


Abstract
The maintenance of the major hepatic cytochrome P450 (CYP) enzymes has been studied in precision-cut human liver slices cultured for up to 72 h in supplemented RPMI 1640 medium. The relative apoprotein levels of 11 CYP enzymes were determined using a panel of antipeptide antibodies. In addition, 7-ethoxyresorufin O-deethylase, tolbutamide methylhydroxylase, debrisoquine 4-hydroxylase, and testosterone 6β-hydroxylase activities were determined as enzymatic markers for CYP1A2, CYP2C9, CYP2D6, and CYP3A4, respectively. There was a large variation in the rate of decline of different CYP levels with time in culture. Based on the rate of decrease, CYP enzymes could be separated into two groups, with CYP2C9, CYP2D6, CYP3A4, and CYP4A11 being relatively stable (half-lives between 70 and 104 h), compared with CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2E1, and CYP3A5, which were relatively unstable (half-lives between 23 and 36 h). Enzyme activities decreased at rates similar to those of their corresponding apoproteins. There was also a large difference in the stability of individual CYP enzymes from different liver donors, particularly for the most rapidly declining CYP enzymes. Similar losses of CYP enzymes were found when human liver slices were cultured in supplemented Williams' medium E for 72 h, except that CYP2E1 apoprotein levels were better maintained. Because of the variable decreases of CYP enzymes, xenobiotic metabolism studies are best performed with freshly cut rather than cultured human liver slices.
Hepatic cytochrome P450 (CYP)1 enzymes are known to have a major role in the metabolism of both xenobiotics (including therapeutic agents) and certain endogenous compounds (Conney, 1986; Nelson et al., 1996; Parkinson, 1996; Pelkonen et al., 1998). In recent years there has been much interest in the development of in vitro systems to study the metabolism and toxicity of new chemical entities. Available liver in vitro models include precision-cut liver slices, hepatocytes, subcellular fractions, heterologous expression systems, and permanent and immortalized cell lines (Wrighton et al., 1995; Paine, 1996; Lake, 1997).
Depending on the desired application, these various liver in vitro models have both advantages and disadvantages. For example, although containing only a limited range of xenobiotic-metabolizing enzymes, preparations of liver microsomes can be stored for extended periods (Wrighton et al., 1995; Pearce et al., 1996). In contrast, hepatocytes and precision-cut liver slices possess a full range of phase I and II xenobiotic-metabolizing enzyme activities. However, total CYP content and individual CYP enzymes are known to decline with time in cultured rodent and human hepatocytes (Paine, 1990; LeCluyse et al., 1996;Maurel, 1996). Although xenobiotic-metabolizing enzymes may be stable for short periods in rat liver slices (Ekins, 1996), a decline in xenobiotic-metabolizing enzymes and total CYP content has been reported in cultured rat liver slices (Wright and Paine, 1992; Lake et al., 1993; Jensen et al., 1997; Müller et al., 1998).
To date, few investigations have examined the stability of xenobiotic-metabolizing enzymes in cultured precision-cut human liver slices. Based on immunoblotting of freshly cut and 72-h cultured human liver slice microsomes, a fall in the levels of CYP1A2 and CYP3A4 has been reported (Lake et al., 1996, 1997, 1998). In addition,VandenBranden et al. (1998) reported the loss of a number of CYP-dependent enzyme activities in human liver slices cultured for up to 96 h.
The aim of this study was to examine the stability of the major hepatic CYP enzymes in cultured human liver slices. Levels of CYP1A2, CYP2A6, CYP2B6, CYP2D6, CYP2E1, CYP3A4, CYP3A5, and CYP4A11 were quantified by immunoblotting with a panel of monospecific antipeptide antibodies (Edwards et al., 1998). In addition, levels of CYP2C enzymes were quantified with an antibody that binds to CYP2C8, CYP2C9, and CYP2C19, these CYP forms being readily separated by SDS-polyacrylamide gel electrophoresis (Edwards et al., 1998). Finally, enzymatic markers for CYP1A2, CYP2C9, CYP2D6, and CYP3A4, i.e., 7-ethoxyresorufinO-deethylase, tolbutamide methylhydroxylase, debrisoquine 4-hydroxylase, and testosterone 6β-hydroxylase, respectively, were also determined.
Materials and Methods
Chemicals.
The sources of the tissue culture materials were as described previously (Beamand et al., 1993). Debrisoquine, 7-ethoxyresorufin, testosterone, tolbutamide, resorufin, 6β-hydroxytestosterone, enzyme cofactors, prestained molecular weight markers, and protein G coupled to horseradish peroxidase were purchased from Sigma-Aldrich Co. Ltd. (Poole, Dorset, UK), and 4-hydroxydebrisoquine and hydroxytolbutamide were obtained from Salford Ultrafine Chemicals and Research Ltd. (Manchester, UK). [Guanidine-14C]debrisoquine (specific activity, 55 mCi/mmol) and [4-14C]testosterone (specific activity, 56 mCi/mmol) were obtained from Amersham International plc (Little Chalfont, Bucks, UK), and SDS-polyacrylamide gel electrophoresis reagents from National Diagnostics (Aylesbury, Bucks, UK).
Preparation of Liver Slices.
Samples of human liver were collected and transported to TNO BIBRA on ice. The donors of the eight human liver samples, designated subjects A to H, were a male aged 2½ years, a female aged 12 years, males aged 15 and 17 years, and females aged 74, 31, 38, and 55 years. The Research Ethics Committee of King's College Hospital granted approval for the use of this tissue, which was surplus to clinical requirements. Tissue cylinders from liver samples were prepared using a 10-mm diameter motor-driven tissue-coring tool. From the cylinders, tissue slices (200–300 μm) were prepared in oxygenated (95% O2/5% CO2) Earle's balanced salt solution containing 25 mM d-glucose, 50 μg/ml gentamicin, and 2.5 μg/ml fungizone at room temperature using a Krumdieck tissue slicer (Alabama Research and Development Corp., Munford, AL).
Culture of Liver Slices.
Liver slices were floated onto Vitron Inc. (Tucson, AZ) type C titanium roller inserts (two slices per insert) and cultured at 2 rpm in glass vials containing 1.7 ml of culture medium using a Vitron dynamic organ culture incubator. The incubator was operated in accordance with the manufacturer's instructions. For studies with liver samples A to E, the culture medium consisted of RPMI 1640 containing 5% (v/v) fetal calf serum, 0.5 mM (final concentration) l-methionine, 1 μM insulin, 0.1 mM hydrocortisone-21-hemisuccinate, 50 μg/ml gentamicin, and 2.5 μg/ml fungizone. Liver slice cultures were maintained at 37°C in an atmosphere of 95% O2/5% CO2. After 1 h, the medium was changed to fresh culture medium, and subsequently the medium was changed every 24 h. Following each medium change and returning the vials to the incubator, the gas flow was increased for 10 s to restore the high oxygen atmosphere. In the studies with liver samples F to H, the culture medium consisted of serum-free Williams' medium E containing 2 mM l-glutamine, 0.1 μM insulin, 0.1 μM dexamethasone, 50 μg/ml gentamicin and 2.5 μg/ml fungizone. Liver slice cultures were maintained as described above with medium changes after 1 h and subsequently every 24 h.
Biochemical Investigations with Liver Slices.
At the end of the culture periods, the liver slices were washed in ice-cold 0.154 M KCl containing 50 mM Tris-HCl, pH 7.4, homogenized in this medium by sonication (Beamand et al., 1993), and stored at −80°C. Liver slice homogenates (prepared from 16–20 slices) were centrifuged at 10,000g av for 15 min to obtain the postmitochondrial supernatant fraction and subsequently at 158,000g average centrifugal force for 40 min to separate the microsomal fraction from the cytosol. Microsomal fractions were resuspended in 0.154 M KCl containing 50 mM Tris-HCl, pH 7.4, and 2 mM EDTA, and aliquots were stored at −80°C. Protein was determined by the method of Lowry et al. (1951) using bovine serum albumin as standard. Liver slice unwashed microsomal fractions were assayed for activities of 7-ethoxyresorufin O-deethylase, tolbutamide methylhydroxylase, debrisoquine 4-hydroxylase, and testosterone 6β-hydroxylase as markers of CYP1A2, CYP2C9, CYP2D6, and CYP3A4, respectively. 7-Ethoxyresorufin O-deethylase activity was determined directly at 37°C in spectrofluorimeter cuvettes containing 2 μM 7-ethoxyresorufin, 0.5 mM NADP+, 7.5 mMdl-isocitric acid, 0.5 U/ml of isocitric dehydrogenase, 5 mM MgSO4, 0.15 to 0.70 mg of microsomal protein, and 50 mM Tris-HCl buffer, pH 8.4, in a final volume of 2 ml. Resorufin formation was monitored at wavelengths of 535-nm excitation and 582-nm emission. Tolbutamide methylhydroxylase was determined in incubation mixtures containing 1 mM tolbutamide, 1 mM NADP+, 7.5 mM dl-isocitric acid, 2 U/ml isocitric dehydrogenase, 5 mM MgSO4, 0.3 to 0.4 mg of microsomal protein, and 0.1 M phosphate buffer, pH 7.4, in a final volume of 0.5 ml. Incubations were terminated after 30 min with 0.02 ml of 70% (v/v) perchloric acid, and the supernatant was analyzed by high performance liquid chromatography (HPLC). Chromatography was performed with a 150- × 4.6-mm column of Supelcosil-5 LC-ABZ protected by a 20- × 4-mm column of Supelcosil-5 LC-ABZ and a mobile phase consisting of 30% (v/v) acetonitrile and 70% (v/v) 20 mM sodium perchlorate, pH 2.5, at a flow rate of 2 ml/min. The eluant was monitored at 230 nm. Debrisoquine 4-hydroxylase was determined in incubation mixtures containing 1 mM [guanidine-14C]debrisoquine (0.5 μCi/tube), 1.0 mM NADP, 7.5 mM dl-isocitric acid, 2 U/ml isocitric dehydrogenase, 5 mM MgSO4, 0.15 to 0.30 mg of microsomal protein, and 0.1 M phosphate buffer, pH 7.4, in a final volume of 0.25 ml. Incubations were terminated after 60 min with 0.02 ml of 70% (v/v) perchloric acid, and the supernatant was taken for HPLC analysis. Chromatography was performed as described for tolbutamide methylhydroxylase, but using a mobile phase of 12% (v/v) acetonitrile and 88% (v/v) 20 mM sodium perchlorate, pH 2.5, with quantitation by liquid scintillation counting. Testosterone 6β-hydroxylase was determined in incubation mixtures containing 0.25 mM [4-14C]testosterone (0.25 μCi/tube), 1 mM NADPH, 0.10 to 0.47 mg of microsomal protein, and 50 mM Tris-HCl buffer, pH 7.4, in a final volume of 1 ml. Incubations were terminated after 30 min with 6 ml of dichloromethane and processed for HPLC analysis with quantitation by liquid scintillation counting as described by Sonderfan et al. (1987).
Immunoblotting.
Immunoblotting was performed using 10 to 100 μg of liver slice unwashed microsomal protein, as appropriate, under conditions previously described (Edwards et al., 1988, 1994). Microsomal proteins were separated by SDS-polyacrylamide gel electrophoresis using 9% (w/v) polyacrylamide gels and electrotransferred onto nitrocellulose filters. The immunoblots were then developed for immunoreactivity using the respective antiserum containing antibodies targeted against CYP1A2 (diluted 1:4000), CYP2A6 (diluted 1:1000), CYP2B6 (diluted 1:4000), CYP2C forms (diluted 1:4000), CYP2D6 (diluted 1:1000), CYP2E1 (diluted 1:4000), CYP3A4 (diluted 1:4000), CYP3A5 (diluted 1:4000), or CYP4A11 (diluted 1:1000). All antibodies had previously been shown to be specific for their target protein (Edwards et al., 1998). Antibody binding was detected using protein G coupled to horseradish peroxidase (12.5 ng/ml), visualized using enhanced chemiluminescence reagents, and recorded on Hyperfilm. The relative intensity of the immunoreactive bands was determined by laser densitometry using an LKB Ultrascan XL Enhanced Densitometer (Pharmacia LKB Biotechnology, St. Albans, Herts, UK).
Additional Studies with Liver Sample Microsomal Fractions.
Liver samples used to prepare the freshly cut human liver slices were stored at −80°C. The samples were thawed, and washed microsomal fractions were prepared as described previously (Lake, 1987). Whole homogenates (0.25 g of tissue/ml) were prepared in ice-cold 0.154 M KCl containing 50 mM Tris-HCl, pH 7.4, using a Potter-type, Teflon-glass, motor-driven homogenizer (A. H. Thomas, Philadelphia, PA). Liver whole homogenates were centrifuged at 10,000g av for 20 min to obtain the postmitochondrial fractions and subsequently at 158,000g av for 40 min to separate the microsomal fraction from the cytosol. The microsomal fraction was resuspended in fresh homogenizing medium and again centrifuged at 158,000g av for 40 min. To mimic the preparation of the liver slices, thin slivers of liver were cut from the liver samples. Whole homogenates were prepared by sonication, and unwashed microsomal fractions were obtained as described above for the freshly cut and cultured liver slices. Liver sample washed liver microsomes and the unwashed microsomal fractions from the sonicated liver slivers were assayed for 7-ethoxyresorufinO-deethylase and testosterone 6β-hydroxylase activities as described above.
Statistical Analysis.
Statistical evaluation of data was performed using two-way ANOVA and Student's t test (PRISM and INSTAT; Graphpad Software Inc., San Diego, CA). CYP enzyme half-lives were derived using semilog plots of CYP levels (either from individual livers or using data pooled from all livers) against time and the equation for the line of best fit established by linear regression analysis (FigP Software Corp., Durham, NC). From this equation, the time at which half-maximal CYP levels occurred was identified and used as the half-life.
Results
Precision-cut human liver slices were prepared from five donors (subjects A to E) and cultured in RPMI 1640 medium containing 5% fetal calf serum and other additions for periods of 1, 24, 48, and 72 h. Microsomal preparations were prepared from freshly cut and cultured liver slices. Because of the small amounts of material available, it was not possible to perform all the measurements on all human liver slice preparations. Moreover, because of the small amounts of tissue, we prepared unwashed microsomal fractions from sonicated liver slice whole homogenates (see Materials and Methods).
CYP Levels in Freshly Cut Slices and Slices Cultured for 1 h.
Immunoblotting of liver slice microsomes was performed to assess the relative levels of CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, CYP3A5, and CYP4A11. Generally, there was little difference between levels of any of the CYP apoproteins measured in microsomes from freshly cut and 1-h cultured human liver slices (Fig.1). The activities of 7-ethoxyresorufinO-deethylase, tolbutamide methylhydroxylase, debrisoquine 4-hydroxylase, and testosterone 6β-hydroxylase were determined as enzymatic markers for CYP1A2, CYP2C9, CYP2D6, and CYP3A4, respectively (Wrighton et al., 1995; Parkinson, 1996; Pelkonen et al., 1998). Although the activities of 7-ethoxyresorufin O-deethylase, tolbutamide methylhydroxylase, and testosterone 6β-hydroxylase were similar in microsomes from freshly cut and 1-h cultured human liver slices, debrisoquine 4-hydroxylase activity was somewhat higher in 1-h cultured liver slice microsomes (Table1).

Relative levels of 10 CYP apoproteins in human liver slices cultured for 1 h in supplemented RPMI 1640 medium, compared with levels obtained from freshly cut slices from the same liver donors.
Microsomes were prepared from liver slices, and immunoblotting was performed as described under Materials and Methods. Results are presented as either mean (n = 2 for CYP2A6) or mean ± S.E. (n = 3 for CYP2C19 and CYP3A5; n = 4 for CYP2D6 and CYP3A4;n = 5 for CYP1A2, CYP2C8, CYP2C9, CYP2E1, and CYP4A11).
Microsomal CYP enzyme activities in freshly cut and 1-h cultured human liver slices
In some additional studies, microsomal fractions were obtained from some of the liver samples used to prepare the freshly cut liver slices. The liver samples were thawed, whole homogenates were prepared with a Potter-type homogenizer, and washed microsomes were prepared as described under Materials and Methods. In addition, to mimic the preparation of precision-cut liver slices, thin slivers of liver were cut, and whole homogenates were prepared by sonication. Unwashed microsomes were prepared from these samples in the same manner as described under Materials and Methods for liver slices. In the liver sample washed microsomal fractions, 7-ethoxyresorufinO-deethylase activity was 48.9 pmol/min/mg of protein (n = 3; range 37.4–60.5 pmol/min/mg of protein), and testosterone 6β-hydroxylase activity was 1186 pmol/min/mg of protein (mean of two liver samples). These enzyme activities are within the ranges observed for other preparations of human liver microsomes used in our laboratories (Edwards et al., 1994, 1998; Renwick et al., 1998). However, in the unwashed microsomal preparations obtained from the sonicated homogenates from the liver slivers, mean 7-ethoxyresorufin O-deethylase and testosterone 6β-hydroxylase activities were 24.6 and 661 pmol/min/mg of protein, respectively. These activities were thus only 50 and 56%, respectively, of the liver sample washed microsome activities. Although the liver slice microsomal enzyme activities reported in Table 1 are at the low end of the range of enzyme activities normally observed in human liver microsomes, we do not consider that this is due to a lack of viability of the liver samples used to prepare the freshly cut liver slices. Rather, as demonstrated by the above additional studies, they are attributable to the homogenization and centrifugation procedures used to prepare unwashed microsomes from the freshly cut and cultured liver slices.
Stability of CYP Enzymes in Cultured Liver Slices.
Human liver slices were cultured for up to 72 h, and CYP apoprotein levels were measured in microsomes prepared from the slices. The level of each CYP apoprotein declined with time in culture, although there was considerable variation in the rates of decrease of the 10 different CYP enzymes (Fig. 2). Figure 3 shows mean data for the 10 CYP enzymes, together with a single experiment where CYP2B6 apoprotein levels were also quantified. The levels of CYP2C9, CYP2D6, CYP3A4, and CYP4A11 were relatively stable (Fig. 3), with 45 to 64% remaining after 72 h in culture and overall half-lives of 70 h or greater (Table 2), whereas CYP1A2, CYP2A6, CYP2C8, CYP2C19, CYP2E1, and CYP3A5 levels declined more rapidly to around 11 to 28% of initial levels after 72 h in culture (Fig. 3), resulting in overall half-lives of between 23 and 36 h (Table 2). Although there was considerable variation in the half-lives of CYP enzymes in liver slices from different donors, there was no relationship between CYP stability and donor liver (P = .135; Kruskal-Wallis nonparametric ANOVA test). Thus, the half-life of each CYP varied independently of other CYP enzymes within each donor liver. As a result of this, variation in the level of each CYP increased progressively during culture. The mean coefficient of variation for all 10 CYP apoproteins was 24% after 1 h in culture, but by 72 h this had increased to 86%. However, there was much less variation in the levels of three of the more stable CYP apoproteins, namely CYP2C9, CYP2D6, and CYP4A11 (Fig.2).

Effect of culture of human liver slices in supplemented RPMI 1640 medium for up to 72 h on levels of 10 CYP apoproteins.
Microsomes were prepared from liver slices, and immunoblotting was performed as described under Materials and Methods. The level of each CYP enzyme was expressed as a percentage of that of the respective freshly cut slice level and is represented as the mean and range (n = 2 for CYP2A6) or the mean and S.D. (n = 3 for CYP2C19 and CYP3A5;n = 4 for CYP2D6 and CYP3A4; andn = 5 for CYP1A2, CYP2C8, CYP2C9, CYP2E1, and CYP4A11). In each case, the line of best fit was determined by linear regression analysis. In some cases (∗), the magnitude of the lower error bar indicating S.D. exceeded the scale of the ordinate.

Comparison of the rate of decrease in the levels of CYP apoproteins in human liver slices after culture.
Data for each of the CYP apoproteins shown in Fig. 2 have been combined, and the scale of the ordinate expanded to allow ready comparison between apoprotein levels. The level of each CYP enzyme was expressed as a percentage of that of the respective freshly cut slice level. The mean values at each time point have been plotted, representing CYP1A2 (▪, n = 5), CYP2A6 (▴,n = 2), CYP2B6 (★, n = 1) CYP2C8 (▾, n = 5), CYP2C9 (⧫,n = 5), CYP2C19 (●, n = 3), CYP2D6 (■, n = 4), CYP2E1 (▵,n = 5), CYP3A4 (▿, n = 4), CYP3A5 (◊, n = 3), and CYP4A11 (○,n = 5) apoproteins with lines of best fit calculated by linear regression analysis.
Half-lives of CYP apoproteins and enzyme activities determined in human liver slices
Enzyme activities declined at similar rates to reductions in the corresponding apoprotein levels (Fig. 4, Table 2). Thus, there were good correlations between 7-ethoxyresorufinO-deethylase activity and CYP1A2 apoprotein content, testosterone 6β-hydroxylase activity and CYP3A4 apoprotein content, and tolbutamide methylhydroxylase activity and CYP2C9 apoprotein content. However, there was a slight divergence between debrisoquine 4-hydroxylase activity and CYP2D6 levels, with activity remaining stable over the culture period, whereas apoprotein levels decreased slightly (P < .001, two-way ANOVA) (Fig. 4).
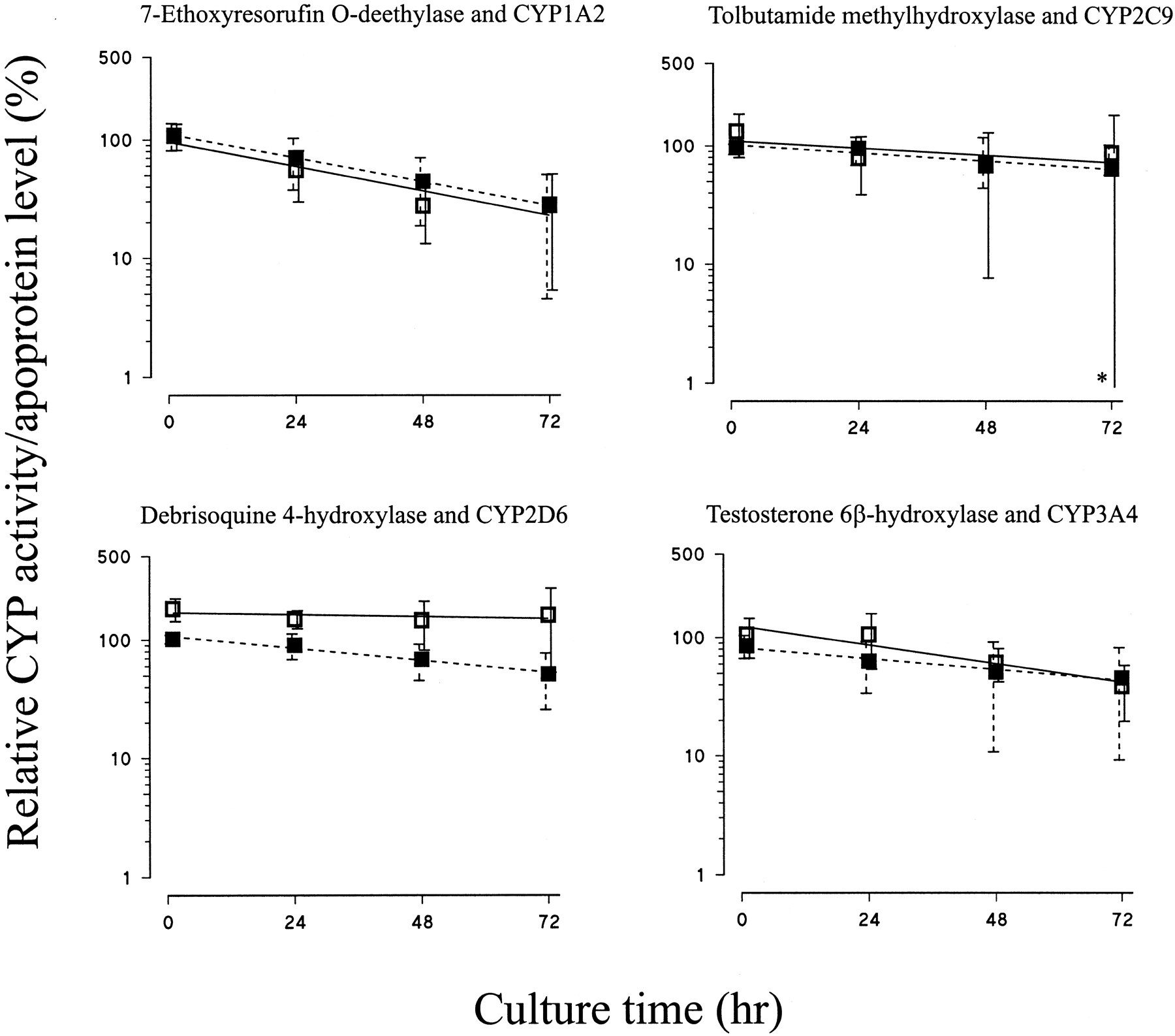
Effect of culture of human liver slices on CYP enzyme activities.
Human liver slices were cultured in complete RPMI 1640 medium for up to 72 h, and microsomal CYP enzyme activities were determined as described under Materials and Methods. The level of each CYP enzyme activity is expressed as a percentage of that of the respective freshly cut slices and is represented as the mean ± S.D. (■) and line of best fit for 7-ethoxyresorufinO-deethylase (n = 3), tolbutamide methylhydroxylase (n = 3), debrisoquine 4-hydroxylase (n = 3), and testosterone 6β-hydroxylase (n = 4). For comparison, data for the levels of the corresponding apoproteins derived from Fig. 2 are shown (▪ and broken lines). In one case (∗), the magnitude of the lower error bar indicating S.D. exceeded the scale of the ordinate.
Comparison of Culture Media.
The effect of culturing human liver slices in serum-free Williams' medium E was investigated using three additional human liver slice preparations (subjects F to H). The liver slice preparations were cultured for 72 h in serum-free Williams' medium E using different hormone concentrations than those used to culture human liver slices from subjects A to E in the RPMI 1640 medium (seeMaterials and Methods for details). The levels of 10 CYP apoproteins, i.e., CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, CYP2D6, CYP3A4, CYP3A5, and CYP4A11, were determined in freshly cut and 72-h cultured human liver slice microsomes (Fig.5). Compared with the corresponding data for the five human liver slice preparations (subjects A to E) cultured in RPMI 1640 medium containing fetal calf serum, there was no significant difference between levels of CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, or CYP4A11 apoproteins using Williams' medium E. In contrast, levels of CYP2E1 apoprotein were significantly higher in liver slices cultured in serum-free Williams' medium E than in liver slices cultured in RPMI 1640 with fetal calf serum (Fig. 5).

Comparison of levels of 10 CYP apoproteins in human liver slices cultured in either supplemented RPMI 1640 medium (for subjects A to E) or in supplemented Williams' medium E (for subjects F to H) for 72 h.
For full details of the media, see Materials and Methods. Levels of CYP apoproteins in 72-h cultured slices are expressed as a percentage of those of the respective freshly cut slices. Results are presented as either mean or mean ± S.E. of three to five experiments using RPMI 1640 medium (see legend to Fig. 1for details) or three experiments using Williams' medium E. Levels of CYP2E1 apoprotein were significantly higher (Student'st test; **P < .01) in liver slices cultured in Williams' medium E than in RPMI 1640 medium.
Discussion
Precision-cut liver slices have been extensively used for studies of xenobiotic metabolism and xenobiotic-induced toxicity (Parrish et al., 1995; Bach et al., 1996; Olinga et al., 1998). A major advantage of this technique is its ready application to the evaluation of species differences in response. Indeed, valuable data have been obtained from comparisons between human liver slices and liver slices from other species.
Previous studies have demonstrated that levels of CYP apoprotein and associated monooxygenase activities decline in cultured rat liver slices as they do in hepatocytes (Paine, 1990; Wright and Paine, 1992;Lake et al., 1993; LeCluyse et al., 1996; Jensen et al., 1997;Müller et al., 1998). Although considered somewhat more stable in human than in rodent hepatocytes, levels of CYP enzymes also decline during culture (Guillouzo et al., 1985, 1993; Maurice et al., 1992; Curi-Pedrosa et al., 1994; Maurel, 1996; Strom et al., 1996). However, these studies also demonstrate that CYP apoprotein levels and enzyme activities are readily detectable in human hepatocytes after several days of culture.
Only a few studies have examined the stability of CYP enzymes in cultured human liver slices. A decline in levels of CYP1A2 and CYP3A4 has been reported, although levels of these two CYP apoproteins and associated enzyme activities were detectable in human liver slices cultured for 72 h in RPMI 1640 medium (Lake et al., 1996, 1997,1998). In contrast, VandenBranden et al. (1998) observed marked losses of CYP enzyme activities in cultured human liver slices.
The current study examined the stability of 11 CYP enzymes in cultured human liver slices by measuring the relative apoprotein levels of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, CYP3A5, and CYP4A11 as well as 7-ethoxyresorufinO-deethylase, tolbutamide methylhydroxylase, debrisoquine 4-hydroxylase, and testosterone 6β-hydroxylase activities. In general, it was found that CYP enzyme levels decrease with time in culture. However, under the conditions used in the present study and contrary to another report (VandenBranden et al., 1998), they were reasonably stable, although there was a large (>4-fold) variation in the rate of decline of different CYP enzymes. The present data, together with previous studies (Lake et al., 1996, 1997, 1998) suggest that, like human hepatocytes, human liver slices can retain detectable levels of CYP enzymes for at least 72 h when cultured under appropriate conditions. Some possible reasons for the apparent discrepancy between the present data and that of VandenBranden et al. (1998) are discussed below.
Initially, a comparison was made between CYP apoprotein levels and enzyme activities in microsomes prepared from human liver slices that had been freshly cut after culturing for a short period (1 h of preincubation). There was a good correspondence between both apoprotein levels and enzyme activities, thus indicating that the initial incubation of human liver slices in culture medium has no immediate effect on CYP levels.
Culturing liver slices for up to 72 h resulted in a progressive decrease in CYP levels with time. Interestingly, CYP enzymes appear to fall into two groups, with CYP2C9, CYP2D6, CYP3A4, and CYP4A11 being relatively stable (half-lives between 70 and 104 h) compared with CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2E1, and CYP3A5, which were relatively unstable (half-lives between 23 and 36 h). Although the majority of this study was carried out by measuring the levels of CYP apoproteins, the data that were obtained on enzyme activities suggest that these conclusions apply to loss of holoenzyme as well as to apoprotein. Hence, similar half-lives were found for 7-ethoxyresorufinO-deethylase activity and CYP1A2 apoprotein content, testosterone 6β-hydroxylase activity and CYP3A4 apoprotein content, and tolbutamide methylhydroxylase activity and CYP2C9 apoprotein content. Although there was a slight deviation between debrisoquine 4-hydroxylase activity and CYP2D6 apoprotein levels, it is clear that both remained relatively stable over the culture period.
As well as variations in the half-life of different CYP enzymes, large differences existed in the stability of individual CYP enzymes in liver slices from different donors. This was particularly so among the most rapidly declining CYP enzymes. This does not appear to be related to the donor or the quality of the liver samples because no relationship was found between maintenance of the levels of different CYP enzymes in individual liver samples.
The reason for the decline in CYP enzyme levels during culture is not known, but a number of possible explanations have been suggested, including cellular dedifferentiation, medium composition, and endogenous degradative processes. For example, studies with cultured human hepatocytes have led to the suggestion that a progressive loss of viability or increased dedifferentiation may be causal in the reduction of CYP levels (Guillouzo et al., 1985, 1993; Maurice et al., 1992;Maurel, 1996), and it is possible that these factors may also be important in cultured liver slices.
Maurel (1996) has also suggested that the loss of inducible CYP enzymes, including CYP1A2 and CYP3A4, is due to the culture of the cells in a chemically defined medium lacking inducers. This would explain the relative stability of CYP2D6 in cultured human hepatocytes (Maurel, 1996) because this CYP enzyme is not believed to be inducible by xenobiotics, unlike members of the CYP1A, CYP2A, CYP2B, CYP2C, CYP2E, and CYP3A subfamilies (Wrighton et al., 1995; Maurel, 1996;Parkinson, 1996; Miners and Birkett, 1998; Pelkonen et al., 1998). Certainly, in the present study, CYP2D6 was relatively stable. However, CYP2C9, CYP3A4, and CYP4A11 were found to have similar stabilities to CYP2D6. Although the inducibility of CYP4A11 in humans is not known, both CYP2C9 and CYP3A4 are inducible by various compounds (Watkins et al., 1985; Maurel, 1996; Parkinson, 1996; Miners and Birkett, 1998;Pelkonen et al., 1998). Hence the loss of CYP enzymes in cultured human liver slices cannot be solely attributed to the absence of CYP enzyme inducers from the culture medium.
Studies with precision-cut rat liver slices have demonstrated that not all culture media adequately maintain liver slice viability (Beamand et al., 1993; Fisher et al., 1995a; Bach et al., 1996; Renwick et al., 1999). In the present study, limited comparisons were made between RPMI 1640 medium with fetal calf serum and serum-free Williams' medium E. Both media were supplemented with insulin and a glucocorticoid (dexamethasone or hydrocortisone). Apart from an apparent higher maintenance of CYP2E1 apoprotein in liver slices cultured in Williams' medium E, no marked differences were observed in the ability of these two media to maintain CYP apoprotein levels in liver slices cultured for 72 h. However, comparisons between culture media and culture media additions are best performed with the same batches of liver slices and not with two separate batches (i.e., subjects A to E and F to H) as shown in Fig. 5. Clearly there is scope for further studies to optimize culture conditions for the maintenance of CYP enzymes in cultured human liver slices.
Studies on the degradation of proteins in the endoplasmic reticulum have shown that rodent CYP enzymes have relatively short half-lives compared with other proteins, including NADPH P450 reductase and cytochrome b5. This suggests that specific biochemical mechanisms exist for their degradation (Correia, 1991), as confirmed by different rates and routes of proteasomal CYP digestion (Roberts, 1997). The routes and relative activities of degradative pathways may account for differences in the half-lives of CYP enzymes and may also contribute to the observed interindividual differences.
Recently, VandenBranden et al. (1998) examined the stability of a range of monooxygenase activities representing CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 in three human liver slice preparations cultured for up to 96 h. Unlike the present study, they reported marked losses of all CYP enzyme activities within 24 h, with some enzyme activities decreasing to undetectable levels after 48 h in culture. There are both apparent similarities and differences between our experimental protocol and that of VandenBranden et al. (1998). In both studies, the liver slices were cultured using a dynamic organ culture system under a high oxygen atmosphere. Certainly investigations with rat liver slices have demonstrated that a high oxygen atmosphere is generally preferable to air as the gas phase (Fisher et al., 1995a,b; Price et al., 1998), and we have utilized such conditions for 72-h CYP enzyme induction studies in human liver slices (Lake et al., 1997, 1998). In both the study of VandenBranden et al. (1998) and the present study, the culture medium was supplemented with gentamicin and fungizone, which suggests that neither of these agents was responsible for the observed differential effects on CYP enzyme stability. Although we used either RPMI 1640 or Williams' medium E andVandenBranden et al. (1998) used Waymouth's medium, other studies with rat liver slices have failed to demonstrate any marked differences between these three media (Fisher et al., 1995b; Renwick et al., 1999). The presence or absence of fetal calf serum in the medium also does not appear to markedly influence CYP enzyme stability, as illustrated by the limited comparisons between RPMI 1640 plus fetal calf serum and serum-free Williams' medium E (Fig. 5). However, unlike VandenBranden et al. (1998), we supplemented our media with insulin and a glucocorticoid (hydrocortisone or dexamethasone). Although additional studies are required to confirm and extend these observations, the present data suggest that these hormones help maintain liver slice viability and levels of CYP enzymes. Certainly, insulin and a glucocorticoid are often added to human hepatocyte cultures for CYP enzyme induction and other studies (Guillouzo et al., 1985; Maurice et al., 1992; Curi-Pedrosa et al., 1994; Strom et al., 1996).
In summary, the fall in levels of CYP enzymes and associated enzyme activities in cultured human liver slices indicates that xenobiotic metabolism studies are best performed with freshly cut rather than with cultured human liver slices. Prolonged culture periods, which may be necessary for slowly metabolized xenobiotics, will result in a differential loss of CYP enzymes, and this may lead to inaccuracies in the prediction of quantitative differences between pathways of metabolism.
Footnotes
-
Send reprint requests to: Dr. Brian G. Lake, TNO BIBRA International Ltd, Woodmansterne Rd., Carshalton, Surrey, SM5 4DS, UK. E-mail: blake{at}bibra.co.uk
-
The provision of financial support by a grant from the Commission of the European Communities (Project “Eurocyp”, BIOMED Contract No. BMH4-CT96-0254) and a Realizing Our Potential Award from the Medical Research Council are gratefully acknowledged.
- Abbreviation used is::
- CYP
- cytochrome P450
- Received May 4, 1999.
- Accepted June 20, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}