active without additional cofactors. Hence, AO activity can be measured purely through the addition of the compound to a mixture of enzyme in a buffered solution. The buffers of choice have consistently been either 100–250 mM potassium phosphate at pH 7.4, or 50 mM Tris buffer (pH 7.5). In the case of purified AO protein, up to 200 mM NaCl has been added to help keep the pure enzyme in solution and mimic physiologic conditions.
Of interest is the mixed use of EDTA as a chelating agent in the reaction milieu. With other metabolizing enzyme incubations (i.e., peroxidases and cytochrome P450), chelating agents have been used to reduce Fenton chemistry or the formation of free radicals with molecular oxygen and free metal ions (Mn2+, Mg 2+, Fe2+), and thus also reduce the misinterpretation of enzyme activity. Therefore, it is believed that the addition of EDTA to AO incubations has been a precautionary step, evidenced by the chemistry being quite different from P450 and peroxidases, and therefore AO would not be affected due to the effective nucelophilic attack. When comparing human cytosolic data for the AO substrate N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide (DACA) from two laboratories, one performed with EDTA (Barr et al., 2013; Choughule et al., 2013) and one without (Zientek et al., 2010), a 5-fold difference (340 vs. 63 pmol/min per mg) was observed. This difference could be explained through the effect of cytosolic protein and binding on clearance rates and/or the batch of human liver cytosol and its preparation. Due to this discrepancy, a direct comparison of the utility of EDTA is warranted.
For many enzymes, the addition of organic solvents to incubation conditions has detrimental effects (Chauret et al., 1998; Easterbrook et al., 2001; Zientek et al., 2008). However, the initial dilution step of the delivery of the compound to an in vitro reaction is often performed using dimethylsulfoxide. This decrease in activity does not seem to be very pronounced for human or other preclinical species using two different aldehyde oxidase substrates in two different laboratories (Obach et al., 2004; Choughule et al., 2013). Levels up to 1–2% have been shown to be compatible with retaining and sustaining AO activity. The effects of other more polar solvents, such as methanol, isopropanol, and acetonitrile, on AO activity have not been published.
Many laboratories in practice have moved toward human hepatocyte or human liver S9 to understand the fraction metabolized associated with AO compared with other routes of metabolism in the liver. Both hepatocyte and S9 fractions possess the full complement of soluble and membrane-bound liver metabolizing enzymes. Unfortunately, they only provide a surrogate of the hepatic drug metabolism, and as stated previously, will not provide information on extrahepatic clearance. With this shortcoming in mind, both matrices are still useful tools to assess hepatic fm(AO), using known specific AO inhibitors. Both raloxifene and menadione are potent uncompetitive inhibitors (Johns, 1967; Obach, 2004; Obach et al., 2004), however, neither is appropriate for use in determining fm(AO) in hepatocytes or S9 fractions because of one or a number of the following: 1) P450 inactivation (Pearson et al., 2007), 2) P450 reversible inhibition (Floreani and Carpenedo, 1990), and/or 3) cytotoxic effects (Price et al., 1996). Recently, Strelevitz and colleagues (2012) provided information on hydralazine as a selective inhibitor of aldehyde oxidase in human hepatocytes, and their work supports hydralazine having the appropriate inhibitory attributes to be used to determine fm(AO) for compounds. Hydralazine was shown to be a mix of competitive (IC50 = ∼5 µM) and time-dependent inhibitor (KI = 83 µM; kinact = 0.063/min) of human liver cytosolic aldehyde oxidase activity. They had proposed a concentration of 25–50 µM in human hepatocytes to selectively inhibit AO metabolism. These concentration levels were supported by monitoring the AO-specific formation of the 5-oxo-zaleplon metabolite, while not affecting the CYP3A-catalyzed zaleplon N-deethylation reaction. Also presented were the effects of the proposed concentrations (25–50 µM) of hydralazine on several drugs with multiple routes of metabolism. At the proposed concentrations, the data showed hydralazine’s selective inhibition of AO with only a mild, relatively inconsequential inhibition of CYP2D6. However, work performed by Brian Ogilvie’s group at Xenotech, LLC indicated hydralazine is a more potent inhibitor of CYP2D6 than first thought, with an IC50 of 18 µM in hepatocytes using dextromethorphan as a probe substrate and monitoring dextrorphan formation (B. Ogilvie, personal communication). Since the IC50 of hydralazine for CYP2D6 is similar to the IC90 obtained for AO by Strelevitz et al. (2012), fm(AO) experiments with CYP2D6 substrates should not use hydralazine.
Polymorphisms of Human AOX1
The work of Fu and colleagues (2013) identified a 3.4-fold range of enzyme abundance among 20 different cytosolic fractions, which could not explain a 90-fold difference in intrinsic clearance of the same cytosolic fractions. This result points to alternative possibilities as to why so much variability in activity is seen among single-donor cytosolic fractions. This in vitro donor-to-donor activity variability in humans (Sugihara et al., 1997; Al-Salmy, 2001; Sahi et al., 2008) may be driven, in part, by documented polymorphisms in the population (Hartmann et al., 2012). The activity variations and polymorphic effects have the potential to impact the interpretation of in vitro reactions using pooled liver fractions or hepatocyte, and thus the ability of drug metabolism and pharmacokinetic scientists to predict fm(AO). Thus, it is important to consider the impact of AO polymorphisms on fm(AO) and recent literature results in context with the available tools when working on compounds metabolized by AO.
Two studies in particular have investigated the potential for AO polymorphism in the population impacting activity, one by Hartmann and colleagues (2012) in which a sampling of genotyped Italian subjects provided information for site-directed mutagenesis in E. coli–expressed recombinant enzymes, and another by Hutzler and colleagues (2014) utilizing genotyped cryopreserved hepatocytes. The investigation by Hartmann et al. described single nucleic acid polymorphisms (SNPs) that resulted in multiple variants, each identified and recorded by frequency. The mutations resulted in the expression of one nonsense mutation (Y126stop), producing a truncated nonfunctional protein, five nonsynonymous SNPs (R802C, R921H, N1135S, S1271L, H1297R), and one synonymous SNP (L1268silent). AOX1 variants that had a high rate of frequency in the population (and thus may have an influence on human metabolism) were prepared and expressed via site-directed mutagenesis of the human AOX1 cDNA and heterologous expression in E. coli. Each clone was tested for activity, molybdenum saturation, terminal sulfide ligand association of the molybdenum cofactor–pterin complex, and characterized as dimeric or monomeric; they were then compared with the human wild-type enzyme prepared in the same way. Comparisons of activity resulted in subpopulations of AO phenotypes of poor metabolizer, extensive metabolizer (similar to wild type), and fast metabolizer. The poor metabolizer R921H was described as having a mutation of alanine to arginine. This mutation occurs in a highly conserved region in all members of the molybdenum-containing oxygenases, and with it, close proximity to the pterin molecule of the molybdenum-pterin cofactor may easily affect the catalytic turnover rate. The two fast metabolizers were identified as N1135S and H1297R, where up to a 2.5-fold increase in activity was observed with the recombinant enzymes compared with the wild-type enzyme. The activity differences in the recombinant enzymes alone do not explain the large variability observed in cytosolic liver fractions. However, along with the observed polymorphism, the increase or decrease in dimerization and/or the lack of conversion of the oxo- to the activating sulfide from the recombinant systems may explain the impact on activity observed in liver fractions.
Recently, Hutzler and colleagues (2014) investigated the fast metabolizer mutations (N1135S and H1297R) in 75 cryopreserved single-donor hepatocyte lots through genotyping, which was then compared with phenotyping by activity to assist in the understanding of population pharmacokinetic variability. Their results infer an influence similar to what Hartmann (2012) reported about these mutations. Hutzler (2014) reports a trend of these mutations towards having a higher enzyme activity compared with wild-type individuals; however, due to the large inherent variability of AO in the population and the lack of statistical power in 75 data points, detection of a statistically relevant difference could not be achieved. These results together with those of Hartmann provide some credibility that these identified polymorphisms do play a part in population variability, and might provide some understanding of populations at risk for over- or underexposure to an AO-mediated cleared compound. However, until there are statistically significant evaluations which are powered sufficiently to reveal a connection between polymorphisms, enzyme abundance, disease state, and enzyme activity, an in vitro strategy to quantify AO’s impact will not be possible and will continue to hamper the evaluation of fm(AO) beyond a general assessment.
Recommended Strategy to Assess the Fraction Metabolized by AO
In vitro assessments suggest, when utilizing any human-derived source of hepatic AO enzyme and scaling the resulting data, an underprediction of systemic clearance is most probable (Zientek et al., 2010; Akabane et al., 2012; Hutzler et al., 2012). Therefore, to minimize the underprediction associated with the AO clearance, donor tissues should be pooled to minimize misleading results, regardless of enzyme source (cytosol, S9 fraction, hepatocytes). Each donor to the pool should be evaluated for the range of activity, and a pool of donors which is optimized for representing a population should be prepared (Akabane et al., 2012). An alternate approach to obtaining a sufficient donor pool is to increase the number of donors which comprise the pool, hence limiting the effect of any low-activity individuals. Any attempt to assess the fraction metabolized by AO should never be conducted with a single-donor liver fraction due to its potential to have unrealistic levels to represent the larger population of AO activity. In practice, it is rare to have only one individual enzyme responsible for all of the metabolism of a compound; therefore, those liver fractions which contain a full complement of metabolizing enzymes are the most appropriate to discern AO contribution from other metabolizing enzymes. The recommended isolated liver fractions consist of either a pooled human liver S9 fraction (Zientek et al., 2010) or cryopreserved hepatocytes (Akabane et al., 2012; Hutzler et al., 2012). Once a reliable source of enzyme has been identified and multiple routes of clearance are suspected to contribute to the overall clearance of a compound, the only verified approach to assess hepatic fm(AO) is via the use of a specific inhibitor.
As stated previously, AO, when assembled properly, is fully functional, and no additional cofactors are needed; therefore, the experimental assay conditions are quite simple. Once preliminary studies in S9 or cytosolic liver fractions (Fig. 6) have identified AO as a contributing route of metabolism, more mechanistic studies can be adopted to ascertain hepatic fm(AO). These more mechanistic studies to assess the fm(AO) are initiated by the delivery of the compound to the reaction in the presence and absence of a specific inhibitor, such as hydralazine, and incubation at 37°C. Kinetic time points are taken via acquiring small aliquots from the reaction vessel throughout the course of the assay and quenched in 4°C polar organic (such as acetonitrile). After each time point is taken, parent depletion is measured via LC-MS, and an elimination rate constant is calculated. If multiple enzymatic pathways are being investigated, all appropriate cofactors and inhibitors will need to be included in the S9-driven reaction for separation of each enzyme’s contribution (Fig. 6). If the source of enzyme is from cryopreserved hepatocytes, a similar kinetic method can be used via replacing the Tris or potassium phosphate buffer used in the S9 fraction experiment for hepatocyte media (such as Williams’ E medium or InVitroGro KHB) after the hepatocytes have undergone the thawing process (Akabane et al., 2012; Hutzler et al., 2012, 2014; Strelevitz et al., 2012). Regardless of enzyme source, the uninhibited hepatic CLint is compared with the inhibition of the enzyme, and the fraction metabolized fm(AO) is then assigned in relation to other metabolizing enzymes responsible for the compound’s clearance (Strelevitz et al., 2012).

Strategy to assess aldehyde oxidase contribution to hepatic clearance. XO, xathine oxidase.
In summary, even for well established enzymes such as the cytochrome P450s, we continue to develop tools to identify specific risks with more precision. For P450, these improved tools have been identified as new and more specific inhibitors. Therefore, as our ability to predict the fraction metabolized by cytochrome P450 has grown, so has our ability to influence drug design, reducing involvement of this family of enzymes in total drug clearance. This knowledge has had both benefits and detriments. The obvious benefit is producing compounds with lower clearance due to P450s, and thus longer duration of action in the body. The detriment is that this understanding may lead to chemical design modifications which shunt the clearance to enzymes for which we are less prepared in our ability to conduct phenotyping and enzyme contribution experiments. These less understood enzymes include the UGT family and AO.
Although still evolving, great strides have been made in the better characterization of both UGT and AO drug-metabolizing enzymes, especially in the science involving the family of UGTs. Scientists have leveraged lessons learned in characterizing the P450s to accelerate the investigation of AO and UGTs, the latter currently indicated as the second most dominant drug-metabolizing enzyme system. Despite recent advances, however, a considerable amount of work still remains to increase the confidence in translation of in vitro estimates to those in the clinic.
This article has highlighted several drug-metabolizing enzyme families and the tools to probe and assess their overall contribution to metabolic drug clearance. The approaches used to investigate these different pathways share a commonality in the general tools and methods used. The optimized details of each enzyme pathway have been represented together with their limitations. Overall, through utilizing a complete system (liver S9 fraction or hepatocytes) and using optimized conditions for each pathway, a cumulative assessment of the fraction metabolized can be estimated for each metabolic enzyme.
Acknowledgments
The authors thank Drs. Deepak Dalvie, Theunis Goosen, Bill J. Smith, and Ellen Wu for their helpful comments and critical evaluation of this work.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Zientek, Youdim.
Footnotes
- Received April 26, 2014.
- Accepted October 8, 2014.
The authors have no conflicts of interest to declare and have not received compensation for the preparation of this manuscript.
Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- AO
- aldehyde oxidase
- BSA
- bovine serum albumin
- CYP3cide
- 1-methyl-3-[1-methyl-5-(4-methylphenyl)-1H-pyrazol-4-yl]-4-[(3S)-3-piperidin-1-ylpyrrolidin-1-yl]-1H-pyrazolo[3,4-d]pyrimidine
- DACA
- N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide
- DDI
- drug-drug interaction
- EMA
- European Medicines Agency
- HLM
- human liver microsome
- ISEF
- intersystem extrapolation factor
- LC-MS
- liquid chromatography–mass spectrometry
- MIP
- 3-(2-methyl-1H-imidazol-1-yl)pyridine
- NCE
- new chemical entity
- OSI-930
- 3-((quinolin-4-ylmethyl)amino)-N-(4-(trifluoromethoxy)phenyl) thiophene-2-carboxamide
- P450
- cytochrome P450
- PPM
- 3-(pyridin-3-yl)-1H-(pyrazol-5-yl)methanamine
- PPP
- 2-phenyl-2-(1-piperidinyl)propane
- RAF
- relative activity factor
- rhP450
- recombinant human cytochrome P450
- rhUGT
- recombinant human UDP-glucuronosyltransferase
- S9
- supernatant fraction 9000 g
- SNP
- single nucleic acid polymorphism
- SR-9186
- 1-(4-imidazopyridinyl-7 phenyl)-3-(4′-cyanobiphenyl) urea
- UDPGA
- uridine diphosphate glucuronic acid
- UGT
- UDP-glucuronosyltransferase
- USFDA
- U.S. Food and Drug Administration
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}