


Abstract
Cytochrome P450 (CYP) 2E1 is implicated in a variety of chemically initiated hepatotoxicities, including alcoholic liver disease. These pathological conditions arise from increased production of reactive intermediates caused by elevated enzyme concentrations. Thus, the ability to detect enhanced CYP2E1 levels would aid in identifying individuals at high risk for xenobiotic-promoted liver injury. With this in mind, the present investigation assessed in vivochlorzoxazone metabolism and compared pharmacokinetic parameters with CYP2E1 expression in blood. Twenty-two subjects were recruited and divided into two groups, control subjects and alcohol abusers, based on responses to two screening questionnaires. Those individuals with higher survey scores, i.e. those who consumed alcohol more frequently, exhibited higher rates of chlorzoxazone metabolism. Indeed, a correlation (r = 0.66, p < 0.01) was obtained when scores were compared with the pharmacokinetic parameter AUC for chlorzoxazone. Lymphocyte microsomes isolated from blood samples obtained from these same individuals were subjected to immunoblot analyses to detect CYP2E1 levels. That lymphocytes contained CYP2E1 was confirmed by reverse transcription-polymerase chain reaction and sequence analysis of the cDNA. Quantification of immunoreactive bands revealed that levels of this P450 were 2.3-fold higher in alcoholics than in control subjects. This increase in lymphocyte CYP2E1 content in alcoholic subjects coincided with a 2.1-fold increase in chlorzoxazone clearance and a 2-fold decrease in the AUC for chlorzoxazone. Importantly, a correlation (r = 0.62,p < 0.01) was observed between CYP2E1 content in lymphocytes and chlorzoxazone clearance rates. Thus, monitoring lymphocyte CYP2E1 expression may provide a substitute for estimating hepatic activity of this P450.
The ethanol-inducible P4501 CYP2E1 is a toxicologically important enzyme because of its unique ability to convert many substrates to cytotoxins (1, 2). Moreover, chemical metabolism by this enzyme produces oxygen radicals, which can ultimately lead to lipid peroxidation (3-6). Ethanol is one such substrate, and its oxidation by CYP2E1 has been implicated in alcoholic liver disease (7, 8). Of particular concern is that CYP2E1 concentrations in humans vary extensively, due in part to pathophysiological conditions (including obesity) and exposure to xenobiotics (such as ethanol or isoniazid) (9-11). Variations in the expression of the enzyme caused by these factors determine the degree of hepatotoxicity elicited by alcohol or other chemicals.
CYP2E1 is primarily an hepatic enzyme; however, evidence suggests that it is also present in other tissues, including lung, kidney, nasal mucosa, and bone marrow (12-14). More recently, CYP2E1 was found in the white cell fraction of peripheral blood from humans (15), rabbits (16), and rats (17). Expression of CYP2E1 in the lymphocyte fraction of white blood cells appears to be influenced by the same factors that affect the concentration of the hepatic enzyme, including xenobiotics and physiological states (15-17). Indeed, insulin-dependent diabetes enhances the expression of lymphocyte CYP2E1 in humans (15), and starvation increases the expression of CYP2E1 protein and mRNA in rodent lymphocytes (17). With regard to xenobiotics, ethanol produced a 10-fold enrichment of lymphocyte CYP2E1 in rabbits, compared with their control counterparts (16). Interestingly, CYP2E1 induction by either ethanol (16) or fasting (17) occurred in both liver and lymphocytes, in a parallel manner. Thus, changes in CYP2E1 content in white blood cells may reflect xenobiotic-promoted alterations in other tissues, including liver.
Because expression of CYP2E1 is altered by many factors affecting chemical metabolism and therefore toxicity, the ability to easily estimate human CYP2E1 content is appealing, particularly for assessment of risks from chemical exposure. Currently, the concentrations of hepatic CYP2E1 can be estimated by determining pharmacokinetic parameters of CZX metabolism. However, application of this procedure may not be suitable for screening large numbers of samples in general clinical settings. Therefore, approaches that are less time consuming and more convenient are currently being investigated. One approach could include estimating hepatic CYP2E1 content from its expression in HPBLs. This may be feasible, given results obtained in animal studies (16, 17). Although several investigations have demonstrated xenobiotic-metabolizing P450 enzymes, including CYP1A1 (18), 3A5 (19), and 2E1 (15), in human white blood cells, there are no direct comparisons with the hepatic content of these P450s. The present investigation was designed to determine whether CYP2E1 levels in human lymphocytes are higher in alcoholic subjects and whether white cell expression reflects hepatic CZX activity.
Materials and Methods
Volunteers.
Twenty-two subjects (10 men and 12 women, ranging in age from 20 to 50 years) participated in this investigation. Volunteers were healthy subjects within 20% of ideal body weight. Individuals with diabetes, individuals receiving prescription medications, or individuals diagnosed with a codependency were eliminated from the study. At the time of recruitment, informed consent was obtained, questionnaires were completed, and a blood sample was drawn for routine blood evaluations (Reference Laboratory, Inc., Albuquerque, NM). Based on laboratory results, subjects demonstrating abnormal liver function values or those diagnosed with hepatic disease were discharged. Volunteers proceeding with the investigation were admitted to the University of New Mexico Clinical Research Center, where they received thorough physical examinations. Subjects remained overnight in the Clinical Research Center to prevent alcohol abusers access to alcoholic beverages and to allow BACs in individuals with elevated levels to return to zero. BACs of <10 mg/dl ensured that competitive inhibition of CZX metabolism did not occur. During the overnight stay, volunteers were placed on caffeine-free diets and alcoholics were carefully monitored for any signs of withdrawal. At 7:30 a.m., blood samples were obtained to determine BACs; all subjects possessed values of <5 mg/dl. Blood (320 ml) was drawn at 8:00 a.m. for lymphocyte isolation from all but two alcoholic subjects; in these two cases the large sample was unobtainable. After the blood drawing, 500 mg of CZX was orally administered. Blood samples (10 ml) were subsequently obtained at 0, 0.25, 0.50, 0.75, 1.0, 1.5, 2, 3, 4, 5, 6, 8, 10, and 12 hr. Plasma was separated by centrifugation at 2300 rpm for 15 min at 4°C, immediately frozen, and stored at −20°C until assayed.
Clinical Evaluations.
Subject screening procedures consisted of two types of evaluations. First, subjects completed a survey developed by our laboratory, composed of questions adapted from the CAGE questionnaire, a common screening instrument used by health care professionals to detect alcohol problems (20, 21). Second, a more detailed personal interview was used to compile alcohol usage profiles for all individuals. The profile incorporated physiological, psychological, and social components and was determined by posing questions compiled in the NLAES. The NLAES is an Alcohol Use Disorders and Associated Disabilities Interview Schedule questionnaire similar to that inDiagnostic and Statistical Manual of Mental Disorders, fourth edition, but it was developed primarily for epidemiological research purposes (22). Both surveys were subjectively scored using a protocol developed by an epidemiologist in our laboratory.
Responses to the questionnaire adapted from the CAGE questionnaire were subjectively assigned weighted scores, with greater weight being given to questions that were more specific. The emphasis of the survey was on the frequency of drinking and the amount of alcohol consumed. A simple yes/no question was given less weight than a question specifically asking how frequently the behavior was exhibited. Responses to how often the subject drank alcoholic beverages were scored from 0 to 2, with 0 being never, not in the last year, or less than 1 day/month. A score of 1 was indicative of behavior that occurred either 1–3 days/month or 1 day/week, and a score of 2 indicated that alcohol was consumed 2–3 days/week, 4–5 days/week, or every day. Greater weight was placed on a score of 2 when summing the raw scores for each question.
The NLAES had three variables for each question that dictated the raw score. The first variable determined whether the subject had ever experienced the event or circumstance stated in the question. For an affirmative response a score of 1 was assigned, whereas a negative response was given a score of 0. The second variable assessed whether the experience had occurred in the last 12 months. The third established a weighted factor for how frequently the circumstance had occurred during the last 12 months and ranged from 1 to 5, with 1 being almost never and 5 being all the time. If the event had not happened in the last 12 months, then the weighted score was assigned a value of 0. The score for a given question was equal to the sum of the first and third variables. The second variable (whether the experience had occurred in the last 12 months) was used as an introduction to a more specific investigation of behavior frequency and as an indicator of whether a weighted factor was present. The total score of the survey was the sum of the raw scores for each question. The severity of behavior was quantified by summing the total scores for the questionnaire developed by our laboratory and the NLAES.
Lymphocyte Isolation.
Lymphocytes were immediately separated from 320 ml of whole blood according to the method of Boyum (23). Briefly, 20 ml of Histopaque-1077 (Sigma Chemical Co., St. Louis, MO) was added to a 50-ml conical centrifuge tube. Whole blood was layered onto Histopaque-1077, and tubes were centrifuged at 400g for 30 min at ambient temperature. After centrifugation, the opaque interface was transferred to a new tube, phosphate-buffered saline was added, and tubes were centrifuged at 250g for 10 min at 4°C. Cells were resuspended in RPMI 1640 medium (Gibco BRL Products, Gaithersburg, MD) containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum and were cultured overnight to separate lymphocytes from monocytes. Cultures were then transferred to 50-ml conical tubes and centrifuged at 250g for 10 min at 4°C. The lymphocyte pellet was either resuspended in 5.0 ml of homogenizing buffer (0.1 M Tris-HCl, pH 7.4), containing 0.5 mM phenylmethylsulfonyl fluoride (Sigma), for microsome preparation or mixed with Trizol (Gibco BRL Products) for RNA isolation. Both samples were frozen in liquid nitrogen and stored at −70°C until use.
Microsome Preparation.
Microsomes were isolated from white cells by previously published procedures (16, 24). Briefly, lymphocytes were sonicated on ice for 3 × 60 sec to disrupt cells. Microsomes were then prepared from disrupted cells, and pellets were resuspended in sucrose assay buffer (25). Lymphocyte microsomes were stored at −70°C until use. Protein values were determined with bovine serum albumin as a standard (26).
Immunoblot Analysis.
Microsomal proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose filters. Filters were subsequently blocked in 5% nonfat dry milk/TBST (20 mM Tris buffer, pH 7.6, 137 mM NaCl, 0.1% Tween-20) for 1 hr at 37°C and allowed to react overnight at 4°C in 5% milk/TBST containing a previously characterized anti-human CYP2E1 IgG (5 μg/ml) (27). Filters were then incubated for 60 min with biotinylated goat anti-rabbit IgG (1:2000 in TBST; Calbiochem, La Jolla, CA), followed by a 60-min incubation with streptavidin-conjugated horseradish peroxidase (1:2000 in TBST; Calbiochem) at room temperature. Immunochemical staining was performed by reaction of the filters with 10 ml of enhanced chemiluminescence detection reagents (Amersham, Arlington Heights, IL) for 1 min at room temperature and exposure to Amersham Hyperfilm for 10–30 sec. Immunoreactive CYP2E1 content in lymphocyte microsomes was quantified with a Microtek Scanmaker IIHR scanner interfaced to ImageQuant software. Lymphocyte microsomal protein was applied in various amounts (5–75 μg) to determine the linear range of signal intensity of immunoblots. The concentration of microsomal protein (25 μg) used for all subsequent immunoblot analyses was within the linear portion of that curve.
Reverse Transcription/Polymerase Chain Reaction.
Total RNA from lymphocytes was isolated using Trizol reagent (Gibco BRL Products) and quantified by measurement of its absorbance at 260 nm; purity was assessed by determination of the 260/280-nm ratio. First-strand cDNA synthesis was performed with RNA as previously described (25). The cDNA was then amplified using oligonucleotide primers that were 21 bp in length and flanked CYP2E1 exons 4 (bp 501–523) and 6 (bp 954–976), as described elsewhere (25). The resulting DNA was ligated into pCRII vector (Invitrogen, San Diego, CA) and transformed into competent (INVαF′) Escherichia colicells. Ultrapure plasmid DNA was isolated from the E. colitransformants and sequenced using the dideoxy-chain termination method (28) and Sequenase (United States Biochemicals, Cleveland, OH).
Analytical Methods.
CZX and 6-OH-CZX concentrations in plasma were determined by a reverse-phase HPLC-based assay (9). Plasma samples (250 μl) were thawed on ice, and 750 μl of 2 M sodium acetate buffer, pH 4.5, was added. Each sample was evaluated in triplicate. Twenty microliters (200 activity units) of Helix pomatia type H-2 β-glucuronidase (Sigma) were added to each tube and incubated overnight at 37°C. After incubation, 200 μl of 0.2 M theophylline was added to each tube, for use as an internal standard. The reaction was terminated by addition of 3 ml of 1 N HCl and centrifuged at 3500g for 10 min at 4°C. The supernatant was removed and placed in separate tubes containing 2 ml of ethyl acetate. Samples were vigorously vortex-mixed for 1 min and centrifuged at 2000 rpm for 8 min at 4°C. The organic layer was removed and the aqueous phase was subjected to a second extraction with 2 ml of ethyl acetate. The extracts were pooled and evaporated to dryness under nitrogen with low heat. The residues were resuspended in 100 μl of mobile phase (20%, v/v, acetonitrile/80%, v/v, 0.5% phosphoric acid) and vigorously vortex-mixed. A 20-μl aliquot of extract was subsequently analyzed by use of a model 504 autosampler (Beckman Instruments, Palo Alto, CA), a model 600 solvent delivery system (Waters Millipore, Milford, MA), and a model 486 detector (Waters Millipore) set at 287 nm. Chromatographic separation was achieved with a C8 Ultrasphere column (5-μm, 4.6 × 250 mm; Beckman Instruments). The mobile phase was pumped at a flow rate of 1 ml/min.
Standard curves of area ratios for CZX/theophylline and 6-OH-CZX/theophylline were constructed from plasma containing various amounts (0.20–8.0 μg/ml) of CZX (Sigma) and 6-OH-CZX (McNeal Consumer Products, Fort Washington, PA). Recoveries of 6-OH-CZX and CZX were 77 and 70%, respectively, with this extraction method. Values for each sample were extrapolated from linear calibration curves (r = 0.99) for the standards. The interday coefficient of variation (N = 22), determined by addition of CZX and 6-OH-CZX to plasma samples, was approximately 15% for each at concentrations of 2, 4, and 8 μg/ml. The limits of detection for both CZX and 6-OH-CZX were 0.09 ± 0.01 μg/ml.
Pharmacokinetic Analysis.
The AUC for CZX and its metabolite were determined using the trapezoid rule. Noncompartmental analysis was used to compare the oral clearance of CZX in control and alcoholic subjects and was determined from the ratio of the administered dose to the total AUC.
Statistical Analysis.
Three separate statistical tests were used to determine significance or possible correlations between study populations. Group comparisons were performed using Student’s t test for independent samples. A one-way analysis of variance was used to determine any statistical significance that might exist between more than two distributions or sample groups. Correlation analyses were also performed, and a curve fit determined the association of the two fields. Statistical calculations were made using GraphPad Instat version 2.05a software. Statistical significance was set at p < 0.05. Values were expressed as the mean ± SE.
Results
Volunteers were randomly recruited with strategically placed flyers. The mean age of the 10 male subjects involved was 33.1 ± 3.16 years, and that of the 12 female subjects was 36.67 ± 2.49 years. Participants were divided into two groups, control subjects and alcohol abusers, based on subjective scores received in the screening surveys. For the 22 individuals participating in this investigation, scores ranged from 0 to 192 (fig. 1). The distribution demonstrates that control subjects possessed scores from 0 to 38, whereas alcohol abusers exhibited scores from 58 to 192. Significant differences in scores based on gender, between alcoholic men (114.4 ± 24.20) and women (99.17 ± 14.56) or between control male (7.6 ± 2.34) and female (4.00 ± 1.24) volunteers, were not apparent, suggesting that alcohol consumption did not exhibit a gender preference. Body weights (men, 75.36 ± 4.73 kg; women, 62.58 ± 3.02 kg) and heights (men, 179.25 ± 3.53 cm; women, 160.20 ± 1.46 cm) for all subjects were within the normal range for healthy individuals.

Distribution of survey scores.
The 22 subjects participating in this investigation were assigned survey scores based on responses to two questionnaires, as described inMaterials and Methods. The scores indicate the frequency and amount of alcohol consumption.
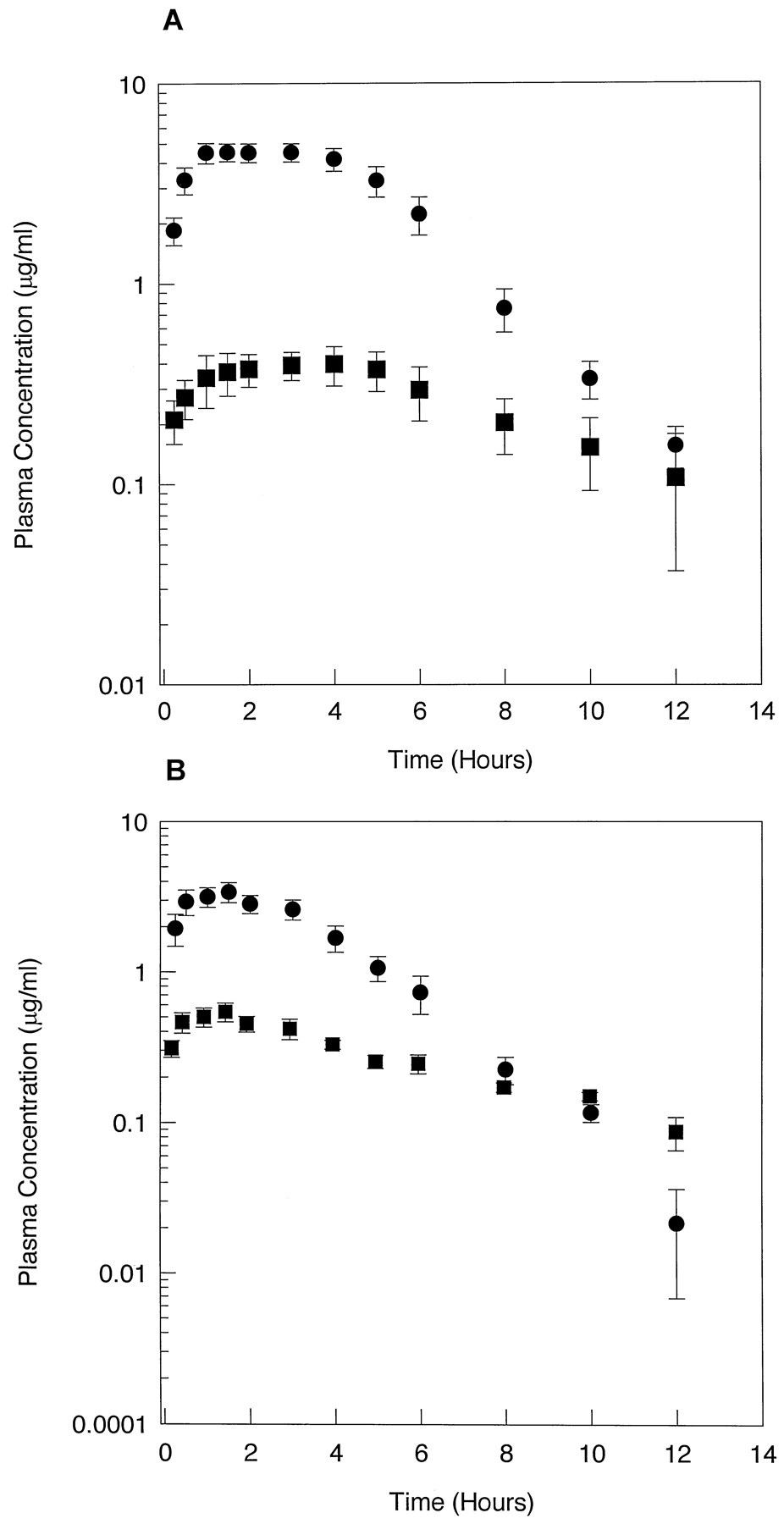
Subjects were given a 500-mg single dose of CZX to assess CYP2E1-mediated metabolism in vivo. Pharmacokinetic parameters for CZX metabolism were determined for each of the volunteers. The mean plasma concentration-time profiles for all control subjects and alcohol abusers are shown in fig.2. In control subjects, CZX was rapidly absorbed, with plasma concentrations peaking at 2 hr and declining to detection limits by 12 hr. Plasma concentrations of the metabolite also peaked at 2 hr in control subjects but declined to undetectable levels after 10 hr. In contrast, plasma concentrations of CZX in alcohol abusers (fig. 2B) peaked at 1.5 hr but at 1.4-fold lower concentrations than observed in plasma from control subjects. The concentration of 6-OH-CZX in alcohol abusers also exhibited a peak at 1.5 hr, but the content was 1.4-fold greater than that observed in samples from control subjects. Furthermore, by 10 hr the parent drug had declined to nearly undetectable levels in alcoholic subjects. These results indicated that peak plasma concentrations of both the parent drug and its metabolite were achieved earlier in alcohol abusers than in control subjects and that CZX levels declined at a faster rate in alcoholics. Additionally, greater plasma concentrations of 6-OH-CZX were obtained in the alcohol abusers, compared with control subjects.

Plasma concentration-time profiles for CZX and 6-OH-CZX.
Plasma concentration-time curves were constructed for control subjects (N = 11) (A) and alcohol abusers (N = 11) (B) after a single 500-mg oral dose of CZX. Each value represents the mean ± SE. •, CZX; ▪, 6-OH-CZX.
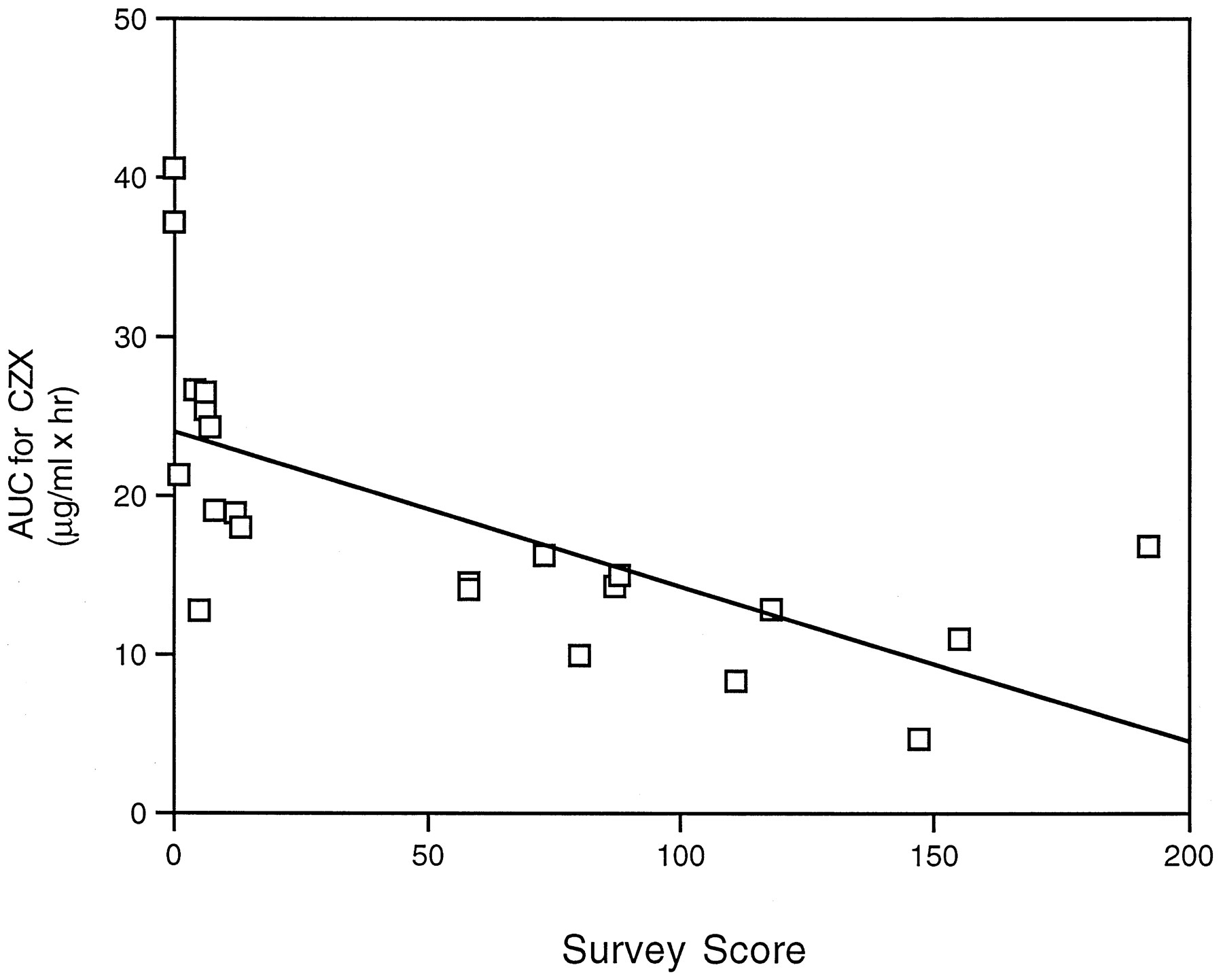
Analysis of pharmacokinetic parameters indicated that theCmax of CZX was 1.4-fold lower in alcohol abusers than control subjects, whereas theCmax for 6-OH-CZX was 1.4-fold higher in alcoholics (table 1). As a result of lower plasma CZX concentrations in alcoholics, oral clearance was significantly increased by 100% (table 1). No statistically significant differences were noted between alcoholics and control subjects in the AUC for 6-OH-CZX. However, there was a significant difference in the AUC for CZX between control subjects (24.64 ± 2.49) and alcohol abusers (12.56 ± 1.11). Furthermore, the 6-OH-CZX/CZX AUC ratio was 2.2-fold greater in samples from alcohol abusers than in those from control subjects (table 1). Parameters were also compared by gender, but no significant differences were observed for CZX clearance or AUC for CZX (table 1). Results indicate that chronic alcohol consumption influences pharmacokinetic parameters for CZX. Indeed, when the AUC for CZX was compared with the frequency of alcohol consumption, as determined by the survey score, a correlation was observed (r = 0.66, p < 0.01) (fig. 3).
Pharmacokinetic parameters for CZX metabolism were assessed for each individual by quantifying CZX and its metabolite, 6-OH-CZX, after oral administration of 500 mg. Quantification was by an HPLC-based assay described in Materials and Methods. Values are mean ± SE of three separate determinations.

Correlation between AUC for CZX values and survey scores.
Correlation analysis was performed on the AUC for CZX values and the survey scores for 22 subjects involved in this study. A correlation coefficient of r = 0.66 (p< 0.01) was obtained.
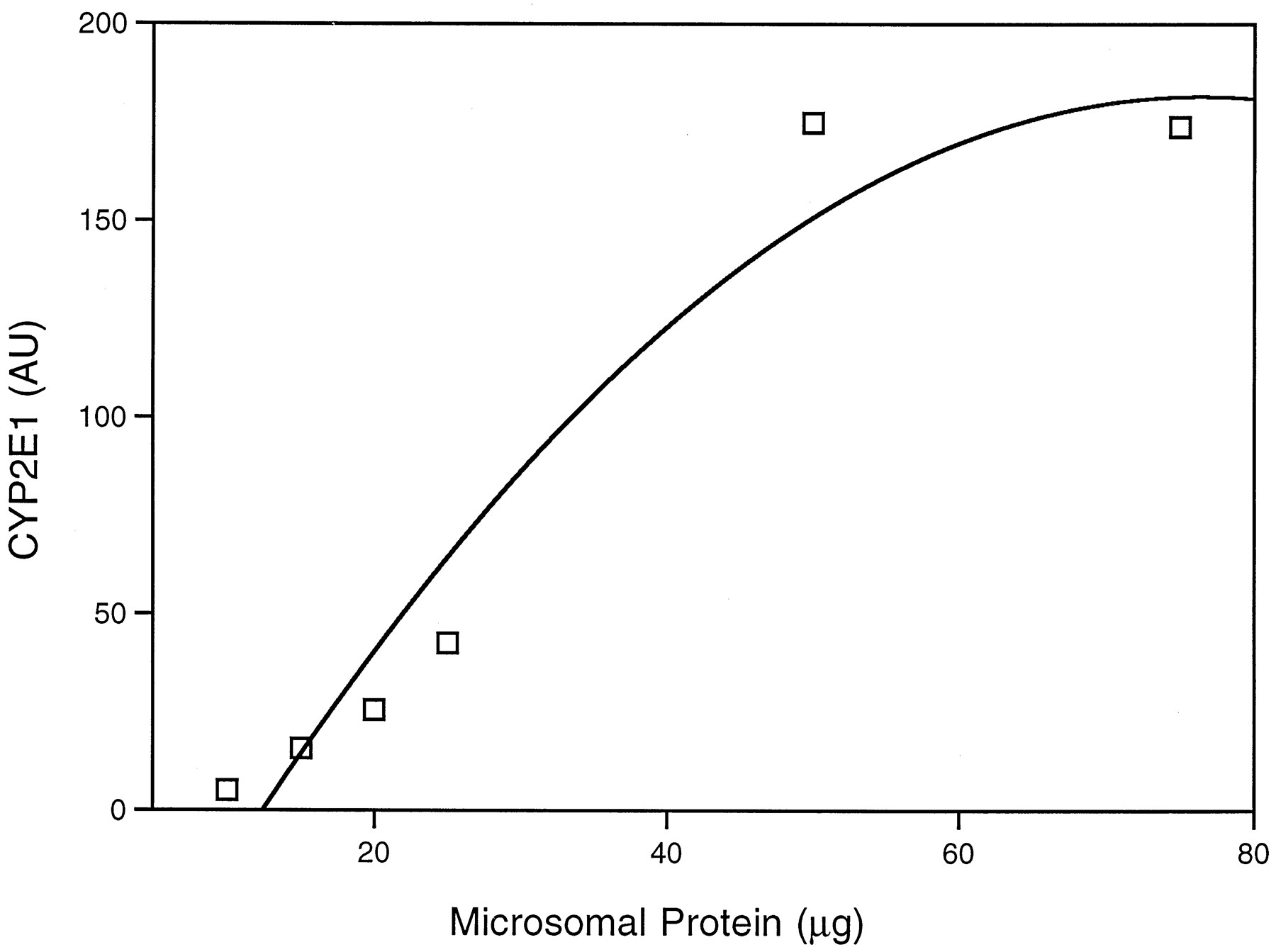
The relationship between the CYP2E1-mediated activity of liver, as determined by in vivo CZX metabolism, and the concentration of the enzyme in HPBLs was explored. Detection of CYP2E1 in lymphocytes was performed by immunoblot analysis of isolated microsomes. Before examination of individual samples, varying amounts (5–125 μg) of lymphocyte microsomal protein were applied to polyacrylamide gels and, after separation, transferred to a nitrocellulose filter, which was stained with anti-human CYP2E1 IgG. Fig.4 is a representative immunoblot containing microsomes from lymphocytes and liver, for comparison. A band migrating 3500 Da below than that of liver is apparent in lanes containing lymphocyte microsomes. The faster migration of the lymphocyte protein may be due to differences in molecular mass between the liver and blood enzymes. A standard curve was constructed by application of 5–75 μg of lymphocyte microsomal protein and subsequent quantification of the 50,500-Da immunoreactive band. From this curve, 25 μg was selected for use on subsequent blots, because this amount provided a readily detectable signal within the linear range of intensity (fig. 5).
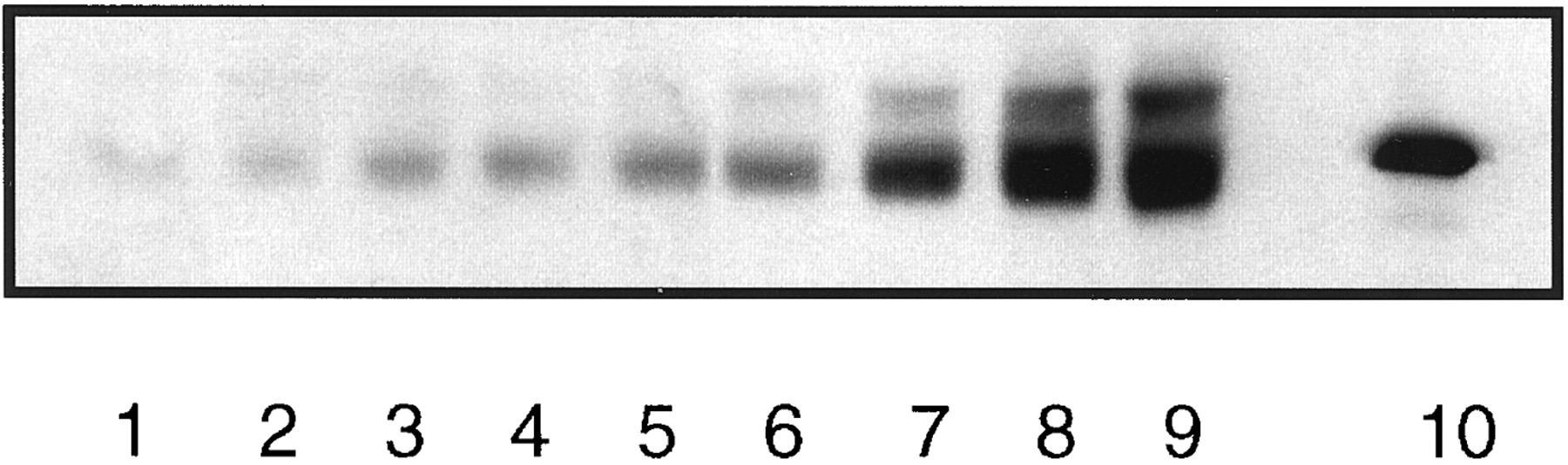
Representative immunoblot of various concentrations of lymphocyte microsomal protein.
Microsomes were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose filter, and the filter was reacted with anti-human CYP2E1 IgG, as described inMaterials and Methods. Lanes 1–9, 5, 10, 15, 20, 25, 50, 75, 100, and 125 μg of lymphocyte microsomal protein;lane 10, human liver microsomes (0.5 μg of protein).

CYP2E1 content in various protein concentrations of lymphocyte microsomes.
Lymphocyte microsomes were prepared from one individual and subjected to immunoblot analysis as described in Materials and Methods. Various amounts of microsomal protein (5, 10, 20, 25, 50, and 75 μg) were applied to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transfer to a nitrocellulose filter and staining with anti-human CYP2E1 IgG. The human anti-CYP2E1-reactive band was quantified as described. Values for CYP2E1 content are expressed as AU and are the means of two determinations.
To verify that CYP2E1 was present in HPBLs, we performed reverse transcription/polymerase chain reaction. Total RNA isolated from lymphocytes of four subjects was used as a template in a reverse transcription reaction to generate cDNA. The cDNA was then mixed with specific primers, as described in Materials and Methods, and amplified for 30 cycles by the polymerase chain reaction. Amplification yielded a 475-bp fragment from lymphocyte RNA that was similar in size to the fragment amplified from a human liver RNA sample. Sequence analysis revealed that the amplified lymphocyte DNA was identical to the corresponding region of human CYP2E1 cDNA previously described by Song et al. (29).
Quantification of immunoblots containing lymphocyte microsomes from 20 subjects in this study revealed a 10.5-fold variability, which ranged from 7.5 to 80 AU/μg microsomal protein. A 2.3-fold greater staining intensity was observed in lymphocyte microsomes from alcoholic subjects (42.57 ± 9.05 AU/μg), compared with control subjects (18.14 ± 3.04 AU/μg) (fig. 6). Additionally, a 2.1-fold increase in CZX clearance and a 2-fold decrease in the AUC for CZX were noted in alcoholic subjects, compared with control subjects (table 1 and fig. 6). To assess whether expression of CYP2E1 in lymphocytes could serve as an indicator of P450 activity in liver, enzyme levels were compared with CZX metabolism. Correlation analysis was performed on immunoquantified lymphocyte CYP2E1 and the pharmacokinetic parameter of CZX clearance. Interestingly, a correlation (r = 0.62,p < 0.01) was observed between these two parameters (fig. 7), suggesting that chronic alcohol consumption enhanced expression of the enzyme in liver and white cells in a parallel fashion.
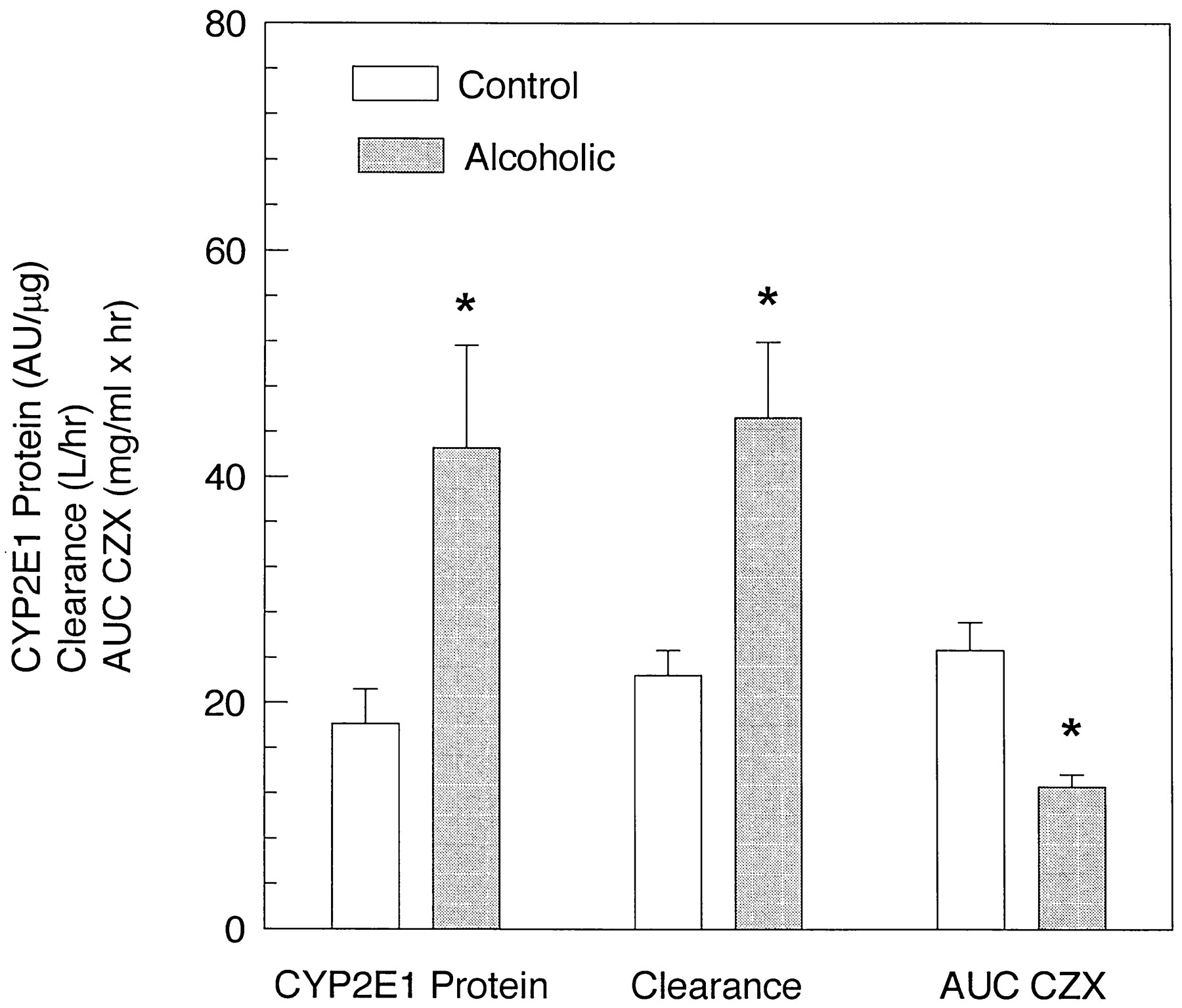
CYP2E1 content in lymphocyte microsomes, CZX clearance, and AUC for CZX values for alcohol abusers and control subjects.
Quantification was performed on immunoblots containing microsomes from lymphocytes of 9 alcoholic and 11 control subjects involved in this study. CZX pharmacokinetic parameters (AUC and clearance) were determined as described in Materials and Methods. Each number represents the mean of alcoholic and control subjects, and the value for each individual is the mean of three determinations. *, statistically significant difference from control subjects,p <0.05.

Correlation between lymphocyte CYP2E1 and CZX clearance.
Correlation analysis was performed on the CYP2E1 content of lymphocyte microsomes and the CZX clearance for 20 subjects (9 alcohol abusers and 11 control subjects). A correlation coefficient ofr = 0.62 (p <0.01) was obtained.
Discussion
The present study compared the expression of the ethanol-inducible P450 CYP2E1 in HPBLs with in vivo activity of the hepatic enzyme. The 6-hydroxylation of CZX was used as a biomarker for hepatic CYP2E1 activity, because several investigations (30-33) have validated its specificity for this reaction in vivo. Other P450 enzymes, including recombinant human CYP1A2 and rat CYP3A, appear to exhibit CZX 6-hydroxylase activity (34, 35). However, the in vivo contribution of recombinant CYP1A2 and the clinical significance of rat CYP3A to CZX hydroxylation have not been confirmed. Indeed, reports suggest that neither CYP1A2 nor CYP3A4 plays a major role in this reaction in human subjects. One such study showed that disulfiram, a specific CYP2E1 inhibitor, significantly decreased metabolism of CZX in vivo when simultaneously administered to volunteers (33). Furthermore, increased CZX metabolism in alcoholics, compared with control subjects (36, 37), suggests that altered pharmacokinetic parameters are due to ethanol-mediated induction of a P450, presumably CYP2E1, because this enzyme is the major ethanol-inducible P450 in humans (2, 38). That CYP2E1 is the primary P450 induced by chronic alcohol exposure was recently challenged, when it was determined that CYP3A4 levels in isolated hepatocytes were increased by ethanol (39). However, confirmation of this induction in vivo remains to be established. At present, CZX appears to be a valid estimate of human CYP2E1 activityin vivo, and chronic alcohol consumption increases its metabolism via enhanced expression of this enzyme.
CYP2E1 content in HPBLs from subjects involved in the present study was assessed by immunoblot analyses. The blots revealed a band in HPBL microsomes that migrated below that in liver (fig. 4). Presently, the 3500-Da difference in the molecular masses of lymphocyte and hepatic CYP2E1 is believed due to either variations in the amount of microsomal protein used for sodium dodecyl sulfate-polyacrylamide gel electrophoresis or structural alterations between the enzymes from the two tissues. Suggestive of the latter argument, previous studies from our laboratory demonstrated that rabbit microsomal CYP2E1 in lymphocytes and liver are the same molecular weight (16), despite the fact that 40-fold more protein from lymphocyte microsomes was applied to the gel. The amounts of liver and lymphocyte microsomal protein loaded onto the gels in the current investigation were similar to those used in the previous study. Further evidence suggesting a structural difference in CYP2E1 of blood and liver was provided by a study where HPBL homogenates from diabetic children exhibited a lower molecular weight form of CYP2E1 than the corresponding enzyme in human hepatic microsomes (15). Taken together, evidence supports the contention that structural differences may exist between the human hepatic and blood enzymes.
Despite the lower molecular weight of CYP2E1 in lymphocytes, enhanced expression of the enzyme was observed in alcoholic subjects. Indeed, the extent of CYP2E1 induction was similar to the increase in CZX metabolism, demonstrating good agreement with studies conducted in experimental animals (16, 17). However, in the present study the magnitude of induction of white cell CYP2E1 in alcoholics was less than observed in rabbits treated with ethanol (16). This lower level of CYP2E1 enhancement may be due to the additional overnight culturing of HPBLs, coupled with the rapid turnover of this enzyme. Recombinant human CYP2E1 transfected into Hep G2 cells displayed a half-life in culture of 2.5–6 hr, whereas in the presence of inducing agents, including ethanol, the recombinant enzyme exhibited a half-life of 37 hr (40). Additionally, results obtained in vivo demonstrated that the 6-OH-CZX/CZX ratio decreased rapidly during ethanol withdrawal in alcoholic subjects (37). This rapid decline in the ratio was attributed to a 2.5-day half-life determined for CYP2E1. Thus, if HPBL CYP2E1 possesses characteristics similar to those of its recombinant or hepatic counterpart, quantification of the ethanol-enhanced enzyme in HPBLs occurred here before turnover, but when concentrations were declining.
Results (data not shown) from our laboratory indicated that CYP2E1 content was reduced by approximately 20% in microsomes from white cells cultured overnight, compared with noncultured cells of alcoholic subjects. The advantage gained by culturing white cells overnight is that monocytes are separated from lymphocytes (41). Elimination of monocytes, where CYP2E1 is absent, provides a more homogeneous cell population, thereby increasing immunoblot sensitivity. Thus, the advantage of increased sensitivity by elimination of monocytes surpassed the decrease in alcohol-induced CYP2E1 content caused by the 12-hr overnight culture.
In summary, this investigation demonstrated that CYP2E1 protein and its corresponding mRNA were present in freshly isolated HPBL microsomes. Before this report, CYP2E1 was identified in whole homogenates of HPBLs cultured for 4 days (15). Eliminating the 4-day culture period simplified the procedure and reduced the time required for detecting CYP2E1 in human blood samples. Importantly, the increase in CYP2E1 protein in lymphocytes was similar to the alterations in CZX clearance and the AUC for CZX, indicating that chronic ethanol consumption produced similar increases in CYP2E1 expression in lymphocytes and liver. Thus, P450 enzymes expressed in HPBLs may be used to predict alterations in hepatic P450-mediated activity caused by xenobiotic exposure. Screening of P450 concentrations in freshly isolated HPBLs, rather than with invasive liver biopsies or time-consuming in vivo metabolic studies, may be an efficient method for identifying individuals at greater risk for chemically promoted hepatotoxicity.
Acknowledgments
The authors thank Gerald Curley, Barbara Mounho, and Margo Lopez for their technical assistance.
Footnotes
-
Send reprint requests to: Dr. Judy L. Raucy, The Agouron Institute, 505 Coast Blvd. South, Suite 400, La Jolla, CA 92037-4696.
-
This work was supported by United States Department of Health and Human Services Grants AA08990 (J.L.R.) and NCRR-GCRC RR00997 (Clinical Research Center, University of New Mexico). Approval for studies involving human subjects was granted by the University of New Mexico Human Research Review Committee (HRRC 90–213).
- Abbreviations used are::
- P450 or CYP
- cytochrome P450
- CZX
- chlorzoxazone
- 6-OH-CZX
- 6-hydroxychlorzoxazone
- AU
- absorbance units
- bp
- base pairs
- CAGE
- cut, annoyed, guilty, and eye opener
- NLAES
- National Longitudinal Alcohol Epidemiological Survey
- TBST
- Tris-buffered saline with Tween-20
- HPBL
- human peripheral blood lymphocyte
- BAC
- blood alcohol concentration
- Received April 29, 1997.
- Accepted August 5, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}