


Abstract
Fluoxetine is one of the most widely prescribed selective serotonin reuptake inhibitors (SSRIs) that is marketed worldwide. However, details of its human hepatic metabolism have been speculative and incomplete, possibly due to the sensitivity of analytical techniques and selectivity of specific in vitro probes and reagents used. Studies with (R)-, (S)-, and racemic fluoxetine were undertaken to determine the stereospecific nature of its metabolism and estimate intrinsic clearance contributions of each CYP for fluoxetine N-demethylation. Measurable fluoxetineN-demethylase activity was catalyzed by CYP1A2, -2B6, -2C9, -2C19, -2D6, -3A4, and -3A5. All enzymes catalyzed this reaction for both enantiomers and the racemate, and intrinsic clearance values were similar for the enantiomers for all CYP enzymes except CYP2C9, which demonstrated stereoselectivity for R- over theS-enantiomer. Scaling the intrinsic clearance values for the individual CYP enzymes to estimate contributions of each in human liver microsomes suggested that CYP2D6, CYP2C9, and CYP3A4 contribute the greatest amount of fluoxetine N-demethylation in human liver microsomes. These data were corroborated with the examination of the effects of CYP-specific inhibitors quinidine (CYP2D6), sulfaphenazole (CYP2C9), and ketoconazole (CYP3A4) on fluoxetine N-demethylation in pooled human liver microsomes. Together, these findings suggest a significant role for the polymorphically expressed CYP2D6 in fluoxetine clearance and are consistent with reports on the clinical pharmacokinetics of fluoxetine.
Fluoxetine is a commonly prescribed antidepressant of the selective serotonin reuptake inhibitor (SSRI)3 class, which is generally clinically effective against many disorders in which serotonin plays a role (Wong et al., 1995). Early in vitro studies suggested that fluoxetine and its metabolite norfluoxetine were potent competitive inhibitors of CYP2D6-mediated metabolic pathways (Crewe et al., 1992; Otton et al., 1993). Both (R)-fluoxetine and (R)-norfluoxetine were also found to be more potent inhibitors of CYP2D6 than theS-isomers (Stevens and Wrighton, 1993). Additionally, fluoxetine has demonstrated varying in vitro inhibitory potency toward CYP2C19 (Kobayashi et al., 1995), CYP3A4 (von Moltke et al., 1994; Ring et al., 1995), and CYP2C9 (Schmider et al., 1997; Hemeryck et al., 1999). However, demonstration of inhibition of a CYP is not necessarily indicative that the same enzyme has a major role in its biotransformation. Some investigators have attempted to identify the enzymes responsible for fluoxetine metabolism using cosegregation studies, recombinant CYPs, CYP isoform-specific chemical inhibitors, and CYP isoform-specific inhibitory antibodies. Findings have been mixed, concluding with CYP2D6 and CYP3A4 (Stevens and Wrighton, 1993) or CYP2C9 (von Moltke et al., 1997) having a role in metabolism. More recently, CYP2C9 has been suggested as the major CYP responsible for metabolism of racemic fluoxetine (based on the inhibition by sulfaphenazole), and CYP2D6 was suggested as unimportant when experiments were conducted at fluoxetine concentrations of 100 μM (von Moltke et al., 1997). These in vitro findings do not agree with clinical pharmacokinetic data that demonstrate a major role for CYP2D6 in the clearance of fluoxetine because the area under the concentration versus time curve for unchanged fluoxetine was higher in CYP2D6-poor metabolizers than extensive metabolizers (Hamelin et al., 1996; Fjordside et al., 1999). Furthermore, in CYP2D6-poor metabolizers, clearance of (R)-fluoxetine is greater than (S)-fluoxetine due to another CYP besides CYP2D6 (Fjordside et al., 1999).
To date, there is not a close correlation between the reported in vivo and in vitro studies for the metabolism of fluoxetine. Some of the lack of agreement between the in vitro and in vivo data could be attributable to the high concentrations of fluoxetine (>50 μM) used in previous assessments of CYP enzyme contribution toN-demethylation in human liver microsomes. In this report, we describe a new sensitive HPLC/MS assay for norfluoxetine that enabled the examination of fluoxetine N-demethylation activity at substrate concentrations below 1.0 μM. Enzyme kinetic parameters were determined for (R)-, (S)-, and racemic fluoxetine in recombinant heterologously expressed human CYP enzymes to estimate contributions of these CYP enzymes in human liver. Additionally, CYP enzyme contribution to fluoxetineN-demethylase activity in human liver microsomes was assessed using CYP-specific inhibitors at a more pharmacologically relevant substrate concentration of 0.5 μM, as opposed to the high concentrations used by other investigators. The data reported herein are discussed in context to findings presented in previous reports (Stevens and Wrighton, 1993; von Moltke et al., 1997) and the clinical pharmacokinetics of fluoxetine (Hamelin et al., 1996; Fjordside et al., 1999). Additionally, because data were collected on the racemate as well as the two fluoxetine enantiomers, a better understanding of similarities and differences in pharmacokinetics between the stereoisomers can be obtained.
Experimental Procedures
Materials.
Baculovirus-expressed human CYP enzymes were prepared at Pfizer (Christopherson et al., 1995) and purchased from PanVera Corp. (Madison, WI) and Gentest Corp. (Woburn, MA). Human liver microsomes were generated at Pfizer using liver tissue from 54 individual donors. The inhibitors were obtained from the following sources: quinidine (Sigma, St. Louis, MO), ketoconazole (RDI, Flanders, NJ), and sulfaphenazole (Gentest). [3-(4-Methanesulfonylphenyl)-3-(4-trifluoromethylphenoxy)propyl]dimethylamine used as the internal standard was obtained from Peakdale Fine Chemicals (Bear, DE). Racemic fluoxetine and norfluoxetine were synthesized at Pfizer Central Research, and the individual fluoxetine enantiomers (−)-(R)-fluoxetine and (+)-(S)-fluoxetine were separated by chiral preparative HPLC on a Chiralcel OD column (Chiral Technologies, Exton, PA). Enantiomeric purity was >99%.
Recombinant Human CYP Incubations.
Incubation mixtures (0.2 ml) contained recombinant CYP at various microsomal protein concentrations (Table1) in a solution of potassium phosphate buffer (100 mM, pH 7.4) and MgCl2 (3.3 mM). Substrate concentration ranges are listed in Table 1. Each concentration was run in duplicate for each substrate [(R)-, (S)-, and racemic fluoxetine] and covered a 100-fold range. Incubations were initiated by the addition of NADPH (1.3 mM) and were allowed to proceed over a previously determined time period for which reaction velocity linearity had been demonstrated. Incubations were conducted at 37°C with shaking open to the air. Incubations were terminated by the addition of NaOH (50 μl, 1 M).
Summary of assay conditions for fluoxetine N-demethylation in heterologously expressed recombinant human CYP enzymes
Inhibition of Fluoxetine N-Demethylase in Human Liver Microsomes.
Fluoxetine (0.5 μM) was incubated with pooled human liver microsomes (1.0 mg/ml) in 100 mM potassium phosphate (pH 7.4) containing 3.3 mM MgCl2. Quinidine (0–10 μM), ketoconazole (0–10 μM), and sulfaphenazole (0–30 μM) were tested as inhibitors. Incubations were commenced with the addition of NADPH (1.3 mM), conducted at 37°C for 10 min as described above, and terminated with the addition of NaOH (50 μl, 1 M). Each data point represents the average of duplicate determinations.
Liquid-Liquid Extraction of Norfluoxetine.
To terminated incubation mixtures was added 0.1 ml of internal standard solution {25 μg/ml [3-(4-methanesulfonylphenyl)-3-(4-trifluoromethylphenoxy)propyl]dimethylamine in water}, and the sample was vortexed. Norfluoxetine and the internal standard were extracted with 3 ml of methyl-t-butyl ether. Samples were mixed on a multitube vortex mixer and spun in a centrifuge (2000g, 5 min), the aqueous layer was frozen in an acetone/dry ice bath, and the organic layer was decanted into a clean tube and evaporated under N2 at 30°C. Residues were reconstituted with 50 μl of HPLC mobile phase (10 mM ammonium formate, 0.1% formic acid/acetonitrile; 80:20) and transferred to injection vials.
Norfluoxetine HPLC/MS/MS Assay.
Concentrations of norfluoxetine were determined using atmospheric pressure chemical ionization tandem triple quadrupole mass spectrometry. A Hewlett Packard Series 1100 quad channel pump provided solvent delivery of an isocratic mobile phase consisting of 10 mM ammonium formate, 0.1% formic acid/acetonitrile (70:30). Separation of internal standard and norfluoxetine was achieved with a Phenomenex LUNA C18, 30 × 2 mm, 3-μm column (Phenomenex, Torrance, CA). Flow was 0.5 ml/min, and total run time was 3.5 min. Under these conditions internal standard and norfluoxetine had retention times of 1.2 and 2.3 min, respectively. Injection volume was 25 μl. Detection was achieved using a Sciex model 2000 LC/MS/MS mass spectrometer (Sciex, Thornhille, Ontario, Canada). Effluent was split before introduction into the turbo ion spray source so that flow into the mass spectrometer was 50 μl/min. Ionization was conducted in the positive ion mode at a source temperature of 150°C and using nitrogen for nebulizing and heating gas. Ion spray voltage was 4.0 kV, the orifice voltage was optimized at 20 eV, and the collision energy was adjusted to −15 eV. Internal standard and norfluoxetine were analyzed using multiple reaction monitoring—m/z 402 = >240 andm/z 296 = >134 for the internal standard and norfluoxetine, respectively. For norfluoxetine, this reaction followed the protonated parent mass [M + H]+ = 296 to its corresponding collision-induced dissociated fragment atm/z 134, which corresponded to the 3-phenylpropylamine. Eight standards prepared in inactive microsomal systems were analyzed in duplicate for each analytical run. The assay had a linear dynamic range of 0.01 to 2.5 μM. Calibration and regression of the curve were completed using 1/X2 weighting. Interassay accuracy was determined to be 102 and 123% at the lower and upper limits of quantitation, and corresponding precision values were 5 and 8%.
Data Analysis.
Substrate saturation data were plotted on Eadie-Hofstee plots to check for linearity or nonlinearity of the data. After this examination, the data were plotted on substrate saturation curves and fitted to the Michaelis-Menten equation using Deltagraph (version 4.0.3; SPSS Inc., Chicago, IL) to yield apparent KM andVmax values. Estimates of the contributions of the individual CYP enzymes to fluoxetine N-demethylation were accomplished by multiplying the intrinsic clearance values by the amounts of the CYP enzymes per milligram of liver microsomal protein, as described by Rodrigues (1999). Inhibition curves for quinidine, sulfaphenazole, and ketoconazole were fitted using Deltagraph using the function:
Results and Discussion
Since the introduction of fluoxetine (Wong et al., 1995) and other similarly acting molecules, SSRIs have become the treatment of choice in most cases of depression and related illnesses. This is despite the fact that several were quickly discovered to have drug-drug interaction issues with multiple CYPs as outlined in the many recent reviews on the topic of drug-drug interactions and SSRIs (Mitchell, 1997; Baker et al., 1998; Brosen, 1998; Greenblatt et al., 1998; Preskorn, 1998;Richelson, 1998).
Enzyme kinetic examination of fluoxetine N-demethylation in seven recombinant heterologously expressed human CYP enzymes demonstrated that all seven had measurable activity (Table2). All kinetic data appeared to behave in a classical Michaelis-Menten fashion, which contrasted with previous observations of substrate inhibition at high fluoxetine concentrations in human liver microsomes (Stevens and Wrighton, 1993; von Moltke et al., 1997). Intrinsic clearance values for (R)-, (S)-, and racemic fluoxetine were greatest for CYP2D6, with a rank order of CYP2D6 > CYP2C9 > CYP3A4 > CYP2C19 for (R)-fluoxetine, and CYP2D6 > CYP3A4 > CYP2C9 > CYP2C19 for (S)-fluoxetine. The other CYP enzymes examined had substantially lower intrinsic clearance values (Table 2). Apparent KM values were fairly similar for the two enantiomers for each of the CYP enzymes with the exception of CYP2C9, in which the KM for the R-enantiomer was 13-fold lower than that for theS-enantiomer and the intrinsic clearance was 5-fold greater (Table 2). CYP2B6 also demonstrated an enantiomeric difference with aKM for the R-enantiomer 4-fold lower than the S-enantiomer. Enzyme kinetic data for the racemate was generally represented by a “blending” of the kinetic data for the individual enantiomers. Substrate saturation plots for CYP2D6, CYP2C9, and CYP3A4 are shown in Fig.1.
Summary of average (standard error) enzyme kinetic parameters for fluoxetine N-demethylation in recombinant heterologously expressed human CYP enzymes2-a
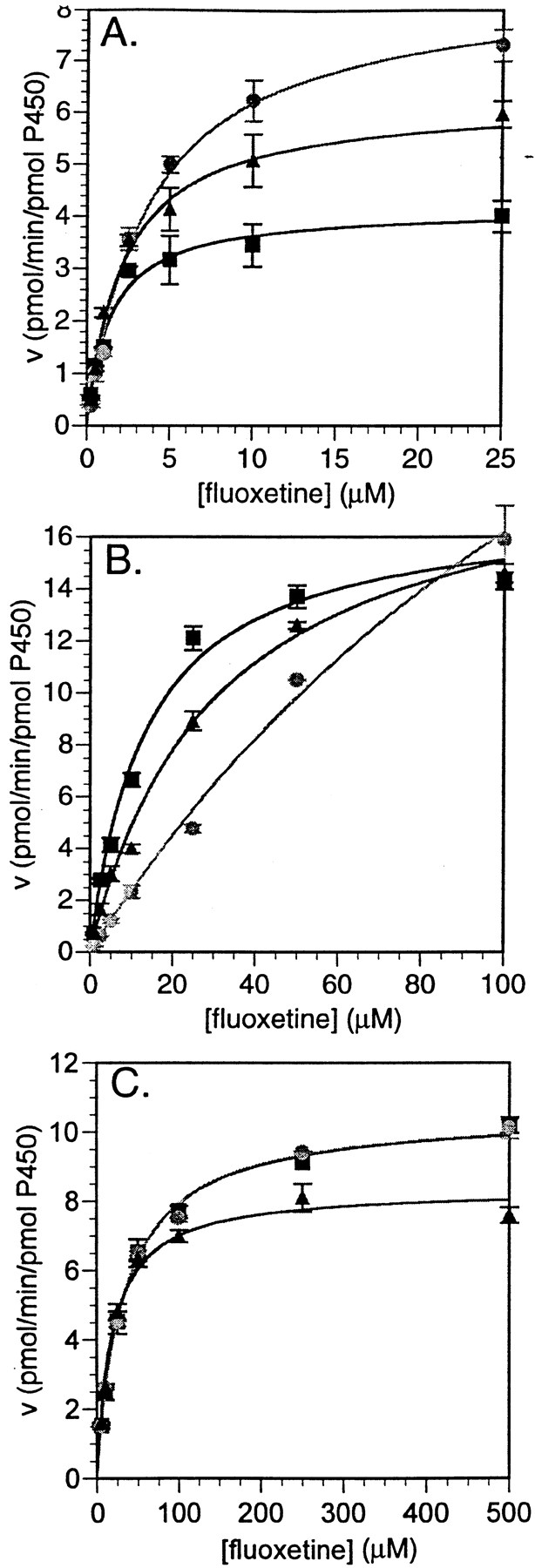
Substrate saturation plots for fluoxetine N-demethylase activities catalyzed by human CYP enzymes.
A, CYP2D6; B, CYP2C9; C, CYP3A4. Symbols represent (R)-fluoxetine (▪), (S)-fluoxetine (●), and racemic fluoxetine (▴). Data represent the average values of two determinations.
When these intrinsic clearance data were adjusted to reflect expression levels of individual CYP enzymes in human liver, it appeared that CYP2D6, CYP3A4, and CYP2C9 were the predominant enzymes involved in fluoxetine N-demethylation (Table3). Despite CYP2D6 possessing the greatest intrinsic clearance value on a per CYP basis, the expression level of this enzyme in the liver is substantially lower than that of CYP3A4 and CYP2C9. Thus, the latter two enzymes would be predicted to also contribute substantially to fluoxetine N-demethylation in vivo. Additionally, it would be estimated that CYP2C9 is associated with a greater impact on the metabolism of the R-enantiomer than the S-enantiomer.
Estimates of relative contribution of CYP enzymes to fluoxetine N-demethylation in human liver microsomes using recombinant CYP intrinsic clearance data
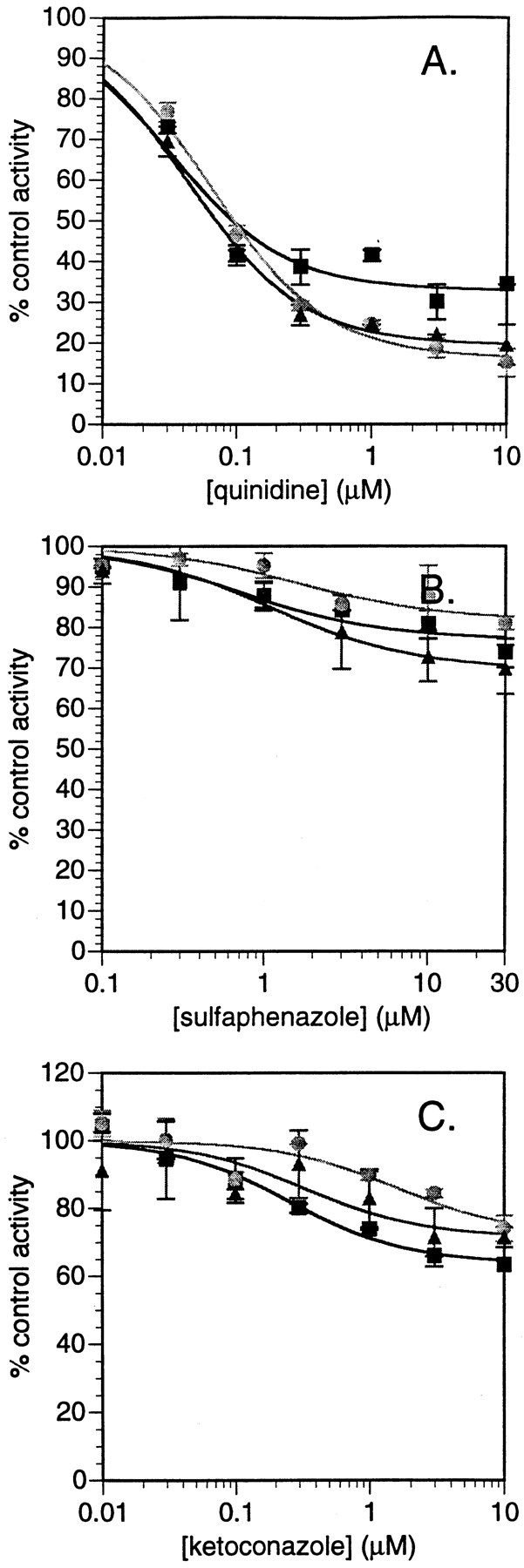
Based on these findings using recombinant CYP enzymes, experiments were undertaken to examine the effect of the CYP-specific inhibitors quinidine (CYP2D6), sulfaphenazole (CYP2C9), and ketoconazole (CYP3A4) on fluoxetine N-demethylation in pooled human liver microsomes. Key to this approach was not only the determination of whether the inhibitory potency on fluoxetine N-demethylation of these inhibitors was consistent with their known inhibitory potencies for CYP isoform-specific marker activities, but also determination of the “maximum inhibition” calculated by fitting the data to inhibition functions. This latter value should be indicative of the percentage contribution of the various CYP enzymes to the reaction in human liver microsomes. Importantly, these experiments were conducted at a lower fluoxetine concentration (0.5 μM) than had been previously examined in this type of experiment (50 and 100 μM were used previously; Stevens and Wrighton, 1993; von Moltke et al., 1997). The increased sensitivity achieved through the LC/MS/MS assay described herein allowed us to pursue these studies at subsaturating substrate concentrations. Complete inhibition was not observed with any of the three inhibitors (Fig. 2). Quinidine demonstrated the greatest inhibition of fluoxetineN-demethylase for (R)-, (S)-, and racemic fluoxetine with maximal inhibition values of 67, 84, and 81%, respectively (Table 4). Because the apparent KM for fluoxetineN-demethylation catalyzed by CYP2D6 was 1.4 to 3.8 μM, any examination of quinidine inhibition would be misleading if the substrate concentrations used in such an examination exceed these lowKM values (i.e., CYP2D6 activity will be saturated). Furthermore, clinically efficacious systemic concentrations of fluoxetine are in the range of 15 to 50 ng/ml (0.05–0.16 μM), with free systemic concentrations even lower (due to 94% plasma protein binding) (Benet et al., 1996), which are values well under theKM for CYP2D6-mediated fluoxetineN-demethylation. Thus, it would be expected that CYP2D6-mediated systemic clearance of fluoxetine would not be saturated in vivo. Maximal inhibition values for ketoconazole and sulfaphenazole ranged around 20 to 40% each, indicating a lesser role for these CYP enzymes in fluoxetine N-demethylation in human liver microsomes (Table 4). The contribution of CYP2D6, CYP3A4, and CYP2C9 estimated from the inhibition data and the intrinsic clearance data from recombinant CYP do not precisely agree. This is most likely due to imprecision of measurements of CYP isoform content in human liver microsomes used in the scaling of the intrinsic clearance data measured in recombinant CYP enzymes as well as some overlap of inhibitory activity of the CYP isoform-specific inhibitors at elevated inhibitor concentrations. Nevertheless, the same overall conclusion of the importance of CYP2D6 in fluoxetine N-demethylation can be made.
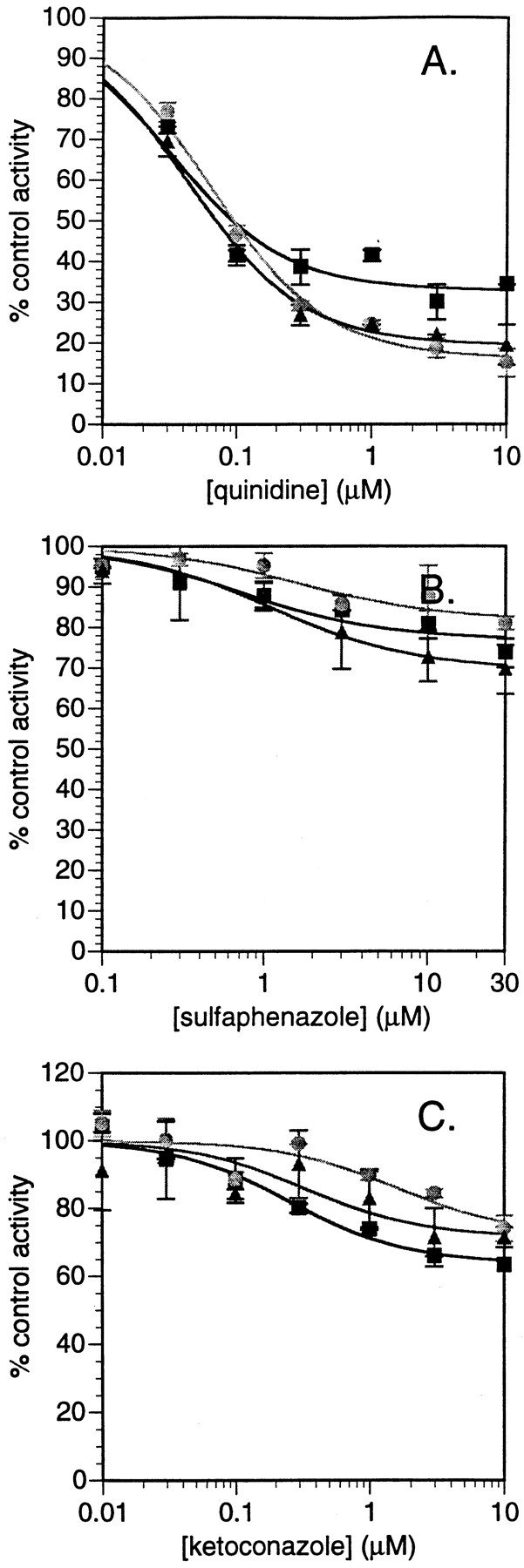
Inhibition of fluoxetine N-demethylase activity in human liver microsomes by CYP-specific inhibitors.
A, quinidine; B, sulfaphenazole; C, ketoconazole. Symbols represent (R)-fluoxetine (▪), (S)-fluoxetine (●), and racemic fluoxetine (▴). Data represent the average values of two determinations.
Inhibition of fluoxetine N-demethylation in human liver microsomes by CYP specific inhibitors
In conclusion, the in vitro data presented herein support a role for CYP2D6, CYP3A4, and CYP2C9 in the N-demethylation of fluoxetine in human liver microsomes. The major role of CYP2D6 in fluoxetine N-demethylation in vitro is consistent with data generated in CYP2D6 extensive and poor metabolizer subjects (Hamelin et al., 1996; Fjordside et al., 1999), and clarifies previously reported in vitro findings on the CYP enzymes involved in fluoxetineN-demethylation (Stevens and Wrighton, 1993; von Moltke et al., 1997). The importance of the N-demethylation pathway in the overall clearance of fluoxetine in humans has not been unequivocally and quantitatively determined with14C-labeled clinical drug disposition studies as >70% of drug-related material in excreta was not identified (Lemberger et al., 1985). However the importance of CYP2D6 in theN-demethylation pathway demonstrated in these in vitro studies, combined with the reported CYP2D6 extensive metabolizer/poor metabolizer differences in fluoxetine pharmacokinetics (Hamelin et al., 1996; Fjordside et al., 1999), is highly suggestive of fluoxetine N-demethylation as a major clearance mechanism for this drug.
Acknowledgments
We gratefully acknowledge the efforts of Melissa Rockwell, Christina Lydon, Dan Virtue, and Dr. Roderic O. Cole for preparative scale chiral separation of fluoxetine enantiomers and David Raunig for assistance with statistics.
Footnotes
-
Send reprint requests to: R. Scott Obach, Drug Metabolism Department, Candidate Synthesis, Enhancement, and Evaluation, Central Research Division, Pfizer, Inc., Groton, CT 06340. E-mail: obachrs{at}groton.pfizer.com
-
↵1 Present address: University of Connecticut Health Center, Farmington, CT 06030.
-
↵2 Present address: Lilly Research Laboratories, Eli Lilly and Co., Indianapolis, IN 46285.
- Abbreviations used are::
- SSRI
- selective serotonin reuptake inhibitor
- CYP
- cytochrome P450
- HPLC/MS/MS
- high pressure liquid chromatography-tandem mass spectrometry
- Received May 1, 2000.
- Accepted July 5, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}