


Abstract
Nevirapine (NVP), a non-nucleoside inhibitor of HIV-1 reverse transcriptase, is concomitantly administered to patients with a variety of medications. To assess the potential for its involvement in drug interactions, cytochrome P-450 (CYP) reaction phenotyping of NVP to its four oxidative metabolites, 2-, 3-, 8-, and 12-hydroxyNVP, was performed. The NVP metabolite formation rates by characterized human hepatic microsomes were best correlated with probe activities for either CYP3A4 (2- and 12-hydroxyNVP) or CYP2B6 (3-and 8-hydroxyNVP). In studies with cDNA-expressed human hepatic CYPs, 2- and 3-hydroxyNVP were exclusively formed by CYP3A and CYP2B6, respectively. Multiple cDNA-expressed CYPs produced 8- and 12-hydroxyNVP, although they were produced predominantly by CYP2D6 and CYP3A4, respectively. Antibody to CYP3A4 inhibited the rates of 2-, 8-, and 12-hydroxyNVP formation by human hepatic microsomes, whereas antibody to CYP2B6 inhibited the formation of 3- and 8-hydroxyNVP. Studies using the CYP3A4 inhibitors ketoconazole, troleandomycin, and erythromycin suggested a role for CYP3A4 in the formation of 2-, 8-, and 12-hydroxyNVP. These inhibitors were less effective or ineffective against the biotransformation of NVP to 3-hydroxyNVP. Quinidine very weakly inhibited only 8-hydroxyNVP formation. NVP itself was an inhibitor of only CYP3A4 at concentrations that were well above those of therapeutic relevance (Ki = 270 μM). Collectively, these data indicate that NVP is principally metabolized by CYP3A4 and CYP2B6 and that it has little potential to be involved in inhibitory drug interactions.
Nevirapine (NVP)2, a non-nucleoside HIV-1 reverse transcriptase inhibitor, has been available for use in combination with nucleoside HIV-1 reverse transcriptase inhibitors (e.g., zidovudine, didanosine, etc.) since August 1996. More recently, NVP has received U.S. Food and Drug Administration approval for use in combination with HIV-1 protease inhibitors (e.g., saquinavir, ritonavir, indinavir, etc.). Because of its administration to patients with AIDS, NVP is also given concomitantly with a variety of medications for the treatment of opportunistic infections. Some of the drugs commonly coadministered with NVP have been implicated in clinically significant drug interactions. For example, ketoconazole, erythromycin, saquinavir, and ritonavir have each elevated the plasma concentrations of concomitantly administered drugs due to inhibitory interactions (Spinler et al., 1995; Zylber-Katz, 1995; Merry et al., 1997; Van Cleef et al., 1997; Albengres et al., 1998; Greenblatt et al., 1998). Clearly, the potential exists for NVP to be involved in clinically significant drug interactions with drugs from several therapeutic classes. The drug interaction potential of NVP is exacerbated by its capacity to induce the cytochromes P-450 (CYPs) that are responsible for its own biotransformation (Cheeseman et al., 1995;Havlir et al., 1995; Lamson et al., 1995). Therefore, an essential aspect of the safe and efficacious use of NVP must be the circumvention and management of drug interactions.
The prevention and management of drug interactions within patients receiving NVP concomitantly with other drugs requires a thorough understanding of the enzymes that are involved in their biotransformation pathways. To date, there are no published reports that comprehensively describe the biotransformation pathways of NVP in humans. A recent report (Riska et al., 1999) provides an extensive discussion of the in vivo biotransformation and elimination of NVP in humans. Briefly, the authors report that, in humans, NVP is eliminated primarily in the urine as glucuronide conjugates of 2-, 3-, 8-, and 12-hydroxyNVP. It is the aim of this report to perform CYP reaction phenotyping of the in vitro biotransformation of NVP to 2-, 3-, 8-, and 12-hydroxyNVP by human hepatic microsomes. The CYPs responsible for the biotransformation of NVP, and correspondingly, the CYPs that are likely to be induced by NVP in humans, are identified. Thereafter, the potential for NVP to be involved in clinically relevant drug interactions is discussed.
Experimental Procedures
Materials.
Erythromycin, ketoconazole, quinidine, troleandomycin, NADPH, Tris HCl, Tris base, sucrose, BSA, and EDTA were purchased from Sigma Chemical Co. (St. Louis, MO). NVP and 2-, 3-, 8-, and 12-hydroxyNVP were synthesized at Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT) according to published methods (Hargrave et al., 1991;Klunder et al., 1998). The identities of the metabolite standards were confirmed with mass spectrometry and NMR at Boehringer Ingelheim Pharmaceuticals, Inc. Using authentic standards of the metabolites, the identity of each metabolite produced by in vitro incubations with human hepatic microsomes was verified by comparison of HPLC retention times, diode array UV spectra, and mass spectra.
Microsomal Preparation.
For human hepatic microsomes that were prepared at Boehringer Ingelheim Pharmaceuticals, Inc. (Ridgefield, CT), donor tissue was purchased from either the Medical College of Wisconsin (Milwaukee, WI) or from the National Disease Research Interchange (Philadelphia, PA). Liver tissue was stored frozen at −80°C until microsomes were prepared. The liver tissue was processed into microsomes using a modification of the method of Guengerich (1994). Modifications included ultracentrifugation at 145,000g to obtain the microsomal pellet and suspension of microsomes into 66 mM Tris buffer (pH 7.4, containing 250 mM sucrose and 5.4 mM EDTA). Microsomes were stored either in liquid nitrogen or in a freezer maintained at −80°C.
For human hepatic microsomes prepared at the University of Washington (Seattle, WA), donor tissue was obtained from the Solid Organ Transplant Program (University of Washington Medical Center) and the Northwest Organ Procurement Agency (Seattle, WA). Liver tissue was stored at –70°C until microsomes were prepared. Microsomes were prepared according to the method of Thummel et al. (1993) and were suspended into 100 mM potassium phosphate buffer containing 1 mM EDTA (pH 7.4). Microsomes were stored at –70°C until used for in vitro metabolic studies.
All microsomal total protein concentrations were calculated using the method of Lowry et al. (1951), using BSA as a standard. Characterized human hepatic microsomes were obtained from Human Biologics, Inc. (Phoenix, AZ) as an Hepatoscreen Test Kit. Microsomes containing cDNA-expressed human CYPs were purchased from Gentest Corp (Woburn, MA) and were prepared by Gentest from human lymphoblastoid AHH-1 cells.
NVP Hydroxylation Assays.
NVP hydroxylation rates were determined in vitro by incubation of NVP at various concentrations with 2 mg of microsomal protein and 2.5 mM NADPH at 37°C in 66 mM Tris buffer (pH 7.4) for 30 min (1 ml total assay volume). Preliminary studies had demonstrated that in vitro metabolism rates were constant for at least 45 min under these conditions. All NVP hydroxylation studies were performed using microsomes that were prepared at Boehringer Ingelheim Pharmaceuticals, Inc. or with microsomes that were provided with the Hepatoscreen Test Kit. NVP was added to culture tubes as a solution in methanol. Unless otherwise indicated (see Chemical Inhibition of the Biotransformation of NVP), methanol was evaporated from the tubes before the other assay components were introduced, so that no organic solvent was present in the microsomal incubations. Metabolism in the assays was initiated by the addition of NADPH and terminated by the addition of 50 μl of 2 N aqueous sodium hydroxide. NVP and its metabolites were extracted into 5 ml of ethyl acetate that was subsequently evaporated at 40°C under a stream of nitrogen. The dried residue was dissolved into 200 μl of HPLC mobile phase, 100 μl of which was injected onto HPLC. The HPLC system consisted of a Hewlett-Packard (Wilmington, DE) 1050 pump and autoinjector and a Hewlett-Packard 1040A diode-array detector set to monitor UV absorbance between 200 and 400 nm and to record a chromatogram at 240 nm. Several different reversed phase C18 HPLC columns maintained in a column oven at 40°C were used in the HPLC system and were found to be suitable for these analyses. In most cases, either a Waters (Milford, MA) Nova-Pak C18 (300 × 3.9 mm) or Symmetry C18 (100 × 4.6 mm) column was used with a flow rate of 1 ml/min. The Nova-Pak C18 column was used with an isocratic mobile phase of 17:83 (v/v) acetonitrile/0.05 M phosphate buffer (pH 4.6) containing 0.1% triethylamine (v/v). The Symmetry C18 column was used with a mobile phase gradient of 10:90 to 14:86 (v/v) acetonitrile/water containing 0.1% glacial acetic acid (v/v) over 20 min. NVP and its metabolites were quantitated using calibration curves that were constructed using authentic standards.
Correlations with Characterized Human Hepatic Microsomes.
These studies were performed using characterized human hepatic microsomes (Human Biologics, Inc., Phoenix, AZ) and the NVP hydroxylation assay described previously. The assays were performed at two NVP concentrations (25 and 100 μM). Data analysis was performed using computer-generated simple linear correlations of NVP metabolite formation rates with the CYP-specific enzyme activities provided with the characterized human hepatic microsomes. The CYP-specific activities were 7-ethoxyresorufin-O-deethylase (CYP1A2), caffeineN-demethylase (CYP1A2), coumarin 7-hydroxylase (CYP2A6), tolbutamide methyl-hydroxylase (CYP2C9), S-mephenytoin 4-hydroxylase (CYP2C19), dextromethorphan O-demethylase (CYP2D6), chlorzoxazone 6-hydroxylase (CYP2E1), testosterone 6β-hydroxylase (CYP3A4), and lauric acid 12-hydroxylase (CYP4A). XENOTECH LLC (Kansas City, MO) provided the enzyme activities of these microsomes for 7-ethoxy-4-trifluoromethylcoumarin deethylase (EFCOD) and S-mephenytoin N-demethylase (MND) (both CYP2B6).
Biotransformation of NVP by cDNA-Expressed CYPs.
These studies were performed using microsomes containing cDNA-expressed human CYPs (Gentest) and the NVP hydroxylation assay described previously. The assays were performed at a 50 μM NVP concentration. The specific CYPs that were examined for their capacity to metabolize NVP were CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4. The metabolism of NVP by CYP3A5 was also studied, but at a NVP concentration of 100 μM. Data analysis was performed by monitoring the enzymes that were capable of metabolite formation and calculating the biotransformation rates as picomoles per minute per nanomole of CYP.
Antibody Inhibition of NVP Biotransformation.
These studies were performed using either pooled human hepatic microsomes prepared at Boehringer Ingelheim Pharmaceuticals, Inc. or microsomes containing cDNA-expressed human CYPs (Gentest). The pooled human hepatic microsomes were prepared from two separate livers from which good activity for 3-hydroxyNVP formation had been observed from their individual microsomal preparations. The assays were performed at two NVP concentrations, 25 and 400 μM. Antibodies to CYP3A4 (order no. A334, lot no. 1) and CYP2B6 (order no. A326, lot no. 1) were obtained from Gentest and were used according to the manufacturer's specifications. Data analysis was performed by comparing the NVP metabolite formation rates in the presence of antibody to those in assays containing no antibody.
Chemical Inhibition of the Biotransformation of NVP. The studies for the inhibition of 2-, 3-, and 12-hydroxyNVP formation were performed with pooled human hepatic microsomes prepared at Boehringer Ingelheim Pharmaceuticals, Inc. These microsomes were a pool of microsomes containing high levels of activity for the 3-hydroxylation of NVP. The inhibition studies of 2-, 3-, and 12-hydroxyNVP formation were also done with microsomes containing cDNA-expressed human CYP3A4 or CYP2B6 (Gentest). The studies for the inhibition of 8-hydroxyNVP formation were performed with pooled human hepatic microsomes prepared by Human Biologics, Inc. and included with their kit of characterized microsomes. These microsomes were a pool of equal amounts of their microsomes coded as samples 2, 3, 5, 6, and 11. All assays were performed at 100 μM NVP. Ketoconazole (0.5 and 2.5 μM), troleandomycin (50 μM), erythromycin (50 μM), and quinidine (15 μM) were tested for their ability to inhibit NVP metabolite formation. Assays with troleandomycin and erythromycin required metabolic activation for 15 min before the addition of NVP in a methanol solution (<5 μl/ml) to the incubations. The incubations containing no inhibitors were also preincubated for 15 min before the addition of NVP in methanol. Data analysis was performed by comparing the NVP metabolite formation rates in the presence of chemical inhibitors to those in assays containing no chemical inhibitors.
Inhibition of CYP-Specific Biotransformation Rates by NVP.
These studies were carried out at the University of Washington using three separate human hepatic microsomal preparations from different donors. The microsomes were prepared at the University of Washington. Various in vitro CYP-specific biotransformations were performed in the presence of 0, 25, 100, or 250 μM NVP. Incubations for each substrate probe were performed in microsomes prepared from three different donors chosen to represent a range of activities, but all with sufficient activity to catalyze the specific reaction. The seven substrate probes (and CYPs) that were monitored were (R)-warfarin 6-hydroxylase (CYP1A2, Bush et al., 1983), coumarin 7-hydroxylase (CYP2A6, Miles et al., 1990), (S)-warfarin 7-hydroxylase (CYP2C9, Rettie et al., 1992), (S)-mephenytoin 4′-hydroxylase (CYP2C19, Wrighton et al., 1993; Goldstein et al., 1994), bufuralol 1′-hydroxylase (CYP2D6, Boobis et al., 1985; Kronbach et al., 1987; Yamazaki et al., 1994), p-nitrophenol hydroxylase (CYP2E1, Tassaneeyakul et al., 1993a,b), and (R)-warfarin 10-hydroxylase (CYP3A4, Brian et al., 1990;Rettie et al., 1992). Data analysis was performed by comparison of the metabolite production rates of probe compounds in the presence of NVP to those observed in incubations containing no NVP. When possible, aKi was calculated using a Dixon plot.
Results
Calculation of Michaelis-Menten kinetics for the in vitro formation of 2- and 3-hydroxyNVP by human hepatic microsomes, cDNA-expressed CYP2B6, and cDNA-expressed CYP3A4 has been performed from data acquired using the NVP hydroxylation assay (data not shown). The analysis of 2-hydroxyNVP formation yielded apparentKm values of 212 and 279 μM in human hepatic microsomes and in microsomes containing cDNA-expressed CYP3A4, respectively. The analysis of 3-hydroxyNVP formation yielded apparentKm values of 609 and 834 μM in human hepatic microsomes and in microsomes containing cDNA-expressed CYP2B6, respectively. The Michaelis-Menten kinetics for the in vitro formation of 8-hydroxyNVP could not be calculated due to analytical problems, i.e., detection limits, and those of 12-hydroxyNVP formation could not be fit to a Michaelis-Menten curve.
Correlations with Characterized Human Hepatic Microsomes.
Four metabolites, 2-, 3-, 8-, and 12-hydroxyNVP (Fig.1) were produced during the in vitro biotransformation of NVP by ten characterized human hepatic microsomal preparations (Table 1). Typically, the two major metabolites produced in the microsomal incubations were 2- and 12-hydroxyNVP. 3-HydroxyNVP was a major metabolite in half of the microsomal incubations and 8-hydroxyNVP was always a minor metabolite.

Structure of NVP indicating the specific sites of metabolic hydroxylation.
The in vitro rates of formation (picomoles per minute per milligram of microsomal protein) of metabolites by characterized human hepatic microsomes in incubations with 25 or 100 μM NVP
The activities of the CYP-specific probe substrates for the characterized human hepatic microsomes are listed in Table2. The biotransformation rates of the four NVP metabolites in these incubations were correlated against the rates of eleven different CYP-specific probe compounds (Table3). Very good correlations were obtained with probe activities for CYP3A4 and CYP2B6. For 2- and 12-hydroxyNVP, the best correlations were with 6β-hydroxytestosterone formation (CYP3A4). For 3-hydroxyNVP, the best correlations were with furafylline-inhibited EFCOD and MND (both are probes for CYP2B6). 8-HydroxyNVP formation rates were equally well correlated with 6β-hydroxytestosterone formation, furafylline-inhibited EFCOD, and MND in incubations containing 100 μM NVP. In the 25-μM NVP incubations, the best correlations of 8-hydroxyNVP formation were with furafylline-inhibited EFCOD, MND, and coumarin 7-hydroxylase (CYP2A6). However, 8-hydroxyNVP formation rates were only measurable in five of ten in vitro incubations at 25 μM.
The in vitro CYP-specific enzyme activities (picomoles per minute per milligram of microsomal protein) of the characterized human hepatic microsomes used in the NVP hydroxylation assays
The coefficients of determination (r2) for the in vitro rates of CYP-specific enzyme activities versus in vitro rates of nevirapine metabolite formation by characterized human hepatic microsomes in incubations with 25 or 100 μM NVP
Biotransformation of NVP by cDNA-Expressed CYPs.
In incubations of NVP with microsomes overexpressing ten different human CYPs, 2- and 3-hydroxyNVP were exclusively formed by CYP3A and CYP2B6, respectively (Fig. 2). Three enzymes, CYP3A4, CYP3A5, and CYP2D6, were capable of forming 12-hydroxyNVP reasonably well and traces (<0.1 nmol/min/mg protein) of 12-hydroxyNVP were formed in incubations with CYP2C9 (not shown). Although CYP3A4 was able to form traces (<0.1 nmol/min/mg protein) of 8-hydroxyNVP, only CYP2D6 was able to form this metabolite well. There was no 8-hydroxyNVP formation observed in incubations with microsomes containing either CYP2A6 (not shown) or CYP2B6.

In vitro biotransformation of 50 μM NVP (except CYP3A5, where 100 μM NVP was used) by microsomes from human B-lymphoblastoid cells expressing specific human CYPs.
Antibody Inhibition of NVP Biotransformation.
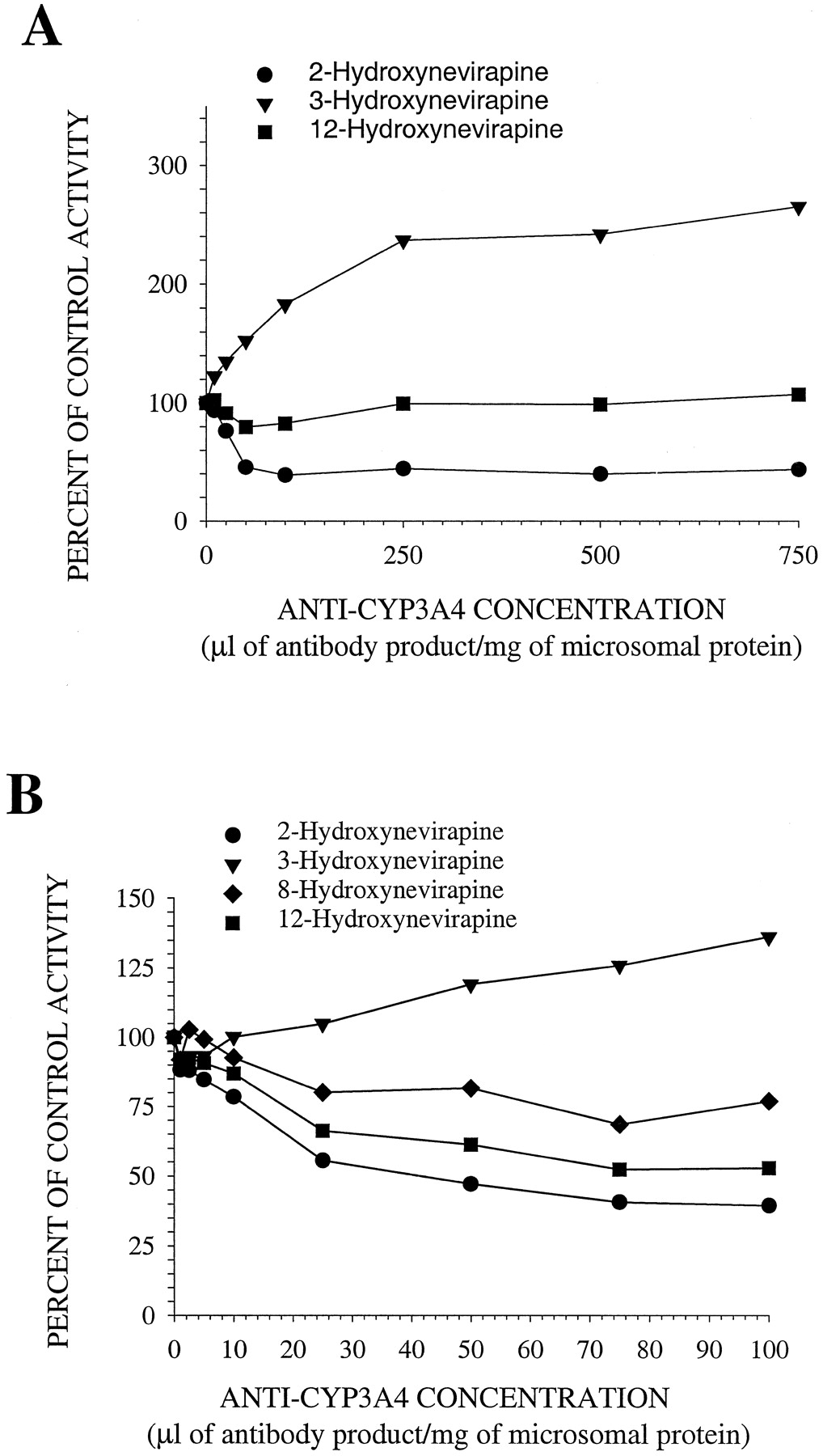
Antibody to CYP3A4 inhibited 2-hydroxyNVP formation by 60% in pooled human hepatic microsomes and stimulated 3-hydroxyNVP formation in a concentration-dependent manner (Fig. 3). The formation of 12-hydroxyNVP in the 25-μM NVP incubations was inhibited by 15 to 20% with <100 μl of antibody product/mg protein, but inhibition of 12-hydroxyNVP formation was not apparent at or above 250 μl of antibody product/mg protein. In the 400 μM NVP incubations, antibody to CYP3A4 inhibited the rate of 12-hydroxyNVP formation by 50%. 8-HydroxyNVP formation could not be measured in the 25 μM NVP incubations, but at 400 μM NVP, it was reduced by 25% in the presence of anti-CYP3A4.

The effect of monoclonal mouse antibody to CYP3A4 on the in vitro rates of formation of metabolites in incubations of pooled human hepatic microsomes with 25 μM (A) or 400 μM (B) NVP.
Control activities: 2-hydroxyNVP, 1.83 and 41.5 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively; 3-hydroxyNVP, 1.65 and 33.1 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively; 8-hydroxyNVP, 11.0 pmol/min/mg microsomal protein; 12-hydroxyNVP, 1.38 and 43.1 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively.
Antibody to CYP2B6 in pooled human hepatic microsomal incubations inhibited the formation of 3-hydroxyNVP by 80 to 85%, whereas the formation rates of 2- and 12-hydroxyNVP were unaffected (Fig.4). 8-HydroxyNVP was not detected in the 25 μM NVP incubations, but anti-CYP2B6 inhibited its formation by 30% in incubations containing 400 μM NVP.

The effect of monoclonal mouse antibody to CYP2B6 on the in vitro rates of formation of metabolites in incubations of pooled human hepatic microsomes with 25 μM (A) or 400 μM (B) NVP.
Control activities: 2-hydroxyNVP, 3.79 and 67.4 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively; 3-hydroxyNVP, 3.82 and 48.2 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively; 8-hydroxyNVP, 16.3 pmol/min/mg microsomal protein; 12-hydroxyNVP, 3.08 and 74.0 pmol/min/mg microsomal protein for 25 and 400 μM NVP, respectively.
Chemical Inhibition of the Biotransformation of NVP.
The effect of CYP-specific chemical inhibitors, i.e., quinidine, ketoconazole, troleandomycin, and erythromycin, on the rates of formation of the four NVP metabolites by human hepatic microsomes was investigated (Table 4).
The effects of chemical inhibitors on the in vitro rates of NVP metabolite formation by human hepatic microsomes and cDNA-expressed CYP in incubations with 100 μM NVP
Ketoconazole (0.5 or 2.5 μM) was a potent inhibitor of the formation of 2-, 3-, and 12-hydroxyNVP, inhibiting metabolite formation by 25 to 95%. 2-hydroxyNVP formation was most susceptible to inhibition by ketoconazole and 3-hydroxyNVP formation was the least susceptible. The slow rate of 8-hydroxyNVP formation did not allow an assessment of its inhibition by ketoconazole.
Troleandomycin (50 μM) was an effective inhibitor of the in vitro biotransformation of NVP to 2-hydroxyNVP, completely inhibiting its formation in human hepatic microsomes and cDNA-expressed CYP3A4. As with ketoconazole, troleandomycin was a more potent inhibitor of 2-hydroxyNVP formation (100%) than of 3- or 12-hydroxyNVP formation (25 and 70%, respectively) in human hepatic microsomes. Troleandomycin was unable to inhibit 3-hydroxyNVP formation in incubations of NVP with cDNA-expressed CYP2B6, but completely inhibited 12-hydroxyNVP formation by cDNA-expressed CYP3A4. As with ketoconazole, slow metabolism rates prevented the determination of the inhibition of 8-hydroxyNVP by troleandomycin.
Erythromycin (50 μM) was an effective inhibitor of 2- and 12-hydroxyNVP formation, reducing their rates of formation in incubations with human hepatic microsomes by 60 and 50%, respectively. Erythromycin was unable to inhibit the biotransformation of NVP to 3-hydroxyNVP in incubations containing either human hepatic microsomes or cDNA-expressed CYP2B6. Again, slow metabolism rates did not allow the determination of the inhibition of 8-hydroxyNVP by erythromycin.
Quinidine (15 μM) was unable to inhibit the rates of formation of 2-, 3-, or 12-hydroxyNVP in human hepatic microsomal incubations. The use of different human hepatic microsomes in this analysis allowed the measurement of the 8-hydroxyNVP formation rates and its formation was inhibited by 7%.
Inhibition of CYPs by NVP.
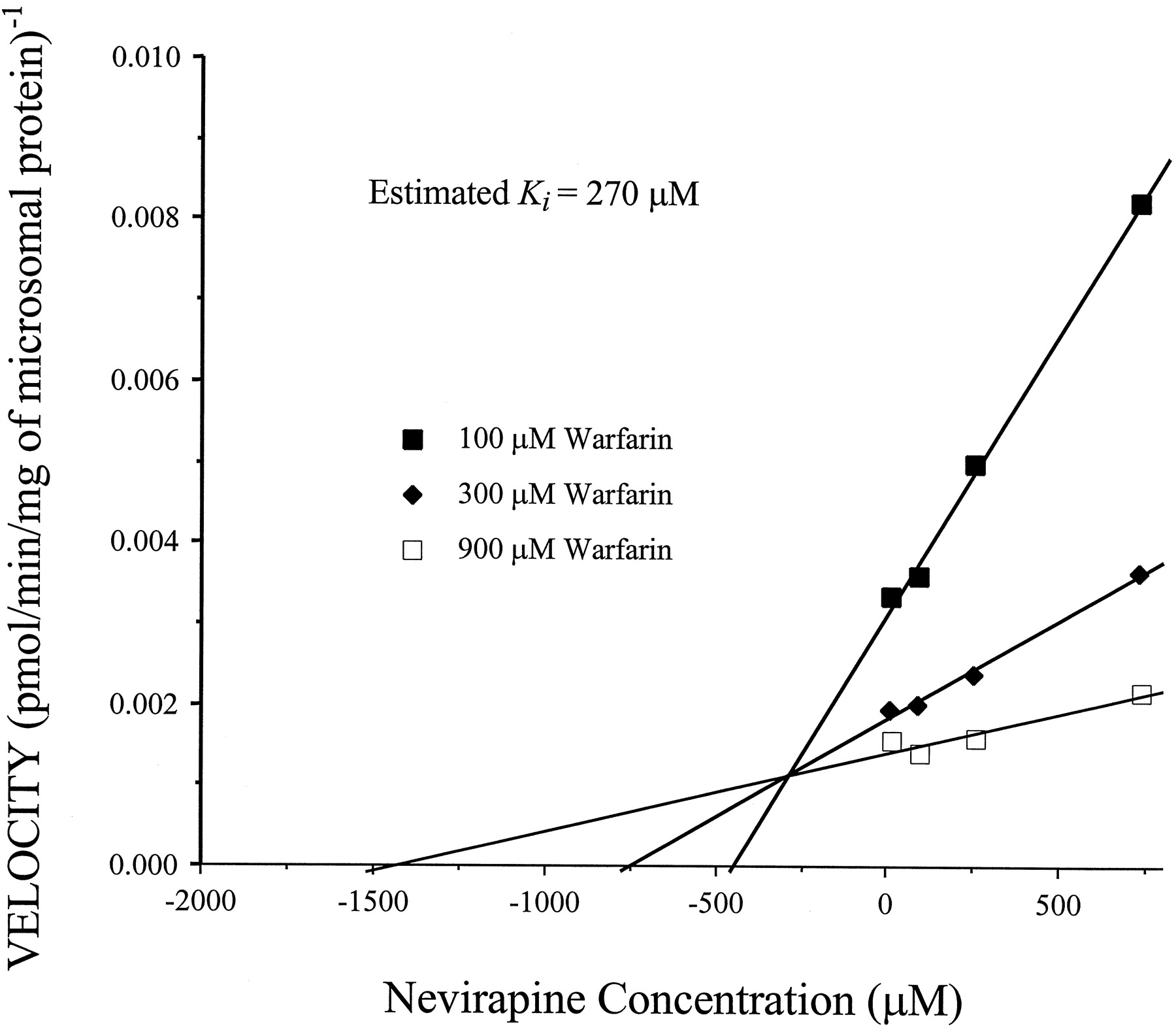
NVP was ineffective as an inhibitor of CYP1A2, 2A6, 2C9, 2C19, 2D6, or 2E1 at concentrations up to 250 μM. Only the CYP3A4-mediated 10-hydroxylation of (R)-warfarin was inhibited in human hepatic microsomal incubations in the presence of NVP. The apparentKi of this inhibition was estimated to be 270 μM (Fig. 5).

Dixon plot of the inhibition of (R)-warfarin 10-hydroxylase activity (CYP3A4) by NVP.
Discussion
The rates of formation of NVP metabolites were correlated against eleven different CYP-specific enzyme activities in characterized human hepatic microsomes at NVP concentrations of 25 and 100 μM. Two substrate concentrations were used because the in vitro rates of metabolism were more readily measured at 100 μM, but 25 μM NVP is more relevant to observed therapeutic plasma concentrations of NVP (therapeutic range <25 μM; Havlir et al., 1995). The two substrate concentrations also allowed an assessment to be made of the involvement of more than one CYP in the formation of the various metabolites. Similarly, two concentrations of NVP (25 and 400 μM) were used in the assays monitoring the effects of inhibitory antibodies to CYPs.
2-HydroxyNVP.
At both substrate concentrations, 2-hydroxyNVP formation was best correlated with 6β-hydroxytestosterone formation (CYP3A4). Only cDNA-expressed CYP3A4 and CYP3A5 were capable of forming 2-hydroxyNVP. Antibody to CYP3A4 inhibited 2-hydroxyNVP formation by 60%. The lack of complete inhibition of 2-hydroxyNVP formation could suggest the involvement of other CYP(s) in this biotransformation. However, Gelboin et al. (1995) have observed a 60 and 25% inhibition ofp-nitrophenol formation by anti-CYP3A4 in incubations with cDNA-expressed CYP3A4 and CYP3A5, respectively, and attributed the lack of complete inhibition to the poor affinity ofp-nitroanisole as a substrate. Accordingly, the 60% inhibition of 2-hydroxyNVP formation in human hepatic microsomal incubations by anti-CYP3A4 could have been due to the poor affinity of NVP for CYP3A4. The Km values of 212 and 279 μM for 2-hydroxyNVP formation in human hepatic microsomes and in microsomes containing cDNA-expressed CYP3A4, respectively, are supportive of this suggestion. Alternatively, the involvement of other CYP3A isoforms (e.g., CYP3A5) may be responsible for the lack of complete inhibition. Ketoconazole, troleandomycin, and erythromycin were all very effective inhibitors of 2-hydroxyNVP formation. Collectively, these data suggest that 2-hydroxyNVP is formed exclusively by the CYP3A subfamily.
3-HydroxyNVP.
At both concentrations of NVP, 3-hydroxyNVP formation was best correlated with furafylline-inhibited EFCOD and MND (CYP2B6) and only cDNA-expressed CYP2B6 formed 3-hydroxyNVP. TheKm values of 609 and 834 μM for 3-hydroxyNVP formation in human hepatic microsomes and in microsomes containing cDNA-expressed CYP2B6, respectively, are in good agreement with each other, suggesting that CYP2B6 is the enzyme responsible for 3-hydroxyNVP formation in human hepatic microsomes. Anti-CYP2B6 inhibited the formation of 3-hydroxyNVP by 80 to 85%. Only ketoconazole was an effective inhibitor of 3-hydroxyNVP formation and its inhibitory effect was similar in incubations with either expressed CYP2B6 or human hepatic microsomes. Troleandomycin moderately inhibited 3-hydroxyNVP formation in human hepatic microsomal incubations, but was ineffective as an inhibitor in cDNA-expressed CYP2B6. The reason for this is not clear, but the involvement of CYP3A4 in this biotransformation is not supported by other data. Collectively, these data suggest that CYP2B6 is the predominant (or only) enzyme forming 3-hydroxyNVP.
8-HydroxyNVP.
At 100 μM NVP, 8-hydroxyNVP formation was well correlated with furafylline-inhibited EFCOD, MND, and 6β-hydroxytestosterone. At 25 μM NVP, it was best correlated with furafylline-inhibited EFCOD, MND, and 7-coumarin hydroxylase (CYP2A6), but 8-hydroxyNVP was measurable in only five of the ten in vitro incubations. A role for CYP2A6 in 8-hydroxyNVP formation was not supported in studies with cDNA-expressed CYP2A6. Only cDNA-expressed CYP2D6 formed 8-hydroxyNVP well, but small amounts were formed by CYP3A4. The formation of 8-hydroxyNVP was only detectable in the 400 μM NVP incubations and was reduced by 25% in the presence of anti-CYP3A4. Although 8-hydroxyNVP formation was not observed in incubations with cDNA-expressed CYP2B6, inhibition of 8-hydroxyNVP formation by 30% in the presence of anti-CYP2B6 suggests a role for this enzyme in its formation. In most of the chemical inhibitor studies, analytical problems prevented the determination of the rate of 8-hydroxyNVP formation. However, a 7% inhibition of the formation of 8-hydroxyNVP was observed in human hepatic incubations containing 15 μM quinidine, supporting a role for CYP2D6 in 8-hydroxyNVP formation. Collectively, these data suggest that multiple enzymes, including CYP3A4, CYP2B6, and CYP2D6, are involved in 8-hydroxyNVP formation.
12-HydroxyNVP.
At both concentrations of NVP, 12-hydroxyNVP formation was best correlated with 6β-hydroxytestosterone formation (CYP3A4). cDNA-expressed CYP3A4, CYP3A5, and CYP2D6 were all capable of forming 12-hydroxyNVP in reasonable quantities and small amounts of 12-hydroxyNVP were formed in incubations with cDNA-expressed CYP2C9. The formation of 12-hydroxyNVP was slightly (if at all) inhibited by anti-CYP3A4 in incubations with 25 μM NVP. However, 12-hydroxyNVP formation was inhibited by 50% by anti-CYP3A4 in incubations containing 400 μM NVP. Ketoconazole, troleandomycin, and erythromycin were all effective inhibitors of 12-hydroxyNVP formation. Quinidine was unable to inhibit 12-hydroxyNVP formation in human hepatic microsomes. Collectively, these data suggest a role for the CYP3A4 subfamily in the formation of 12-hydroxyNVP, but imply that several enzymes, possibly including CYP2D6 and CYP2C9, may be capable of its formation, particularly at lower and more clinically relevant substrate concentrations.
Summary of CYP Reaction Phenotyping Studies.
Summarizing the data characterizing the enzymes involved in the biotransformation of NVP, 2- and 3-hydroxyNVP appear to be exclusively formed by CYP3A and CYP2B6, respectively. Whereas CYP3A4 plays a significant role in the biotransformation of both 8- and 12-hydroxyNVP, these two metabolites appear to also be formed by several other CYPs (i.e., CYP2B6, CYP2D6, and CYP2C9).
Clinical Implications.
In clinical trials, nearly all NVP was eliminated in the urine as glucuronides of 2-, 3-, 8-, and 12-hydroxyNVP (Riska et al., 1999). In vitro microsomal data was in good agreement with the clinical observations. 8-HydroxyNVP was present in human subjects as a minor metabolite whereas 2- and 12-hydroxyNVP were present as major metabolites. However, 3-hydroxyNVP was always present as a major metabolite in human subjects, but was a major metabolite in only half of the in vitro incubations. Because the clinical data were obtained from patients receiving NVP daily over the course of 4 weeks (Riska et al., 1999) and NVP has been suggested to be an inducer in humans of the CYPs responsible for its own metabolism (Cheeseman et al., 1995; Havlir et al., 1995; Lamson et al., 1995), a possible explanation for the consistent appearance of 3-hydroxyNVP as a major metabolite in human subjects may be that NVP is an inducer of CYP2B6, normally a minor constituent of total CYP in humans. NVP may also be an inducer of CYP3A4, and known inducers of CYP3A4 in humans, such as rifampin and rifabutin, have caused significant reductions of NVP plasma concentrations in the clinic (37 and 16%, respectively; VIRAMUNE package label, 1999). Although much less is known about either the role of CYP2B6 in the metabolism of drugs or its inducibility in humans, plasma concentrations of NVP may also be susceptible to reduction in individuals receiving inducers of CYP2B6. The significance of induction of CYP2B6 with respect to drug interactions may not be fully appreciated until the role of this enzyme in drug metabolism is more completely understood. Nevertheless, plasma concentrations of drugs that are substrates for CYP3A4 or CYP2B6 may be susceptible to reduction in individuals receiving NVP.
Apart from the significance of NVP as an inducer of CYPs in humans, the clinical significance of its potential to inhibit CYPs must also be considered. Among human hepatic CYPs, NVP was capable of inhibiting only the 10-hydroxylation of (R)-warfarin (CYP3A4). Thus, NVP should not affect the plasma concentrations of drugs that are substrates of CYPs 1A2, 2D6, 2A6, 2E1, 2C9, or 2C19. The estimatedKi for the inhibition of CYP3A4 was 270 μM, a concentration that is unlikely to be achieved in patients (therapeutic range <25 μM; Havlir et al., 1995). Therefore, NVP should have little inhibitory effect on other substrates of CYP3A4. Preliminary studies exploring the possible inhibition of MND (CYP2B6) by NVP have suggested that NVP does not inhibit this reaction at concentrations up to 100 μM (data not shown). Thus, it is unlikely that NVP would reduce the clearance of concomitantly administered drugs due to its low potential to inhibit hepatic CYPs.
Because NVP appears to be predominantly metabolized by two enzymes, CYP3A and CYP2B6, NVP clearance in patients is not likely to be reduced by coadministered medications. However, ketoconazole was a potent inhibitor of both CYP3A4 and CYP2B6 in microsomal incubations, suggesting that it could be involved in drug interactions with NVP. The potential for a clinically significant drug interaction between ketoconazole and NVP is presently under investigation.
In summary, NVP and concomitantly administered drugs, with the possible exception of ketoconazole, have little potential for inhibitory interactions. However, the capacity for NVP to induce CYP2B6 and/or CYP3A4 warrants caution when coadministering drugs that are primarily cleared by these metabolic pathways.
Because 2- and 3-hydroxyNVP appear to be formed exclusively by CYP3A and CYP2B6, respectively, NVP hydroxylation may be a suitable probe for the simultaneous determination of the expression of these enzymes in vitro. Presently, highly specific enzyme probes for CYP2B6 are rare. Furthermore, NVP may be useful as a clinical probe for the simultaneous determination of CYP2B6 and CYP3A4 activity in human subjects, although the presence of 2- and 3-hydroxyNVP in human urine primarily as glucuronide conjugates may limit its utility.
Acknowledgments
We thank Drs. Andrew Parkinson and Ajay Madan of XENOTECH LLC (Kansas City, MO) for providing EFCOD and MND data for the characterized human hepatic microsomes. We also thank Dr. Thomas Ebner and Veronika Diesch of Boehringer Ingelheim Pharma KG (Biberach, Germany) for providing preliminary data on the potential for NVP to inhibit MND in human hepatic microsomes and Dr. Maurice Morelock of Boehringer Ingelheim Pharmaceuticals, Inc., for the calculation of the Michaelis-Menten kinetics of NVP metabolite formation.
Footnotes
-
Send reprint requests to: David A. Erickson, M.Sc., Dept. of Drug Metabolism and Pharmacokinetics, R&D Center, Boehringer Ingelheim Pharmaceuticals, Inc., 175 Briar Ridge Rd., Ridgefield, CT 06877. E-mail: derickso{at}rdg.boehringer-ingelheim.com
-
↵1 Present address: Cedra Corp., 8609 Cross Park Dr., Austin, TX 78754.
- Abbreviations used are::
- NVP
- nevirapine
- CYP
- cytochrome P-450
- EFCOD
- 7-ethoxy-4-trifluoromethylcoumarin deethylase
- MND
- S-mephenytoin N-demethylase
- Received March 17, 1999.
- Accepted September 7, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}