


Abstract
Simvastatin (SV) is a lactone prodrug used for the treatment of hypercholesterolemia. Upon incubation of SV with liver microsomal preparations from human donors, four major metabolic products were formed (3′-hydroxy SV, 6′-exomethylene SV, 3′,5′-dihydrodiol SV, and the active hydroxy acid, SVA), together with several minor unidentified metabolites. The 3′,5′-dihydrodiol SV, a new metabolite, was inactive as an inhibitor of HMG-CoA reductase. Kinetic studies of SV metabolism in human liver microsomes suggested that the major NADPH-dependent metabolites (3′-hydroxy SV, 6′-exomethylene SV, and 3′,5′-dihydrodiol SV) were formed with relatively high intrinsic clearances, consistent with the extensive metabolism of SV observed in vivo. Based on four different in vitro approaches, namely 1) correlation analysis, 2) chemical inhibition, 3) immunoinhibition, and 4) metabolism by recombinant human P450, it is concluded that CYP3A is the major enzyme subfamily responsible for the metabolism of SV by human liver microsomes. Both CYP3A4 and CYP3A5 were capable of catalyzing the formation of 3′,5′-dihydrodiol, 3′-hydroxy, and 6′-exomethylene metabolites. However, CYP3A4 exhibited higher affinity (> 3 fold) for SV than CYP3A5. Also, the studies indicated that CYP2D6, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP1A2, and CYP2E1 did not play significant roles in the metabolism of SV in vitro.
Over the concentration range of 0–40 μM, SV inhibited the activity of CYP3A, but not the activities of CYP2C8/9, CYP2C19, or CYP2D6 in human liver microsomes. The inhibition of hepatic midazolam 1′-hydroxylase, a CYP3A marker activity, by SV was competitive with aKi value of ∼10 μM. SV was > 30-fold less potent than ketoconazole and itraconazole as an inhibitor of CYP3A. Under the same conditions, SVA, the hydrophilic hydroxy acid form of SV, did not inhibit CYP3A, CYP2C8/9, CYP2C19, or CYP2D6 activities. The results suggested that the in vivoinhibitory effects of SV on the metabolism of CYP3A substrates likely would be less than those of ketoconazole and itraconazole at their respective therapeutic concentrations. In addition, metabolic activities mediated by the other P450 enzymes tested are unlikely to be affected by SV.
Simvastatin (SV, or Zocor, Merck & Co. Rahway, NJ )1 is used for the treatment of hypercholesterolemia (1). Following conversion of this lactone prodrug to its hydroxy acid form (SVA), the compound is a potent competitive inhibitor of HMG-CoA reductase, the rate limiting enzyme in cholesterol biosynthesis (2). As is the case with its close analog lovastatin, SV has undergone extensive metabolism to several oxidative products in a series of in vitro and in vivo studies in both animals and humans (3-5). Some of the hydroxy acid forms of these metabolites also are HMG-CoA reductase inhibitors (3). In a recent study, CYP3A was identified as the major enzyme responsible for lovastatin metabolism in human liver microsomes (6). To date, no studies have been reported regarding the in vitro metabolism of SV in human liver microsomes and the identity of the responsible enzymes.
In view of the widespread clinical use of SV and the importance of drug-drug interaction issues, we set out to identify the hepatic enzymes responsible for SV metabolism and to examine the effects of SV on hepatic P450 activities. In the studies described in this article, the in vitro metabolism of SV was characterized in human liver microsomal preparations and compared with that reported earlier from in vivo studies. A combination of four different strategies then was used to identify the responsible enzymes (7), namely 1) correlation analysis, 2) chemical inhibition, 3) immunoinhibition, and 4) metabolism by recombinant human P450. Potential inhibitory effects of SV and SVA, the only synthetic metabolite presently available, were examined using known P450 functional markers. Since SV was found to have inhibitory effects on CYP3A, we also conducted comparative studies using ketoconazole and itraconazole, which are known to act as P450 inhibitors in humans after therapeutic doses.
Materials and Methods
Materials.
SV, [14C]SV and SVA (fig.1) were synthesized at Merck Research Laboratories. Markers and chemical inhibitors of the cytochrome P450 family of enzymes were obtained from Sigma, St. Louis, MO (coumarin, 7′-hydroxycoumarin, chlorzoxazone, testosterone, tolbutamide, DDC, TAO, and quinidine); Steraloids, Wilton, NH (6β-hydroxytestosterone); Research Biochemical International, Natick, MA (6-hydroxychlorzoxazone and SKF-525A); Ultrafine, Manchester, UK (S-mephenytoin, 4′-hydroxymephenytoin, 3-methylhydroxytolbutamide, 1′-hydroxybufuralol and sulfaphenazole); Research Diagnostics, Inc., Flanders, NJ (itraconazole and ketoconazole); and Hoffman-La Roche Inc., Nutley, NJ ((+)-bufuralol, midazolam and 1′-hydroxymidazolam). Furafylline was synthesized at Merck Research Laboratories (Rahway, NJ).

Structures of SV and its metabolites.
* indicates the position of the [14C] label.
Polyclonal antibodies raised against rat CYP3A1, CYP2E1, and monoclonal antibodies for human CYP2A6 were obtained from Xenotech (Kansas City, KS), Oxygene Dallas (Dallas, TX), and Gentest (Woburn, MA), respectively. Human recombinant P450s were purchased from Gentest. All other reagents were of analytical or HPLC grade. Human liver microsomes were prepared from 11 individual subjects (Keystone Skin Bank, Exton, PA and University of New Mexico, Albuquerque, NM), as described previously (8).
In Vitro Metabolism of SV.
A typical incubation mixture, in a final volume of 0.5 ml, contained 0.2 mg liver microsomal protein, 50 μmol sodium phosphate buffer (pH 7.4), 5 μmol MgCl2, and 0.5 μmol NADPH. Unless otherwise specified, the reaction was started by the addition of [14C]SV (substrate) following a 3-min pre-incubation at 37°C, and the reaction was terminated after 10 min by the addition of 2 ml acetonitrile (ACN). The ACN extracts were evaporated and reconstituted for analysis by HPLC (see below). A preliminary experiment showed that the rates of formation of all metabolites were linear during the 10-min incubation period.
For the purpose of isolating SV metabolites for structural identification, incubations were performed using 2 ml incubation mixture and a 15-min incubation period.
Kinetic studies of SV were conducted using 2–100 μM SV in liver microsomal preparations from four different subjects. The experiments with known chemical inhibitors of P450 were performed at a substrate concentration of 100 μM and inhibitor concentrations of 25 μM quinidine, 50 μM furafylline or DDC, 100 μM sulfaphenazole or TAO, or 500 μM SKF-525A. At these concentrations, quinidine, furafylline, sulfaphenazole, and TAO have been demonstrated to be specific for CYP2D6, CYP1A2, CYP2C9, and CYP3A, respectively (9, 10). DDC was shown to inhibit CYP2E1, 2A6, and 2B6 (9-11). In experiments with furafylline, DDC, and TAO, the inhibitors were pre-incubated with liver microsomes and NADPH for 30 min at 37°C before adding the substrate. All other inhibitors were co-incubated with the substrate.
For immunoinhibition studies, microsomes were first incubated at room temperature for 20 min with antibodies or control sera at the ratios of incubation mixture to antibody volume shown to have maximum inhibitory effects (1 mg IgG/mg microsomal protein for preimmune serum and anti-2E1, 0.4 mg IgG/mg for anti-3A1, and 0.1 mg IgG/mg for anti-2A6). The reaction then was carried out as described above.
Incubations with human recombinant P450s were performed using the same conditions as described herein for human liver microsomes, except that the mixture contained 20 pmol P450 (CYP3A4, CYP3A5, CYP2C8, CYP2C9, and CYP2C19 supersomes) or 50–100 pmol P450 (CYP2E1, CYP2A6, CYP2D6, CYP2B6, and CYP1A2 microsomes), and was incubated for up to 40 min. Control incubations using microsomes isolated from the same cell line, containing the vector but without a cDNA insert, also were included. Kinetic studies of SV metabolism by CYP3A4 and CYP3A5 also were conducted using 20 pmol P450/incubation, 2–100 μM SV and 10-min incubation.
Assays for Enzyme Activities.
Assays described previously for CYP3A (testosterone 6β-hydroxylation), CYP2D6 (bufuralol 1′-hydroxylation), CYP2C8/9 (tolbutamide 3-methylhydroxylation), and CYP2E1 (chlorzoxazone 6-hydroxylation) activities (8, 9) were used for the present studies.
Activities of CYP2A6, CYP2C19, and CYP3A also were measured using coumarin (125 μM), S-mephenytoin (400 μM), and midazolam (5 μM), respectively, as marker substrates (11-13). Incubations of 200 μl containing 0.1 mg (coumarin) or 0.2 mg (S-mephenytoin and midazolam) microsomal protein in 100 mM sodium phosphate buffer were incubated with marker substrates at 37°C for 15 min (coumarin), 40 min (S-mephenytoin), or 5 min (midazolam), after the reaction was initiated with 1 mM NADPH. The reaction was stopped by the addition of 50 μl 30% perchloric acid on ice. After microcentrifugation, the supernatant was chromatographed on a C18 column (Waters, 150 × 3.9 mm, 5 μm), preceded by a C18 guard column. A linear gradient of ACN and 0.1% trifluoroacetic acid (12% ACN to 30% ACN in 8 min) was used for the midazolam assay. A mobile phase of 20% ACN in 0.1% trifluoroacetic was employed for the determination of S-mephenytoin. In the case of coumarin, a mixture of 32% ACN in 0.025 M potassium phosphate buffer, pH 4, was used. The eluate was monitored at 210 nm for midazolam andS-mephenytoin and at 313 nm for coumarin.
The inhibitory effects of SV and SVA on CYP activities were examined using 50 μM tolbutamide, 100 μM S-mephenytoin, 10 μM (+)-bufuralol, and 5 μM midazolam. SV or SVA was coincubated with these CYP marker substrates before the reaction was initiated with 1 mM NADPH. The substrate concentrations used were comparable with theirKm values reported elsewhere (13-16). To determine Ki values for the inhibition of midazolam metabolism by SV, SV (0–100 μM) and midazolam (0–100 μM) were coincubated for 5 min at 37°C. Comparative Ki values also were obtained for the inhibition of midazolam hydroxylation by ketoconazole (0–1.2 μM) and itraconazole (0–20 μM).
Analytical Procedures for SV.
An HPLC method was used for the simultaneous determination of SV and its metabolites. The samples were chromatographed on a C8-Zorbax Rx column (250 × 4.6 mm, 5 μm), preceded by a C8 guard column, with a linear gradient of ACN and 0.05% phosphoric acid (33% ACN to 75% ACN in 25 min). The eluate was monitored at 240 nm and by an on-line radioactivity detector.
For identification purposes, metabolites first were isolated using the HPLC conditions described above, except that 10 mM formic acid was used instead of 0.05% phosphoric acid. The isolated fractions, dried under a stream of N2, were subjected to further repurification steps using a mixture of 20–40% ACN in water. The purified metabolites were characterized by mass spectrometry (MS) and NMR spectroscopy. Mass spectral analyses were performed on a Finnigan TSQ 7000 triple quadruple mass spectrometer using electrospray ionization (ESI) in the positive mode, with a spray voltage of 4.5 kV and a capillary temperature at 230°C. MS/MS data were based on collision-induced dissociation (CID) of ions using argon gas at a pressure of 1.6 mtorr and collision energy of -35 eV. NMR spectra were obtained in CDCl3 at 30°C on a Varian Unity 400 MHz spectrometer. Chemical shifts are expressed in ppm using residual chloroform peak at 7.26 ppm.
HMG-CoA Reductase Inhibition Assay.
An enzymatic assay (17) was employed to evaluate the HMG-CoA reductase inhibitory activity of HPLC fractions of a microsomal incubate. An aliquot of each fraction was subjected to alkaline hydrolysis for determination of “total” inhibitors using SVA as a standard (4).
Data Analysis.
Apparent Km andVmax values were estimated using a nonlinear regression program (Enzfit from Biosoft, Ferguson, MO). The CLint was estimated by dividingVmax by Km . Discrimation between type of inhibition was based on visual inspection of 1) the double reciprocal plots of the data, and 2) pattern of changes in Km andVmax values in the presence and absence of inhibitors (18). Ki values then were estimated by fitting nontransformed data to the following equations using a nonlinear regression program (PCnonlin, Scientific Consulting, Apex, NC).
V = (Vmax × S)/[S + (Km (1 + I/Ki ))] for competitive inhibition, and V = (Vmax × S)/[S (1 + I/Ki ) + (Km (1 + I/Ki ))] for noncompetitive inhibition, where S and I represent substrate (midazolam) and inhibitor (SV, ketoconazole, or itraconazole) concentrations, respectively.
Results
Metabolism of SV in Human Liver Microsomes.
Fig. 2 illustrates typical chromatograms derived from incubates of human liver microsomes with SV in the presence of NADPH. Four major radioactive peaks (fig. 2A) and several minor metabolites were apparent (fig. 2B). Except for the hydroxy acid SVA, the formation of all other metabolites was dependent on the presence of both NADPH and microsomal protein. Among the four major metabolites, SVA, 6′-exomethylene SV, and 3′-hydroxy SV have been reported in earlier studies on the metabolism of SV by rat and mouse liver microsomes (3). SVA was characterized by comparing its HPLC retention time and UV spectrum with those of the corresponding reference compound. Identification of the 6′-exomethylene metabolite (fig. 1) was made largely on the basis of its UV spectral characteristics, i.e. λmax = 274 nm, and also by comparison of its HPLC retention time and mass spectral properties to those reported previously (3). Confirmation of the 3′-hydroxy product was made by NMR analysis, showing the presence of a new methyl singlet at 1.7 ppm, together with the absence of both the characteristic 6′-methyl doublet and the 3′ and 4′ vinyl proton signals (3).

A representative HPLC profile of SV metabolites in a human liver microsomal incubate.
Incubations were carried out at 37°C for 10 min, using human liver microsomes (0.4 mg/ml) and SV (100 μM) with NADPH (1 mM).
Identification of the 3′,5′-dihydrodiol SV (fig. 1), a new metabolite, was based on UV, LC-MS/MS, and NMR spectroscopy. This metabolite was devoid of UV absorbance at λ = 238 nm, which is the λmax of SV. As shown in figs. 2A and B, only the radioactivity, but not the UV signal (at λ = 240 nm) of the 3′,5′-dihydrodiol SV was measurable. LC-MS/MS analysis of the metabolite revealed, in its Q1 spectrum, a molecular ion (M+H+) at m/z 453. MS/MS spectrum of this ion displayed a family of product ions consistent with oxidation and hydration of the “hexahydronaphthalene” portion of SV (fig.3). It is proposed that the product ion at m/z 337 resulted from the loss of the ester side chain and that the ions at m/z 319, 301, 283, and 265 resulted from successive dehydration of the m/z 337 ion. The ions at m/z 241 and 215 were likely fragments of the m/z 301 ion, involving the hydroxylactone ring system (fig. 3). The definite identity of this metabolite (m/z 453) was based on NMR studies. The key features of the NMR spectrum of this metabolite were the absence of the characteristic 3′, 4′ vinyl proton signals and the presence of two new signals at 3.76 ppm and 3.84 ppm, diagnostic of CH attached to oxygen (table 1). The TOCSY spectrum showed that the signal at 3.76 ppm was associated with H-2′ and 2′-methyl protons, while the signal at 3.85 ppm was linked to H-6′ and the 6′-methyl group.
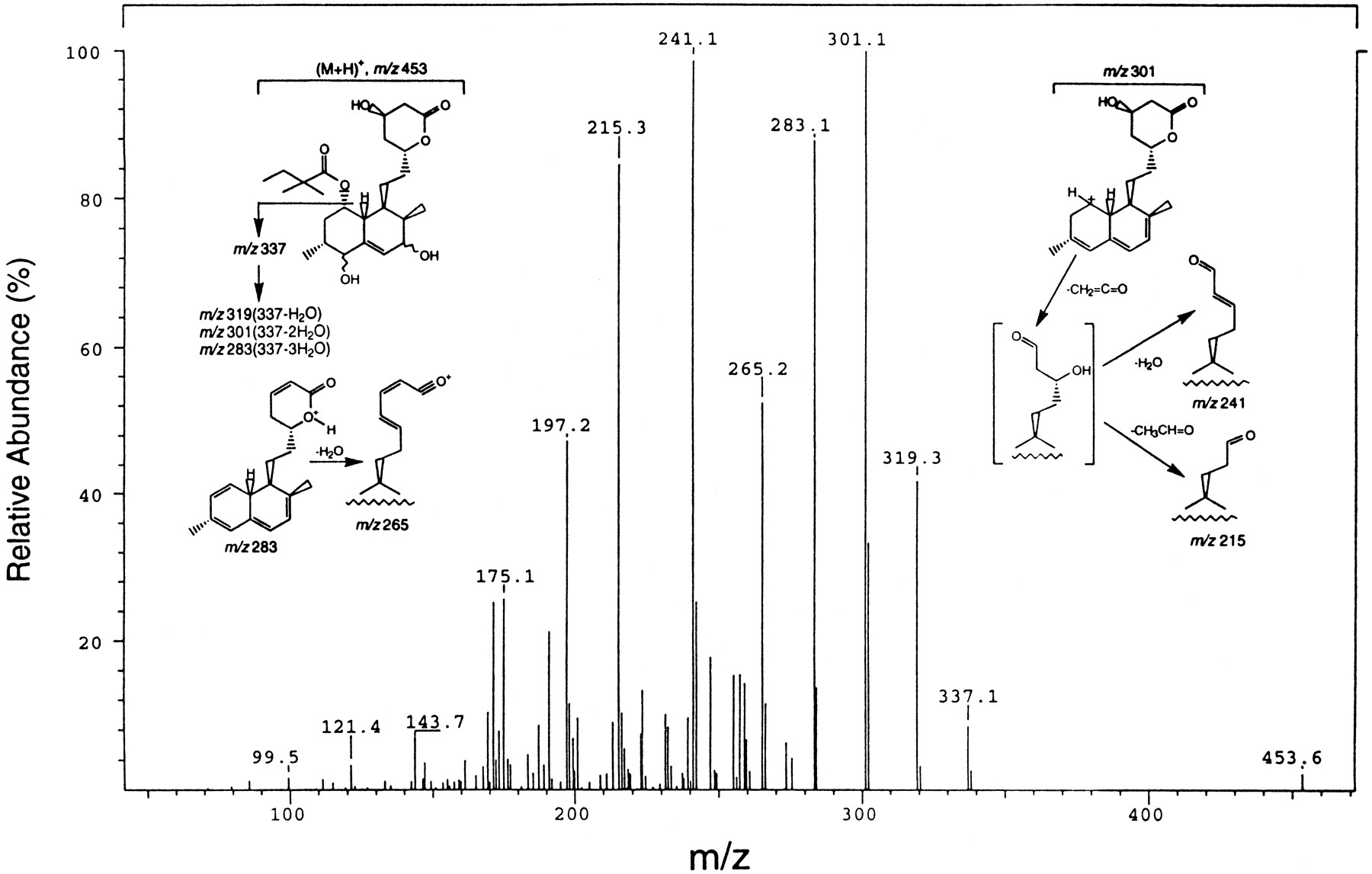
A LC-MS/MS spectrum of 3′,5′-dihydrodiol SV.
NMR parameters of 3′,5′-dihydrodiol SV 1-a
Bioactivity of SV Metabolites.
Since the 3′,5′-dihydrodiol SV was identified as a new metabolite, we examined its inhibitory activity against HMG-CoA reductase. The hydroxy acid form of the 3′,5′-dihydrodiol metabolite proved not to inhibit HMG-CoA reductase (< 5% of SVA activity). The 3′-hydroxy SV also showed minimal inhibitory activity, consistent with an earlier observation (3).
Kinetic Studies.
Kinetic studies of SV metabolism in human liver microsomes suggested that the three major NADPH-dependent metabolites were catalyzed by enzymes of comparable affinity and capacity (table2). Values of the CLint(Vmax/Km ) of metabolite formation were within 2-fold of each other (table 2). The total CLint calculated based on these metabolites was relatively high, consistent with the rapid decrease of SV in the incubations. Since SVA was formed by both chemical and enzymatic hydrolysis, its formation kinetics were not determined.
Kinetics of SV metabolite formation in human liver microsomes
Correlation Studies.
Table 3 summarizes results from correlation studies between SV metabolite formation and several CYP activities. The rates of formation of SV metabolites correlated well with each other (r2 > 0.98, p < 0.0001), suggesting that they might be catalyzed by the same enzyme. Of the P450 marker activities examined, only testosterone 6β-hydroxylase activity (CYP3A) showed a strong correlation (r2> 0.8, p < 0.0005) with total SV metabolism (measured as disappearance of SV) as well as with the formation of the 3′-hydroxy, 6′-exomethylene, and 3′, 5′-dihydrodiol products. The results suggested the possible involvement of CYP3A in these major metabolic pathways. A correlation (r2) of ∼ 0.5 was observed with tolbutamide 3-methylhydroxylase activity, a CYP2C8/9 marker. No correlation was found between SV metabolism and CYP2E1, CYP2C19, CYP2D6, or CYP2A6 marker activities (table 3).
Correlation (r2) between SV metabolism and CYP activities in human liver microsomes (N = 11)
Chemical Inhibition Studies.
Among the P450 inhibitors tested, SKF-525A, TAO, and DDC inhibited the formation of the 3′ hydroxy, 6′-exomethylene, and 3′,5′-dihydrodiol metabolites (fig. 4). The marked inhibition (> 50%) of SV metabolism by SKF-525A and the finding that SV metabolism was dependent on microsomal protein and NADPH suggested strongly that P450 was involved in the metabolism of this drug. The inhibition by TAO (25–70%) indicated the involvement of CYP3A, while that by DDC (≥ 45%) suggested a possible contribution of CYP2E1, CYP2A6, and possibly CYP2B6 in the metabolism (9-11). Quinidine, sulfaphenazole and furafylline did not affect the metabolism of SV (fig. 4), indicating that CYP2D6, CYP2C9, and CYP1A2, respectively, did not play a significant role.

Effects of chemical inhibitors of P450 on the metabolism of SV by human liver microsomes.
Results (mean ± SD) were based on triplicate determinations. All incubations were carried out in the absence or presence of inhibitors, as described in Materials and Methods, at 37°C for 10 min, using human liver microsomes (0.4 mg/ml) and SV (100 μM) with NADPH (1 mM). Control activities (in the absence of inhibitors) = 0.6, 0.3, and 0.6 nmol/min/mg protein for the formation of 3′-hydroxy SV, 6′-exomethylene SV and 3′,5′-dihydrodiol SV, respectively. * indicates statistically significant (p < 0.05) differences from controls.
Immunoinhibition Studies.
Immunoinhibition studies indicated strong inhibitory effects on total SV metabolism by anti-CYP3A, but not by anti-CYP2A6 or anti-CYP2E1 (fig. 5). Anti-CYP3A inhibited the formation of all SV metabolites almost completely, while anti-CYP2E1 inhibited only the formation of 3′-hydroxy SV and 6′-exomethylene SV by ∼20%. Under the conditions used, anti-CYP2A6 and anti-CYP2E1 strongly inhibited (> 80%) the formation of 7′-hydroxycoumarin and 6-hydroxychlorzoxazone, markers for CYP2A6 and CYP2E1 activities, respectively, in human liver microsomes (data not shown).

Effects of anti-P450 antibodies on the human liver microsomal metabolism of SV.
Results were based on duplicate determinations. Human liver microsomes (0.4 mg/ml), preincubated in the absence or presence of IgG, were incubated with SV (100 μM) and NADPH (1 mM), at 37°C for 10 min. Control activities (in the absence of IgG) = 1.5, 0.7 and 1.1 nmol/min/mg protein for the formation of 3′-hydroxy SV, 6′-exomethylene SV and 3′,5′-dihydrodiol SV, and 4.3 nmol/min/mg protein for the disapparance of SV, respectively.
Metabolism of SV by Recombinant P450 Isoforms.
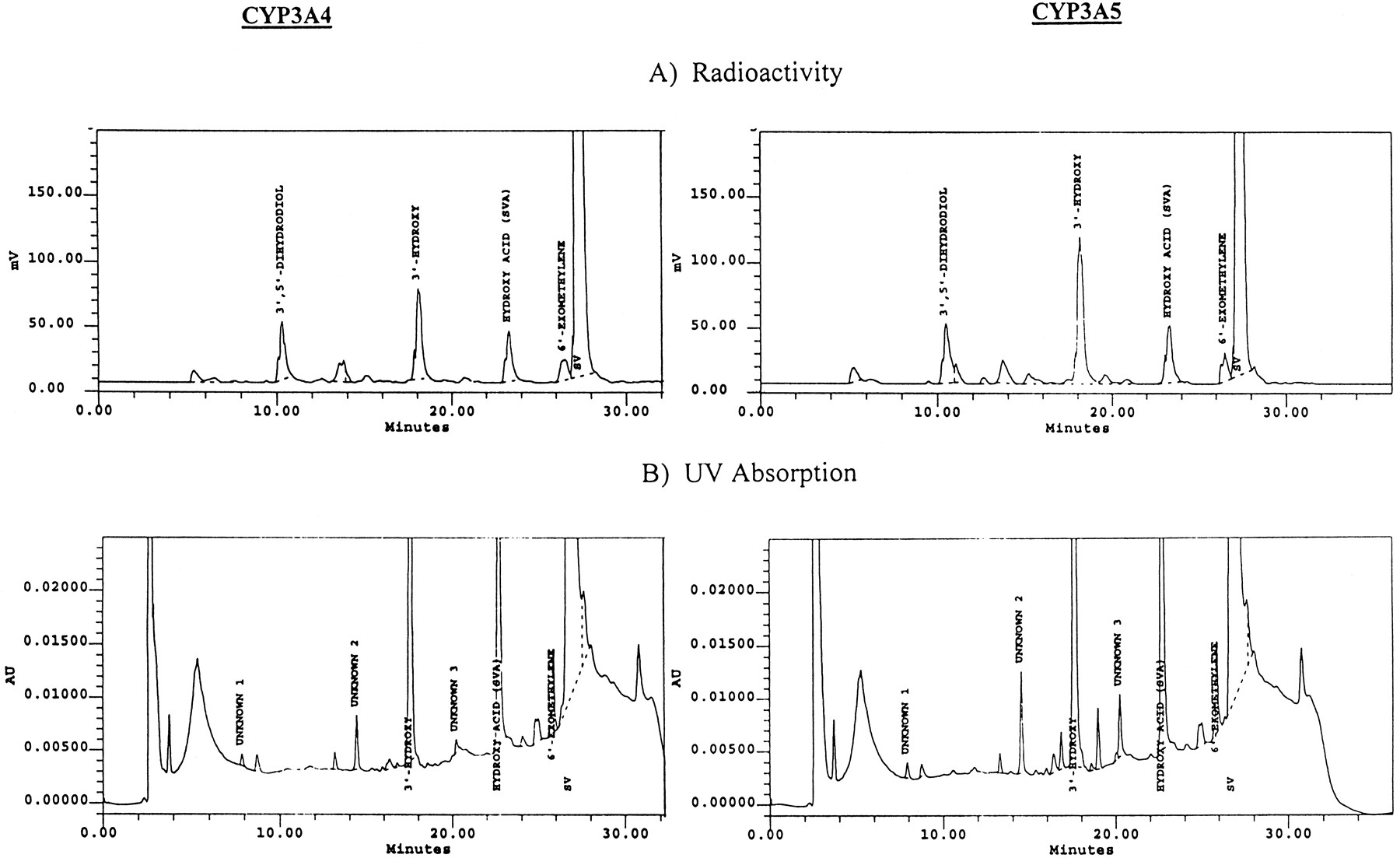
Studies of SV metabolism by human recombinant P450 isoforms indicated that both CYP3A4 and CYP3A5 metabolized SV efficiently (fig.6), further confirming the involvement of this enzyme subfamily in the metabolism of SV. All other P450s tested (CYP2D6, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP1A2, CYP2B6, and CYP2E1) and the control microsomes failed to catalyze the metabolism of SV to any appreciable extent (data not shown). Control experiments ensured that CYP2E1, CYP2A6, CYP2D6, CYP2C9, and CYP2C19 were catalytically active. CYP1A2, CYP2C8, and CYP2B6 were considered active based on the manufacturer’s claims.

HPLC profile of SV metabolites after incubation with recombinant CYP3A4 and CYP3A5.
Incubations were carried out at 37°C for 40 min, using CYP3A4 or CYP3A5 (20 pmol P450) and SV (100 μM) with NADPH (1 mM).
Further studies on the kinetics of SV metabolism by CYP3A4 and CYP3A5 indicated that CYP3A4 possessed relatively higher affinity (3–9 fold) than CYP3A5 in catalyzing the formation of the three major metabolites. The apparent Km ± SE values for formation of 3′,5′-dihydrodiol SV, 3′-hydroxy SV and 6′-exomethylene SV were 30 ± 6.4, 7 ± 2.9, and 25 ± 0.1 μM for CYP3A4 and 91 ± 41, 62 ± 0.9, and 88 ± 0.1 μM for CYP3A5, respectively. Interestingly, the apparentKm values obtained with CYP3A4 were similar to those obtained with human liver microsomes, especially subjects 2, 3, and 4 (table 2), suggesting that CYP3A4 was the major contributor to the metabolism of SV in these subjects.
Inhibitory Effects of SV on Hepatic CYP Activities.
The potential of SV to act as an inhibitor of CYP3A, CYP2C8/9, CYP2D6, and CYP2C19 activities was examined using midazolam, tolbutamide, (+)-bufuralol, and S-mephenytoin as marker substrates, respectively. SV, in a concentration range of 0–40 μM, inhibited only midazolam 1′-hydroxylase activity and did not affect other hepatic enzyme activities (fig. 7A). Under the same conditions, SVA exhibited minimal effects on CYP3A, CYP2C8/9, CYP2C19, and CYP2D6 activities (fig. 7B). The inhibition on hepatic CYP3A activity by SV was competitive (fig.8A), with aKi value of ∼10 μM. Similarly, the inhibition by ketoconazole and itraconazole was determined to be noncompetitive and competitive (figs. 8B and C), with Ki values of 0.1 and 0.3 μM, respectively.

Effects of SV (A) and SVA (B) on CYP activities in human liver microsomes.
Control activities were obtained in the absence of SV or SVA. Results (mean ± SD) were based on triplicate determinations.

Double reciprocal plots for inhibition of midazolam 1′-hydroxylase activity in human liver microsomes by A) SV, B) ketoconazole, and C) itraconazole.
The data points represent the average of duplicate determinations. All incubations were carried out in the absence or presence of SV (4–100 μM), ketoconazole (0.04–1.2 μM) or itraconazole (0.4–20 μM), at 37°C for 5 min, using human liver microsomes (1 mg/ml) and midazolam (4–100 μM) with NADPH (1 mM). V = 1′-hydroxymidazolam formation (pmol/min/mg) and S = midazolam concentration (μM).
Discussion
In the present study, the metabolism of SV was examined in vitro using human liver microsomal preparations. The 3′,5′-dihydrodiol SV, an inactive metabolite, has not been reported previously. This metabolite also has been identified upon incubation of SV with rat liver microsomal preparations (unpublished results). As was reported previously (3), the 3′-hydroxy SV is likely an acid catalyzed isomerization product of the metabolite 6′β-hydroxy SV (fig. 1), considering the present acidic HPLC conditions. A similar metabolic pattern has also been reported with lovastatin in human and rat liver microsomes (6, 19). With the exception of the 3′,5′-dihydrodiol, all SV metabolites have also been observed in the bile of humans treated with SV (3). In human bile, the 3′-hydroxy, 6′β-hydroxy and 6′-carboxy, a product of further metabolism of the 6′-exomethylene SV, were the major metabolites observed (3, 4). Apparently, the present in vitro results agreed well with these in vivo findings.
The relatively high total CLint of metabolite formation, calculated based on the three major NADPH-dependent metabolites of SV, was consistent with the in vivo finding that SV was metabolized to an appreciable extent in humans (4). Extrapolation of the CLint total yielded the hepatic clearance (∼20 ml/min/kg bw), which was comparable with the hepatic blood flow (20, 21). These results suggest that the liver is an important metabolizing organ for SV. Consequently, the results obtained from in vitro hepatic studies likely would be relevant to the in vivo outcome.
Based on four different in vitro approaches, namely correlation analysis, chemical inhibition, immunoinhibition, and metabolism by recombinant P450 isoforms, the present study indicated that CYP3A was the major enzyme subfamily involved in SV metabolism. A similar finding has been reported for lovastatin, a close analog of SV (6). The human CYP3A subfamily is known to be involved in the metabolism of structurally diverse xenobiotics (22). Among the CYP3A enzymes found in adult human livers, CYP3A4 accounts for ∼30% of the total P450 (23), while CYP3A5 was detected in only ∼25% of the samples examined (22). In the present study, both CYP3A4 and CYP3A5 catalyzed effectively the metabolism of SV, although relatively higher affinity was observed with CYP3A4. Similar observations also have been observed with midazolam metabolism (24, data not shown).
The results from chemical- and/or immuno-inhibition studies and those from experiments with recombinant P450 enzymes suggested that CYP2D6, CYP2C8/9, CYP1A2, CYP2E1, and CYP2A6 did not play a significant role in the metabolism of SV. Findings based on correlation studies and the lack of metabolism by recombinant CYP2C19 microsomes indicated that CYP2C19 was not involved in the metabolism of SV. Because of the marked inhibition by DDC, the reason for which presently is not known, the possible involvement of other isozymes in the metabolism of SV could not be ruled out completely.
SV was much less potent (> 30-fold) than ketoconazole or itraconazole in its ability to inhibit human hepatic midazolam 1′-hydroxylase activity. The Ki values of ketoconazole and itraconazole estimated in the present study were comparable with those reported earlier (18, 25, 26). Interestingly, ketoconazole and itraconazole, at therapeutic doses yielding plasma concentrations above their Ki values, have been reported to affect markedly the iv clearance of midazolam and/or the area under plasma concentration-time curves of midazolam obtained after oral administration (27, 28). These results are in good agreement with thein vitro data reported in this and in previous studies (18,25, 26). Considering that plasma concentrations of SV would be < 1 μM (4, 29), much less than its Ki value, and that SV is bound extensively (> 95%) to human plasma proteins (5), the inhibitory effects of SV on CYP3A substrates probably would be minimal as compared with those of ketoconazole and itraconazole. In addition, the inhibitory effects of the polar metabolite SVA were substantially lower than those observed with SV, further supporting this conclusion. The present in vitroresults were in line with those obtained earlier using various CYP3A substrate markers (30, 31). Nevertheless, the in vivoeffects of SV on pharmacokinetics of known CYP3A marker substrates remain to be investigated. In the present study, SV was found not to be an inhibitor of hepatic CYP2D6, CYP2C19, and CYP2C8/9 activities, suggesting that SV is not likely to inhibit the metabolism of substrates for these enzymes. The present findings do not support the recent speculation (32) of a possible interaction between SV and acenocoumarol, a known substrate for CYP2C9 (33).
To conclude, the present in vitro results indicate that SV undergoes extensive oxidative metabolism in human liver, in agreement with the in vivo findings. The major metabolizing enzyme subfamily was identified as CYP3A. CYP2D6, CYP2C8, CYP2C9, CYP1A2, CYP2E1, and CYP2A6 are not involved in the metabolism of SV. SV also was an inhibitor of CYP3A, but not of CYP2C8/9, CYP2C19, and CYP2D6. The inhibition of hepatic CYP3A activity was competitive and was much less pronounced than that observed with the known CYP3A inhibitors ketoconazole and itraconazole.
Acknowledgments
The authors are grateful to Drs. T. A. Baillie, A. H. Y. Lu, J. H. Lin, and J. D. Rogers for their support and invaluable suggestions. We thank Drs. A. Jones and D. Dean for synthesis and purification of [14C]SV, and Ms. D. Walsh for preparation of human liver microsomes.
Footnotes
-
Send reprint requests to: Thomayant Prueksaritanont, Ph.D., Department of Drug Metabolism, WP 26A-2044, Merck Research Laboratories, West Point, PA 19486.
- Abbreviations used are::
- SV
- simvastatin
- SVA
- the hydroxy acid form of simvastatin
- TAO
- troleandomycin
- DDC
- diethyldithiocarbamate
- CLint
- intrinsic clearance
- Received April 15, 1997.
- Accepted June 20, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}