


Abstract
The purpose of this study was to establish bupropion (BUP) hydroxylation as a selective in vitro marker of cytochrome P450 (CYP) 2B6 catalytic activity. Among a panel of 16 human liver microsomes (HLMs), BUP hydroxylase activity varied 80-fold when assayed at 500 μM substrate and significantly correlated with CYP2B6 blotting density (r2 = 0.99) andS-mephenytoin N-demethylase activity (r2 = 0.98). Kinetic analysis of BUP hydroxylation was performed in a subset of seven HLMs representative of the 80-fold range in activity. Sigmoidal kinetics suggestive of allosteric activation was observed in five HLMs exhibiting low or high activity; the mean apparent Km for BUP hydroxylation in these HLMs (130 μM) was similar to theKm for cDNA-expressed CYP2B6 (156 μM). Nonsaturable, biphasic kinetics was observed in two HLMs exhibiting low activity. Among a panel of cDNA-expressed P450 isoforms, CYP2B6 and CYP2E1 demonstrated the highest rates of BUP hydroxylation at 12 mM BUP (7.0 and 2.4 pmol/min/pmol of P450, respectively). The relative contributions of CYP2B6 and CYP2E1 to BUP hydroxylation were estimated by using immunoinhibitory monoclonal antibodies (MAB) to these enzymes. MAB-2B6 produced 88% maximum inhibition of BUP hydroxylation when assayed at 12 mM BUP in a high activity HLM, whereas MAB-2E1 produced 81% maximum inhibition in a low activity HLM. However, negligible inhibition by MAB-2E1 was observed when low and high activity HLMs were assayed at 500 μM BUP. These results demonstrate selectivity of BUP hydroxylation for CYP2B6 at 500 μM BUP, thereby validating its use as a diagnostic in vitro marker of CYP2B6 catalytic activity.
Until recently, cytochrome P450 (CYP)1 2B6 has been thought to play a relatively insignificant role in xenobiotic metabolism due to its presumed low level of expression in human liver (Mimura et al., 1993; Shimada et al., 1994). In addition, previous investigations of the role of CYP2B6 in drug metabolism have been limited by unavailability of selective metabolic probes for this enzyme. However, recent studies utilizing improved immunoquantitation procedures have demonstrated a greater frequency of detection and a higher contribution of CYP2B6 to total P450 content than previously estimated. In addition, 20- to 250-fold variability (Code et al., 1997; Ekins et al., 1998;Stresser and Kupfer, 1999) and ethnic differences (Shimada et al., 1994; Kim et al., 1997) in CYP2B6 expression have been recognized. These findings, along with increased identification of CYP2B6-dependent substrates, have generated increased interest in this understudied and partially characterized enzyme.
The extent to which differential CYP2B6 protein expression reflects phenotypic differences in catalytic activity remains unknown. Interindividual differences in CYP2B6 activity could contribute to clinically significant variability in the systemic exposure, response, and toxicity of CYP2B6-dependent substrates. Examples of such substrates include cyclophosphamide (Roy et al., 1999; Huang et al., 2000), ifosfamide (Granvil et al., 1999; Roy et al., 1999), nevirapine (Riska et al., 1999), efavirenz (Christ et al., 1997), and nicotine (Yamazaki et al., 1999). Validated in vitro and in vivo probes of CYP2B6 catalytic activity are required to relate phenotypic differences in CYP2B6 protein expression to the metabolic disposition of CYP2B6 substrates.
Previously investigated substrate probes of CYP2B6 activity include 7-ethoxy-4-trifluoromethylcoumarin (7-EFC)O-deethylation and S-mephenytoinN-demethylation. 7-EFC has limited utility as a CYP2B6 substrate probe, because its deethylation is catalyzed by multiple enzymes, most notably CYP1A2, CYP2B6, and CYP2E1 (Code et al., 1997;Ekins et al., 1997). In contrast, several in vitro studies support the utility of S-mephenytoin N-demethylation as a selective marker of CYP2B6 catalytic activity (Heyn et al., 1996; Ekins et al., 1998; Ko et al., 1998). However, the selectivity ofS-mephenytoin N-demethylation for CYP2B6 is achieved at substrate concentrations that are approximately 100-fold higher than systemic concentrations attained after a standard single dose of S-mephenytoin. At clinically encountered substrate concentrations, CYP2C9 may significantly contribute toS-mephenytoin N-demethylation, thereby limiting its usefulness as an in vivo probe (Ko et al., 1998). Therefore, alternative catalytic probes of CYP2B6 activity that maintain selectivity at substrate concentrations achieved clinically would offer greater potential for use in vivo.
The antidepressant bupropion (BUP) undergoes extensive hepatic metabolism via oxidative and reductive pathways. The t-butyl hydroxylation pathway yields hydroxybupropion (HBUP), the major metabolite in human plasma (Welch et al., 1987). HBUP achieves peak plasma concentrations (∼500 ng/ml or 2 μM) that are approximately 4-fold greater than those of parent drug after a single 150-mg oral dose of BUP (Ketter et al., 1995; Hsyu et al., 1998). Recently, CYP2B6 was found to demonstrate the highest rate of BUP hydroxylation among a panel of cDNA-expressed P450 enzymes (Wurm et al., 1996). This observation supports further investigation of BUP as a potentially selective in vitro and in vivo probe of CYP2B6 catalytic activity.
The purpose of the current study was to investigate the use of BUP as an in vitro CYP2B6-selective metabolic probe. This study also attempted to identify other P450 enzymes capable of BUP hydroxylation, particularly in human livers with relatively lower CYP2B6 protein expression. Assessment of the relative contributions and the kinetic parameters of the principal enzyme(s) involved in BUP hydroxylation in vitro should provide an estimate of the potential utility of BUP as a selective CYP2B6 catalytic marker in vivo. The present results suggest that BUP hydroxylation is selective for CYP2B6 when assayed at 500 μM BUP. At higher substrate concentrations, CYP2E1 may substantially contribute to the overall rate of BUP hydroxylation in human livers with low CYP2B6 protein expression.
Materials and Methods
Chemicals.
BUP hydrochloride, triprolidine hydrochloride, NADP+, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, magnesium chloride, EDTA, triethylamine, and Tris were purchased from Sigma Chemical Co. (St. Louis, MO). HBUP was graciously supplied by Glaxo Wellcome, Inc. (Research Triangle, NC). Formic acid was obtained from Fischer Scientific (Fair Lawn, NJ) and HPLC-grade acetonitrile and methanol were purchased from Mallinckrodt Baker, Inc. (Paris, KY). All other chemicals were of the highest grade commercially available.
Human Liver Microsomes and cDNA-Expressed Human P450s.
A phenotyped bank of 16 individual (20 mg/ml; product no. H0500, version 4) and a pool of 15 (20 mg/ml; category no. HO610, lot 042099A) human liver microsomes (HLMs) were obtained from XenoTech, LLC (Kansas City, KS). The spectrally determined P450 content of each HLM preparation, expressed as picomoles of P450/mg of protein, was provided by the supplier as follows: 395 (pooled HLM); 544 (HLM 2); 291 (HLM 7); 580 (HLM 11); 302 (HLM 13); 356 (HLM 14); 355 (HLM 15); 633 (HLM 16); 733 (HLM 17); 424 (HLM 18); 351 (HLM 19); 345 (HLM 20); 500 (HLM 21); 478 (HLM 22); 645 (HLM 23); 276 (HLM 24); and 444 (HLM 25).
Microsomes prepared from baculovirus-infected insect cells (SUPERSOMES) were purchased from Gentest Corp. (Woburn, MA). NADPH-P450 oxidoreductase was coexpressed in all microsome preparations, and cytochrome b5 was expressed in microsomes containing cDNA-expressed CYP2E1. The P450 content of each preparation (picomoles of P450/mg of protein) was spectrophotometrically determined by the supplier as follows: 192 (CYP1A2); 286 (CYP2A6); 133 (CYP2B6); 556 (CYP2C9*1); 476 (CYP2C19); 152 (CYP2D6*1); 408 (CYP2E1); and 1053 (CYP3A4). Microsomes containing baculovirus vector only served as controls for experiments with cDNA-expressed enzymes.
Biological Reagents.
Polyclonal antibodies to a 13-residue peptide within human CYP2B6 were used for CYP2B6 immunoquantitation in the HLM bank preparations. The sequence of the CYP2B6 peptide was CIDTYLLHMEKEK (Miles et al., 1989). CYP2B6 antipeptides were raised in New Zealand White rabbits as described (Zhu et al., 2000). Peptides were synthesized and conjugated with keyhole limpet hemocyanin (Gene-med Synthesis, Inc., South San Francisco, CA) before mixture with Freund's complete adjuvant and s.c. injection into rabbits. Cross-reactivity of CYP2B6 antipeptides with other human P450 isoforms was excluded by Western blot analysis using cDNA-expressed P450 enzymes.
Specific immunoinhibitory monoclonal antibodies (MAB) against human CYP2B6 (MAB-2B6, 10 mg/ml) and CYP2E1 (MAB-2E1, 10 mg/ml) were purchased from Gentest Corp. MAB-2B6 was shown to inhibit cDNA-expressed CYP2B6-catalyzed metabolism of diazepam and testosterone by 90% (Yang et al., 1998) and HLM-catalyzed S-mephenytoinN-demethylation by greater than 80% (package information, Gentest Corp.). MAB-2E1 was shown to inhibit chlorzoxazone 6-hydroxylation by 90% in microsomes containing vaccinia-expressed CYP2E1 (Gelboin et al., 1996).
Microsomal Assays.
Rates of HBUP formation were determined in duplicate using insect cell-derived microsomes and pooled or individual HLMs from the HLM bank. Preliminary experiments in pooled HLMs and insect cell-derived microsomes were conducted to identify microsomal protein amounts and incubation times resulting in linear rates of HBUP formation.
Incubation mixtures consisted of 0.1 or 0.2 mg of HLMs or 2.5 to 50 pmol of cDNA-expressed P450 enzyme, 10 μM to 12 mM BUP, 50 mM potassium phosphate buffer (pH 7.4), 1 mM EDTA, 3 mM magnesium chloride, 1 mM NADP+, 5 mM glucose 6-phosphate, and 1 U/ml glucose-6-phosphate dehydrogenase in a total volume of 0.2 ml. For incubations with cDNA-expressed CYP2A6 or CYP2C9, 50 mM Tris buffer (pH 7.4) was substituted for potassium phosphate buffer. All BUP stock solutions were prepared in methanol; 0.01 N HCl was added to stock solutions with concentrations greater than 75 mM to aid solubility.
Reactions in HLMs were initiated at 37°C by addition of the NADPH-generating system and terminated after 30 min by addition of 100 μl of ice-cold acetonitrile and the internal standard triprolidine (20 μg/ml). Incubation mixtures were subsequently vortexed and centrifuged at 3000 rpm for 5 min. The supernatant was then transferred to vials for injections of 100 μl onto the HPLC column. Similar procedures were followed for incubations with microsomes containing cDNA-expressed P450 enzymes, except that reactions were initiated by addition of ice-cold microsomes rather than the NADPH-generating system.
P450 isoform-selective catalytic activities were determined in the HLM bank by XenoTech, LLC according to previously published methods (Pearce et al., 1996). These activities included 7-ethoxyresorufinO-dealkylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6),S-mephenytoin N-demethylation (CYP2B6), paclitaxel 6α-hydroxylation (CYP2C8), tolbutamide methyl-hydroxylation (CYP2C9), S-mephenytoin 4-hydroxylation (CYP2C19), dextromethorphan O-demethylation (CYP2D6), chlorzoxazone 6-hydroxylation (CYP2E1), and testosterone 6β-hydroxylation (CYP3A4). Activity values for these P450-dependent pathways were obtained from product information accompanying the HLM bank (product no. H0500, version 4).
HPLC Analysis.
The HPLC system for detection of HBUP and the internal standard triprolidine consisted of a Hewlett-Packard 1100 liquid chromatograph (Hewlett-Packard, Avondale, PA) connected to a Hewlett-Packard model 1100 UV detector set at 214 nm. Peaks of interest were separated on a 5 μm Waters Symmetry 15 × 0.39 cm C18column (Millipore Corp., Milford, MA). Mobile phases A (0.25% triethylamine and 0.1% formic acid) and B (100% acetonitrile) were pumped at a flow rate of 1 ml/min using a gradient ranging from 15% B at 0 to 15.5 min, 25% B at 16 to 23 min, and 15% B at 23.5 to 30 min. The column temperature was maintained at 40°C. HBUP and triprolidine peaks were integrated using a Hewlett-Packard Chemstation system. Retention times for HBUP and triprolidine were approximately 7.5 and 24.5 min, respectively.
Calibration standards (20–10,333 ng/ml) were prepared by adding known amounts of HBUP to microsomes and reagent stock. HBUP concentrations were calculated from the peak height or area ratio using least-squares linear regression, with weighting by the reciprocal of the squared standard concentrations. Interday coefficients of variation for calibration standards ranged from 22% for the lowest standard to 13% for the highest standard. The lower limit of detection was 20 ng/ml. This concentration is lower than HBUP concentrations observed in human plasma up to 72 h after standard oral doses of BUP (DeVane et al., 1990; Hsyu et al., 1998).
Immunoblotting Analysis.
Western blot analysis of 16 HLM preparations was performed according to previously described methods (Parkinson and Gemzik, 1991). Varying amounts of microsomal protein (10–120 μg) were loaded and electrophoresed on an electrophoresis unit (HSI SE600; Hoefer Scientific Instruments, San Francisco, CA). At the end of each electrophoretic run, microsomal protein was transferred to nitrocellulose according to previously published methods (Towbin et al., 1979) and incubated with polyclonal antipeptide antibodies to human CYP2B6 for primary detection. Alkaline phosphatase-conjugated goat anti-rabbit antibody was used for secondary detection. The dilutions for the primary and secondary antibodies were 1:500 and 1:800, respectively. Immunoreactive CYP2B6 protein was visualized by immersing the nitrocellulose membrane in a solution containing nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate. CYP2B6 band intensity was quantified by densitometry (AAB Software, Fullerton, CA) without background adjustment and corrected for the amount of microsomal protein loaded onto each lane.
Statistical Analyses.
Statistical analyses were performed with JMP version 3.2 software (SAS Institute, Inc., Cary, NC). The correlation of BUP hydroxylase activity with P450 catalytic activities and with the CYP2B6 blotting density for the HLM preparations was assessed by linear regression analysis. Significance of the correlations was determined by an Ftest. The acceptance limit for statistical significance was set at α = 0.05.
Enzyme Kinetic Analyses.
After an initial determination of BUP hydroxylase activity with the HLM bank at 500 μM BUP, a subset of HLMs (HLMs 13, 14, 16, 17, 20, 24, and 25) was selected for detailed kinetic experiments. These seven HLMs included preparations with relatively low and high BUP hydroxylase activity that spanned the range of activity observed during the initial determination. The kinetics of BUP hydroxylation for these seven HLMs, and for microsomes containing cDNA-expressed CYP2B6, were examined over the concentration range of 10 to 2000 μM. An expanded range of BUP concentrations (0.075–12 mM) was employed for kinetic experiments with cDNA-expressed CYP2E1 and CYP3A4 and when saturable kinetics was not observed with some HLM preparations (HLMs 20 and 24).
Kinetic parameter estimates were selected by visual inspection of Michaelis-Menten, Lineweaver-Burk, and Eadie-Hofstee plots of experimental data. Weighted kinetic data were fit to one- and two-component Michaelis-Menten models and to the Hill equation via iterative nonlinear regression analysis using initial parameter estimates (WinNonlin 1.1, Scientific Consulting, Inc., Apex, NC). The apparent Km andVmax values of HBUP formation were estimated from the fitted data. In addition, the value of n(number of binding sites) was estimated for experimental data fit to the Hill equation. Determination of appropriateness of fit to each kinetic model was accomplished by examination of the sum of squares of residuals, the size of the coefficients of variations and standard errors of the parameter estimates, and Akaike's information criterion.
Immunoinhibition of BUP Hydroxylase Activity.
The effects of immunoinhibitory MABs to CYP2B6 (MAB-2B6) and CYP2E1 (MAB-2E1) on BUP hydroxylase activity were evaluated in HLMs with high (HLM 16) or low (HLM 20) enzyme activity. Immunoinhibition experiments were conducted with 10 to 200 μg of MABs at 500 μM and 12 mM BUP for MAB-2E1 and at 12 mM for MAB-2B6. In addition, the extent of MAB-2E1 inhibition of BUP hydroxylase activity at 12 mM substrate was assessed in microsomes containing cDNA-expressed CYP2E1. This pilot experiment was performed to confirm the ability of MAB-2E1 to inhibit the potential contribution of CYP2E1 to BUP hydroxylation.
The indicated amounts of MABs in 25 mM Tris buffer were preincubated on ice with 0.2 mg HLMs for 15 min before reactions were initiated by addition of reagent stock consisting of 500 μM or 12 mM BUP, 50 mM potassium phosphate (pH 7.4), and the NADPH-generating system. Control incubations containing 25 mM Tris buffer and 1% methanol without antibody were performed in parallel. Rates of HBUP formation in the presence of antibody were expressed as the percentage of control activity.
Results
Correlation of BUP Hydroxylase Activity with Diagnostic P450 Activities and CYP2B6 Protein Expression in HLMs.
Initial experiments in pooled HLMs demonstrated linearity in rates of HBUP formation over the range of 0.25 to 2 mg/ml protein and 10- to 40-min incubations. BUP hydroxylase activity was initially determined in 16 HLMs at a BUP concentration of 500 μM to ensure saturation of the principal enzyme(s) contributing to HBUP formation. This substrate concentration was selected on the basis ofKm estimates from preliminary kinetic experiments. In addition, rates of HBUP formation were assessed at 10 μM BUP, a concentration that is near the range of systemic plasma concentrations achieved after a standard oral dose of BUP.
Rates of BUP hydroxylation varied approximately 80-fold (20–1623 pmol/min/mg of protein) among the 16 HLMs when assayed at 500 μM BUP (Fig. 1). When assayed at 10 μM BUP, BUP hydroxylase activity was below the limit of detection for 10 of the 16 HLMs. However, because this experiment was preliminary in nature, no attempt was made to modify assay conditions to obtain measurable rates at this substrate concentration. Only six HLMs with the highest BUP hydroxylase activity at 500 μM BUP had detectable activity at a substrate concentration of 10 μM using standard assay conditions. For these six HLMs, similar rank orders and ranges in BUP hydroxylase activity of 13-fold (10–127 pmol/min/mg of protein) and 10-fold (156–1623 pmol/min/mg of protein) were observed at 10 μM and 500 μM BUP, respectively. The results obtained with these six HLMs indicate a similar kinetic behavior for the rates of HBUP formation between different HLMs and suggest that the extent of variability in the rates of HBUP formation observed at 500 μM in vitro may be similarly observed at the lower plasma concentrations of BUP attained in vivo.
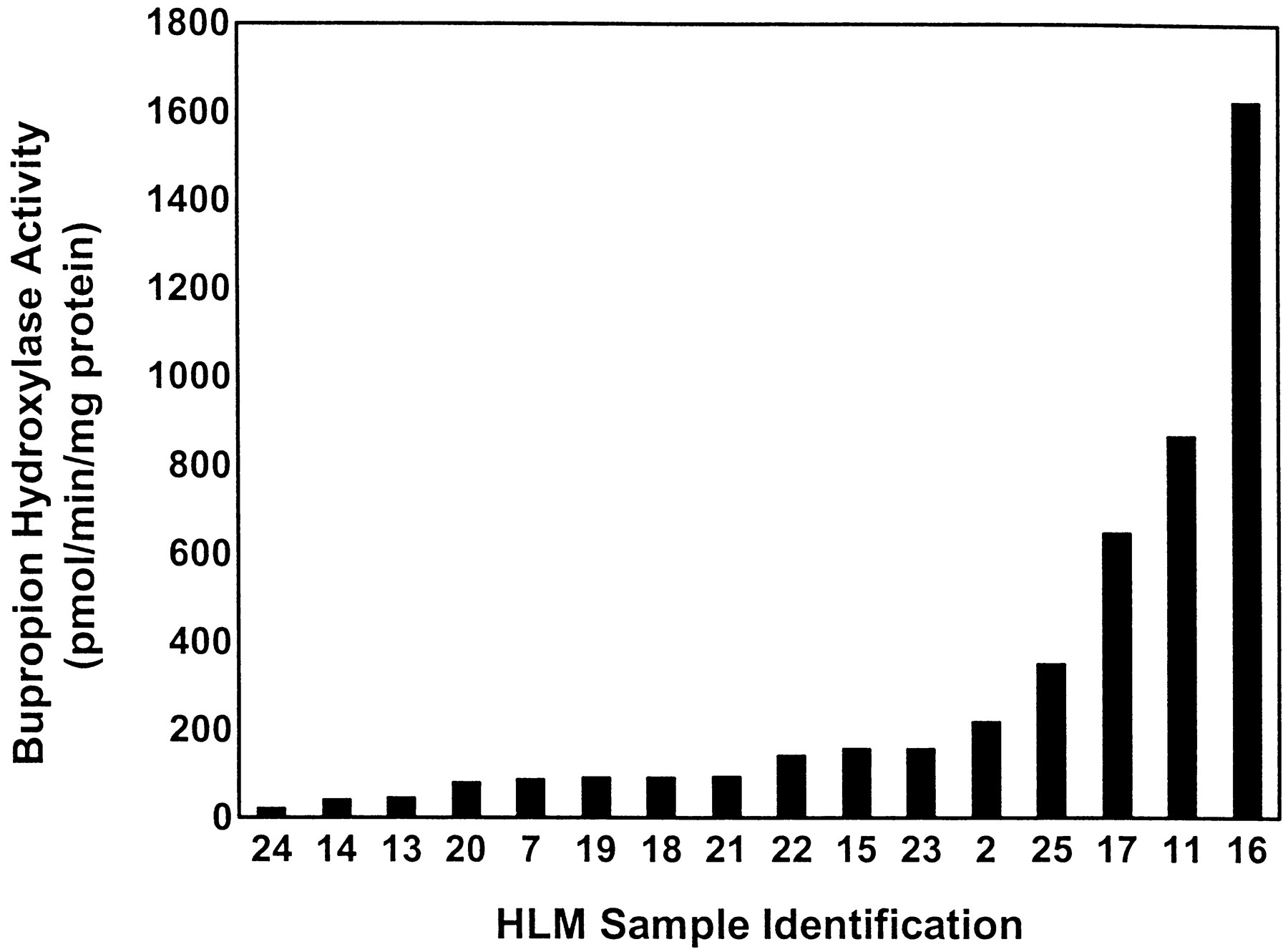
Rates of HBUP formation among HLM bank (n = 16).
Depicted rates of HBUP formation represent the mean of three independent determinations performed in duplicate. Rates were determined by incubating 500 μM BUP, 200 μg of microsomal protein, 50 mM KPO4 (pH 7.4), 1 mM EDTA, 3 mM MgCl2, and NADPH-generating system for 30 min before termination of incubations by a mixture of ice-cold acetonitrile and internal standard. HBUP formation was quantitated by HPLC and normalized for microsomal protein and incubation time.
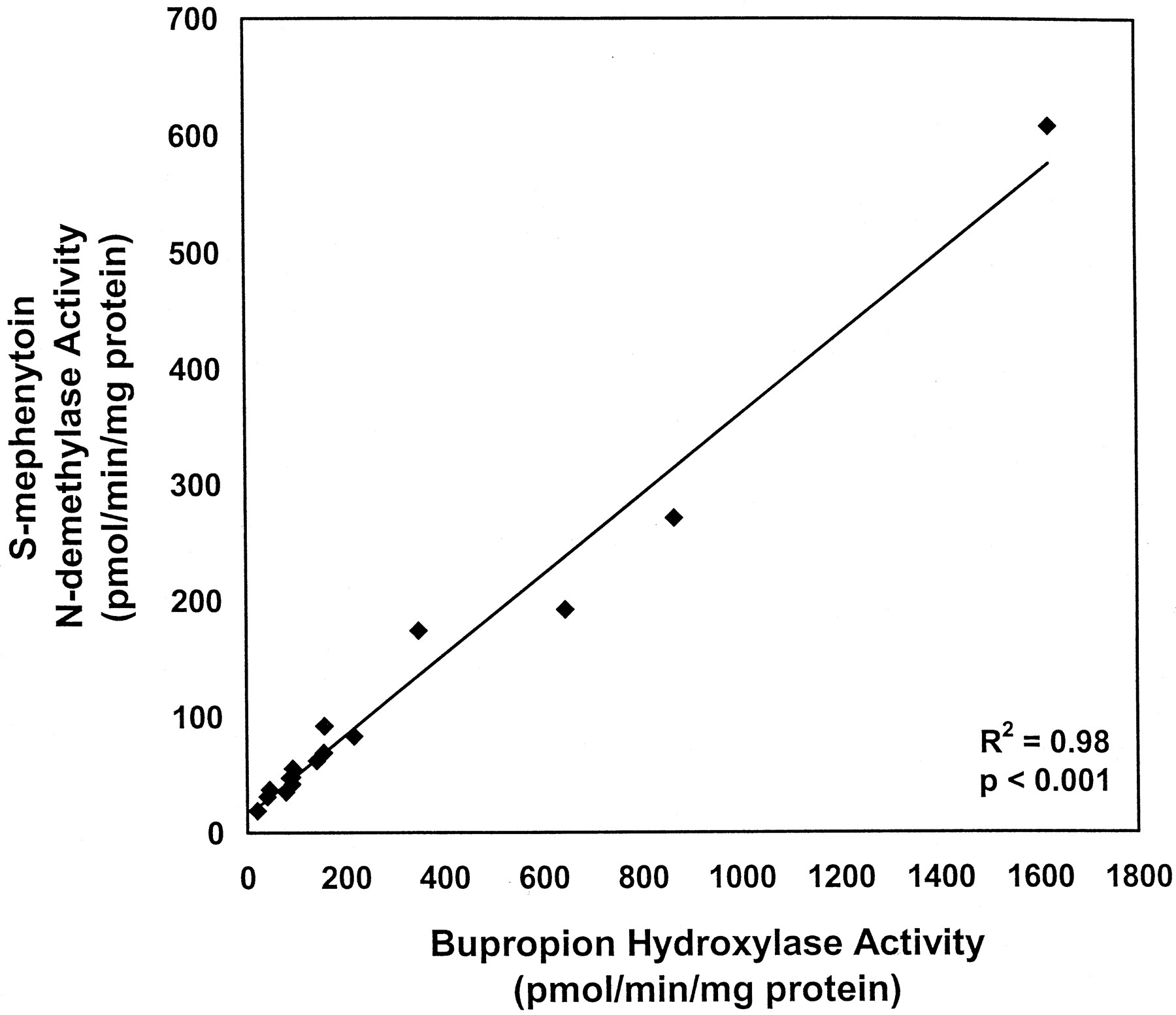
Correlations between P450 isoform-selective catalytic activities and BUP hydroxylase activity were examined in the 16 HLMs assayed at a substrate concentration of 500 μM BUP. BUP hydroxylation significantly correlated with S-mephenytoinN-demethylation (19–609 pmol/min/mg of protein;r2 = 0.98, P < .001; Fig. 2). Weaker but statistically significant correlations were observed with coumarin 7-hydroxylation (r2 = 0.39, P < .01), paclitaxel 6α-hydroxylation (r2 = 0.38, P < .05), and testosterone 6β-hydroxylation (r2 = 0.36, P < .05). No significant correlations were observed between BUP hydroxylase activity and selective catalytic activities for CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP2E1 (data not shown).

Correlation between BUP hydroxylase and S-mephenytoin N-demethylase activities in HLMs (n = 16).
BUP hydroxylase activity was determined as described in Fig. 1.S-mephenytoin N-demethylase activity was determined by Xenotech, LLC (Kansas City, KS). The correlation of the two activities was evaluated by linear regression analysis. Statistical significance was assessed by an F test.
Polyclonal antibodies against a unique CYP2B6 peptide sequence were used for immunodetection of CYP2B6 protein in the HLM bank preparations. These antibodies cross-reacted with cDNA-expressed CYP2B6 but not with the other P450 isoforms tested (Fig.3). CYP2B6 protein was detected in 15 of 16 HLM bank preparations (Fig. 4). No immunodetectable CYP2B6 protein was observed in HLM 24 despite measurable BUP hydroxylase activity, suggesting the possible contribution of other enzymes to BUP hydroxylation in HLMs with undetectable or low levels of CYP2B6 protein. Blotting intensity varied 100-fold among the 15 HLMs with detectable CYP2B6 (25,583–2,591,100 blotting density/mg of microsomal protein) and significantly correlated with BUP hydroxylase activity assayed at 500 μM BUP (r2 = 0.99, P < .001; Fig. 5).

Western blot confirming specificity of polyclonal CYP2B6 antipeptide antibodies.
Specificity of the polyclonal anti-CYP2B6 antibodies was evaluated by testing reactivity of the antibodies with cDNA-expressed 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4. Immunoblotting was performed as described under Materials and Methods. The amount of enzyme loaded onto each lane was 0.5 pmol for lanes 1–8 and 2 pmol for lanes 9–16. Lane 1, CYP1A2; lane 2, CYP2A6; lane 3, CYP2C9; lane 4, CYP2C19; lane 5, CYP2D6; lane 6, CYP3A4; lane 7, CYP2B6; lane 8, CYP2E1; lane 9, CYP1A2; lane 10, CYP2A6; lane 11, CYP2C9; lane 12, CYP2C19; lane 13, CYP2D6; lane 14, CYP3A4; lane 15, CYP2E1; lane 16, CYP2B6; lane 17, blank.

Immunoblot analysis of HLMs (n = 16) with polyclonal CYP2B6 antipeptide antibodies.
Varying amounts of HLM protein (10–120 μg) were loaded onto a SDS-polyacrylamide gel in order of increasing BUP hydroxylase activity, electrophoresed, transferred to nitrocellulose, and incubated with anti-CYP2B6 antibodies for primary detection and alkaline phosphatase-conjugated goat anti-rabbit antibody for secondary detection. Lane 1, HLM 24; lane 2, HLM 14; lane 3, HLM 13; lane 4, HLM 20; lane 5, HLM 7; lane 6, HLM 19; lane 7, HLM 18; lane 8, HLM 21; lane 9, HLM 22; lane 10, HLM 15; lane 11, HLM 23; lane 12, HLM 2; lane 13, HLM 25; lane 14, HLM 17; lane 15, HLM 11; lane 16, HLM 16; lane 17, blank; lane 18, cDNA-expressed CYP2B6 (0.1 pmol).

Correlation between BUP hydroxylase activity and CYP2B6 blotting density of HLMs (n = 16).
Immunodetectable CYP2B6 was quantitated by densitometry and corrected for microsomal protein loaded onto each lane of the SDS-polyacrylamide gel. BUP hydroxylase activity was determined as described in Fig. 1. Data were analyzed by linear regression analysis, with statistical significance determined by an F test.
Kinetic Analyses of BUP Hydroxylation.
Kinetic studies were conducted with a subset of seven HLMs with relatively low or high catalytic activity to determine whether the 80-fold difference in BUP hydroxylase activity with the HLM bank, assayed at 500 μM BUP, reflected the contribution of a single enzyme or multiple enzymes. Of the HLMs selected for detailed kinetic studies, HLMs 16, 17, and 25 exhibited the highest rates and HLMs 13, 14, 20, and 24 exhibited the lowest rates of BUP hydroxylation when assayed at 500 μM BUP. The kinetics of BUP hydroxylation was initially examined over the substrate concentration range of 10 to 2000 μM with the subset of HLMs and with cDNA-expressed CYP2B6. BUP hydroxylation by cDNA-expressed CYP2B6 and HLMs 13, 14, 16, 17, and 25 was characterized by single enzyme kinetics and was saturable (Eadie-Hofstee plots of kinetic data for HLMs 25 and 14 are shown in Fig.6A and 6B). A characteristic hook, suggestive of sigmoidal (allosteric) kinetics, was noted on Eadie-Hofstee plots of kinetic data for HLMs 13, 14, 16, 17, and 25 at the low end of the BUP concentration range. In contrast, no evidence of allosteric activation of BUP hydroxylation by cDNA-expressed CYP2B6 was observed (data not shown).
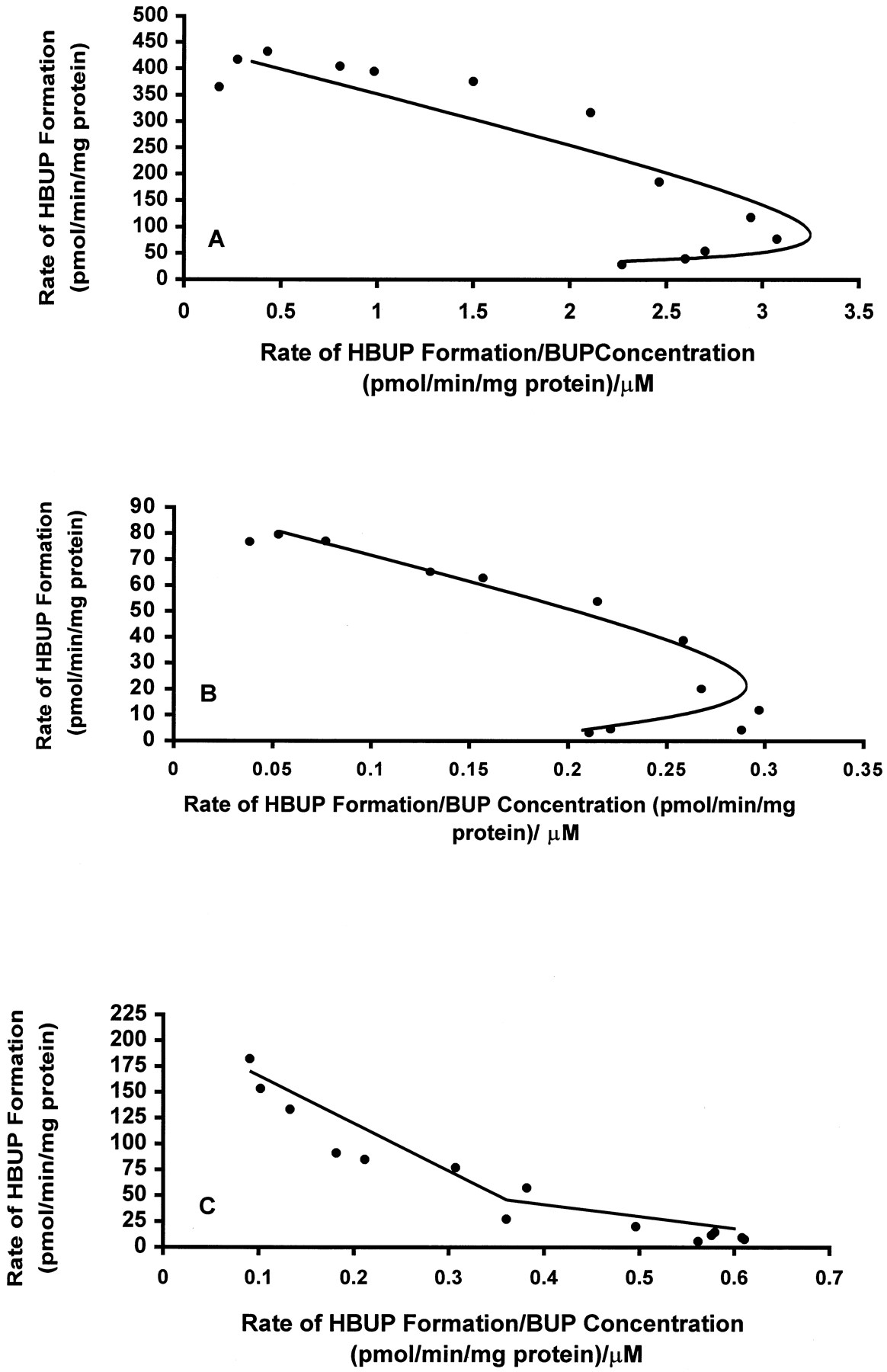
Representative Eadie-Hofstee plots of HBUP formation in select HLM preparations (A, HLM 25; B, HLM 14; C, HLM 20).
Rates of HBUP formation in HLMs 14, 20, and 25 were determined in duplicate at 10 to 2000 μM and plotted against the ratio (rate of formation)/(concentration). HLMs 14 and 20 were relatively low activity preparations, and HLM 25 was a relatively high activity preparation. Eadie-Hofstee plots demonstrated atypical allosteric kinetics for HLMs 14 and 25 and two-component Michaelis-Menten kinetics for HLM 20.
Kinetic parameters of BUP hydroxylation were estimated by fitting kinetic data with HLMs 13, 14, 16, 17, and 25 to the Hill equation and by fitting kinetic data with cDNA-expressed CYP2B6 to the single enzyme Michaelis-Menten equation. The apparent Kmand Vmax for BUP hydroxylation in the HLMs with single enzyme kinetics varied 2-fold (75–168 μM) and 30-fold (82–2451 pmol/min/mg of protein), respectively (Table1). The significant variability in the apparent Vmax for BUP hydroxylation in these select HLMs, coupled with the lack of significant variability in the apparent Km, implies that differential expression of a single enzyme accounts for the 80-fold range of BUP hydroxylase activity when assayed at 500 μM BUP in the HLM bank. The likelihood that this single enzyme is CYP2B6 is supported by similarity between the mean apparent Km for BUP hydroxylation in HLMs (130 μM) and the apparentKm for BUP hydroxylation by cDNA-expressed CYP2B6 (156 μM; Table 1).
Kinetic parameters of BUP hydroxylation in select HLM preparations and in microsomes containing cDNA-expressed CYP2B6
In contrast to HLMs 13, 14, 16, 17, and 25, the low activity HLMs 20 and 24 demonstrated biphasic kinetic behavior when assayed for BUP hydroxylation over the substrate concentration range of 10 to 2000 μM (Eadie-Hofstee plot of kinetic data for HLM 20 is shown in Fig. 6C). In addition, saturable rates of BUP hydroxylation were not achieved over the concentration range of 10 to 2000 μM BUP with these HLMs. Consequently, BUP hydroxylation by HLMs 20 and 24 was assayed at an expanded concentration range of 0.075 to 12 mM. However, rates of BUP hydroxylation were also nonsaturable over this expanded concentration range. Rates of HBUP formation could not be determined at substrate concentrations greater than 12 mM because of solubility limitations. Because HLMs 20 and 24 did not attain saturable rates of HBUP formation, kinetic data for these HLMs could not be reliably fitted to kinetic models. Therefore, kinetic parameters for BUP hydroxylation by HLMs 20 and 24 could not be estimated. However, the inability to saturate BUP hydroxylation over this expanded concentration range suggests that other enzyme(s) capable of BUP hydroxylation are characterized by a higher Km (lower affinity) than CYP2B6.
Evaluation of BUP Hydroxylation by Individual cDNA-Expressed Human P450s.
Because the biphasic kinetic data observed with low activity HLMs (HLMs 20 and 24) indicated the contribution of more than one enzyme to BUP hydroxylation at higher substrate concentrations, a panel of eight cDNA-expressed enzymes (SUPERSOMES) was screened for BUP hydroxylase activity at 12 mM to identify enzymes requiring further investigation (Fig. 7). This substrate concentration was selected to ensure saturation of any highKm isozyme capable of catalyzing BUP hydroxylation. cDNA-expressed CYP2B6 demonstrated the highest rate of HBUP formation (7.0 pmol/min/pmol of P450). cDNA-expressed CYP2E1 and CYP3A4 catalyzed BUP hydroxylation at the second and third highest rates, although at 3- and 30-fold lower rates than cDNA-expressed CYP2B6 (2.4 and 0.23 pmol/min/pmol of P450, respectively). Of note, CYP2E1 was the only cDNA-expressed enzyme in which cytochromeb5 was coexpressed. Rates of BUP hydroxylation by CYP1A2, CYP2A6, CYP2C9, CYP2C19, and CYP2D6 were greater than 30-fold lower than CYP2B6 (Fig. 7). HBUP formation in control microsomes was negligible.
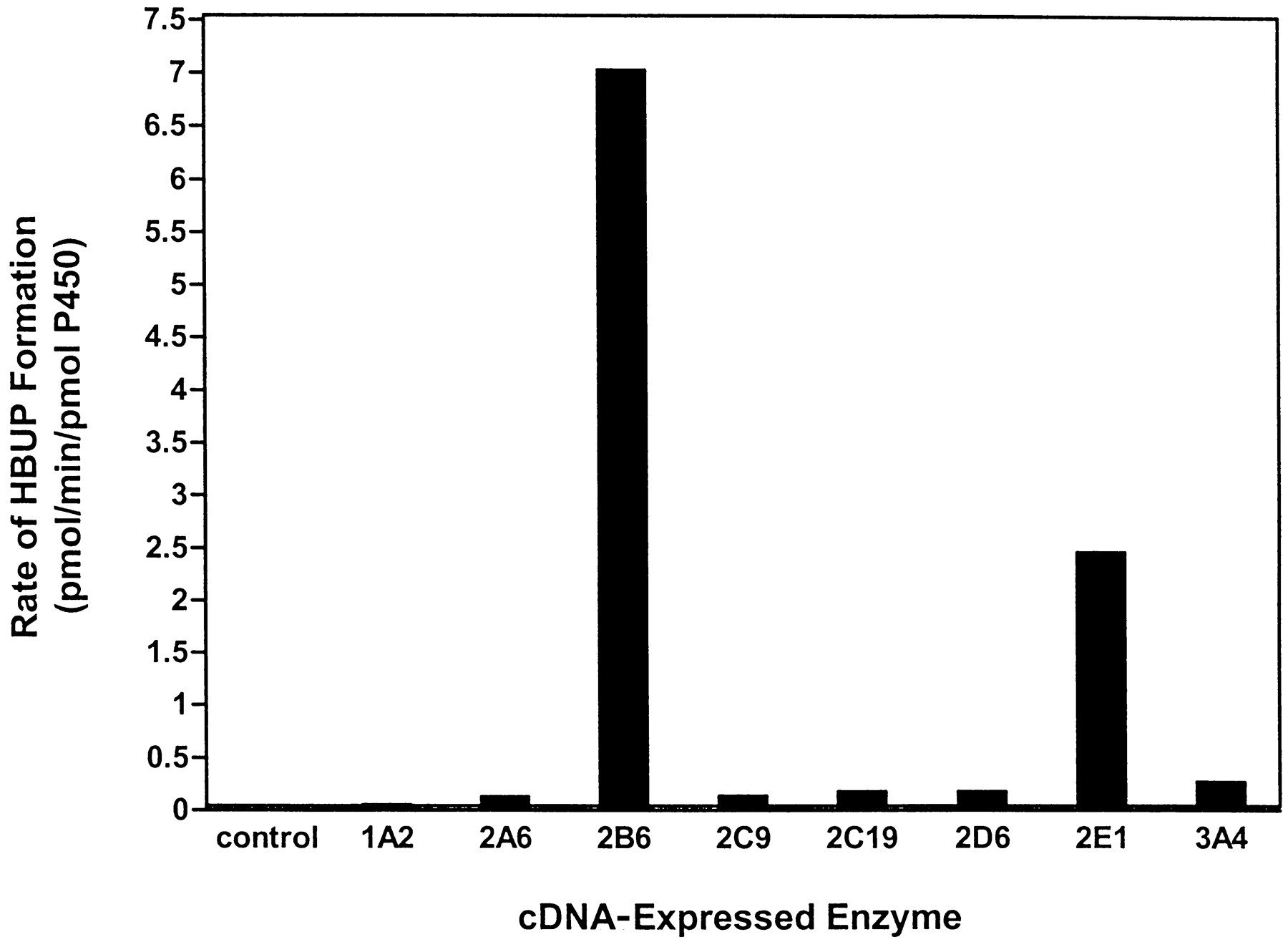
Evaluation of BUP hydroxylation in microsomes expressing cDNAs for the major P450 enzymes.
Rates of HBUP formation were assessed in microsomes from baculovirus-infected insect cells (SUPERSOMES) that expressed cDNA for NADPH-P450 oxidoreductase and CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4. BUP concentration, P450 amount, and incubation time were 12 mM, 50 pmol, and 30 min, respectively. Control incubations were also conducted with insect cell-derived microsomes transfected with baculovirus only. Data are expressed as pmol/min/pmol of P450 and represent the mean of duplicate determinations.
Because cDNA-expressed CYP2E1 and CYP3A4 demonstrated the next highest, albeit lower, rates of BUP hydroxylation compared with CYP2B6 at high substrate concentrations, and because of the greater relative abundance of these enzymes in human liver compared with CYP2B6, the kinetics of cDNA-expressed CYP2E1 and CYP3A4-catalyzed BUP hydroxylation was evaluated to determine whether these enzymes might serve as highKm catalysts of the reaction. As with HLMs 20 and 24, saturable rates of BUP hydroxylation were not achieved by cDNA-expressed CYP2E1 and CYP3A4 over the substrate concentration range of 0.075 to 12 mM (data not shown), suggesting that these enzymes have at least 20-fold higher Km values than CYP2B6. CYP2E1 and CYP3A4 are not likely to substantially contribute to the overall rate of BUP hydroxylation at lower substrate concentrations encountered clinically, because the cDNA-expressed forms of these enzymes failed to catalyze detectable rates of HBUP formation at substrate concentrations less than 500 μM (data not shown).
Immunoinhibition Experiments.
The relative contributions of CYP2B6 and CYP2E1 to BUP hydroxylation were estimated by conducting immunoinhibition studies with MAB-2B6 and MAB-2E1 in HLMs with low (HLM 20) and high (HLM 16) enzyme activity. A preliminary experiment indicated that MAB-2E1 (100 μg) resulted in 80% inhibition of BUP hydroxylation by cDNA-expressed CYP2E1 when assayed at 12 mM BUP (data not shown).
Over the range of 20 to 200 μg, MAB-2B6 inhibited BUP hydroxylation by greater than 80% in the high activity HLM (HLM 16) assayed at 12 mM BUP. In contrast, the same range of MAB-2E1 produced ≤20% inhibition of BUP hydroxylation in HLM 16 when assayed at 12 mM (Fig.8A). In the low activity preparation (HLM 20), BUP hydroxylation was inhibited by greater than 70% when assayed at 12 mM BUP in the presence of 40 to 200 μg of MAB-2E1, but by ≤30% in the presence of MAB-2B6 (Fig. 8B). These results suggest that the maximum contribution of CYP2E1 to the overall rate of BUP hydroxylation is achieved in HLMs with relatively low expression of CYP2B6 protein.

Differential inhibition of BUP hydroxylase activity by MAB-2E1 and MAB-2B6 in HLMs 16 (A) and 20 (B).
Inhibition experiments were performed with monoclonal antibodies against CYP2E1 and CYP2B6 to determine the relative contribution of these isozymes to BUP hydroxylation in HLMs with high (HLM 16) or low (HLM 20) catalytic activity. HLMs (200 μg) were preincubated with antibody (10–200 μg) for 15 min before initiation of the 30-min incubation by addition of BUP 12 mM, buffer, and the NADPH-generating system. Control mixtures consisting of HLMs and 25 mM Tris buffer in the absence of antibody were also performed. Rates of HBUP formation in the presence of antibody were determined in duplicate and expressed as the percentage of control activity.
Although MAB-2E1 inhibited BUP hydroxylation when assayed at 12 mM in a low activity HLM, negligible inhibition of BUP hydroxylation by MAB-2E1 was observed when HLM 16 and HLM 20 were assayed at a substrate concentration of 500 μM (Table 2). The lack of MAB-2E1 inhibition of BUP hydroxylation when assayed at 500 μM BUP is consistent with the observation that cDNA-expressed CYP2E1 failed to catalyze BUP hydroxylation when assayed at substrate concentrations less than 500 μM. Thus, the contribution of CYP2E1 to BUP hydroxylation in HLMs with low CYP2B6 expression appears significant only at higher substrate concentrations (12 mM), supporting the notion that CYP2E1 is a low affinity (highKm) catalyst of BUP hydroxylation in human liver.
MAB-2E1 immunoinhibition of BUP hydroxylase activity at 500 μM in HLMs 16 and 20
Discussion
CYP2B6 is emerging as a more significant component of the P450 enzyme system than previously recognized. The availability of selective probes of CYP2B6 catalytic activity would facilitate further examination of the role of this enzyme in xenobiotic metabolism. The in vitro studies described herein demonstrate that CYP2B6 is the predominant catalyst of BUP hydroxylation in human liver and expand on a preliminary report in which CYP2B6 demonstrated the highest rate of BUP hydroxylation among a panel of cDNA-expressed P450 isozymes (Wurm et al., 1996). The evidence, showing that CYP2B6 is the principal contributor to BUP hydroxylation in HLMs and supporting the use of BUP as a selective in vitro marker of CYP2B6 catalytic activity, consists of the following: 1) similarity in the apparentKm for BUP hydroxylation between HLMs and cDNA-expressed CYP2B6; 2) the rate of BUP hydroxylation was highest with CYP2B6 among a panel of cDNA-expressed P450 isozymes; 3) correlation of BUP hydroxylation with CYP2B6 blotting intensity andS-mephenytoin N-demethylation, and a lack of correlation with other P450 isoform-selective catalytic activities; and 4) inhibition of BUP hydroxylase activity in HLMs using an immunoinhibitory antibody to CYP2B6.
Several lines of evidence support the validity of using BUP as a selective probe of CYP2B6 catalytic activity. For example, the apparentKm for BUP hydroxylation by cDNA-expressed CYP2B6 in this study is similar to the Kmvalue reported elsewhere (Ekins et al., 1999). Although two relatively more abundant P450 isozymes, CYP2E1 and CYP3A4, were also found to be catalytically competent among a panel of cDNA-expressed P450 isozymes, kinetic analyses of BUP hydroxylation using HLMs failed to reveal other catalysts of BUP hydroxylation with higher affinity (lowerKm) than CYP2B6. In addition, cDNA-expressed CYP2E1 and CYP3A4 did not catalyze detectable rates of HBUP formation at substrate concentrations less than 500 μM, and saturable rates of HBUP formation by these cDNA-expressed enzymes were not observed up to 12 mM BUP in kinetic studies, suggesting that CYP2E1 and CYP3A4 have lower affinities than does CYP2B6. The ability of CYP2E1 to catalyze BUP hydroxylation at higher substrate concentrations (12 mM) was supported by data obtained with cDNA-expressed CYP2E1 and by the immunoinhibitory effects of MAB-2E1 on BUP hydroxylase activity. However, the contribution of CYP2E1 to the overall rate of BUP hydroxylation was significant only in one HLM expressing a low level of CYP2B6 protein when assayed at 12 mM BUP. When low or high activity HLMs were assayed at a lower substrate concentration of 500 μM, the contribution of CYP2E1 to the overall rate of BUP hydroxylation was negligible. Therefore, it can be inferred from these findings that CYP2E1 will not significantly contribute to the overall rate of BUP hydroxylation at the lower plasma concentrations of BUP encountered clinically (∼0.5–1 μM). This presumption is supported by an in vivo study that demonstrated no effect of ethanol, a CYP2E1 competitive inhibitor, on the systemic exposure of BUP (Posner et al., 1984). CYP3A4 is also not likely to significantly contribute to BUP hydroxylation at clinically relevant substrate concentrations. Negligible inhibition of BUP hydroxylation by troleandomycin was observed in HLMs assayed at 500 μM BUP (Faucette et al., 2000). Additional data that support the lack of CYP3A4 contribution to the overall rate of BUP hydroxylation will be presented in a subsequent report.
In contrast to the evidence outlined above that supports the selectivity of BUP hydroxylation for CYP2B6, limited evidence indicating a contribution of additional enzymes to BUP hydroxylation was obtained only when BUP hydroxylase activity was assayed at higher BUP concentrations. This limited evidence includes the observation of biphasic kinetics in low activity HLMs, and low but quantifiable HBUP formation in one HLM without immunodetectable CYP2B6.
In summary, these data suggest that BUP is similar to probe substrates that are characterized by relatively higherKm values but that maintain their P450 selectivity due to the absence of other significantly contributing P450 isozymes. Examples of other selective probe substrates with higherKm values include caffeine and tolbutamide for CYP1A2 (Km ∼ 1.2 mM) and CYP2C9 (Km ∼ 200 μM), respectively (Hickman et al., 1998). An even higher Km has also been estimated from experiments conducted with cDNA-expressed enzyme and HLMs using S-mephenytoin N-demethylation, another purported probe of CYP2B6 activity (Heyn et al., 1996). However, compared with BUP, the use of S-mephenytoin as an in vivo probe of CYP2B6 activity is likely to be confounded by the potential contribution of CYP2C9 at the low plasma substrate concentrations observed clinically (Ko et al., 1998). Therefore, BUP may ultimately offer greater utility as a CYP2B6 probe in vivo, because other enzymes are not likely to contribute to its overall rate of hydroxylation at clinically encountered substrate concentrations.
Although these observations suggest a potential use of BUP as an in vivo probe of CYP2B6 catalytic activity, the 80-fold range in BUP hydroxylase activity when assayed in vitro at 500 μM BUP is discordant with the 3- to 10-fold range in HBUP systemic exposure that has been reported in vivo (Laizure et al., 1985; DeVane et al., 1990;Ketter et al., 1995). Factors that could account for this apparent in vitro-in vivo discrepancy include potential interindividual variability in other major competing pathways of BUP metabolism and in the oxidative and conjugative metabolism of HBUP, which would not be observed with microsomal incubations. In addition, other processes affecting the disposition of drugs that exhibit high first pass metabolism, such as hepatic uptake and liver blood flow, could influence the in vivo rate of BUP hydroxylation. Alternatively, the correlation between CYP2B6 protein expression and catalytic activity may be less strong in vivo than observed in vitro, because BUP concentrations that are necessary to reachVmax (>450 μM BUP, or ∼3×Km) are not likely achieved in vivo. Therefore, it is possible that, at the lower BUP concentrations observed clinically, other P450 isozymes capable of BUP hydroxylation, albeit at lower rates than CYP2B6 (see Fig. 7), contribute variably according to their relative expression in human liver, such that absolute differences in CYP2B6 catalytic activity in vivo are not easily observed. Finally, autoactivation of BUP hydroxylase activity was observed at low substrate concentrations in the majority of HLMs selected for kinetic experiments, suggesting another potential confounding variable when correlating CYP2B6 protein expression with catalytic activity. Although autoactivation of cDNA-expressed CYP2B6-catalyzed BUP hydroxylation was not observed, as previously reported (Ekins et al., 1999), other cDNA-expressed CYP2B6-mediated reactions such as 7-EFC O-deethylation (Ekins et al., 1997), testosterone 16β-hydroxylation (Ekins et al., 1998), and verapamilO-demethylation (Ekins et al., 1999) have been previously characterized by allosteric kinetics. The discordant kinetics of BUP hydroxylation between HLMs and insect cell-derived microsomes may reflect differences in the phospholipid environment of the respective in vitro systems. Differences in the NADPH-P450 oxidoreductase to P450 ratios between these in vitro systems might also account for these findings, because it has been postulated that CYP activators affect coupling of NADPH-P450 oxidoreductase with P450s (Shou et al., 1994).
In various studies, CYP2B6 catalytic activity has varied 26- to 80-fold using BUP hydroxylation (present study) or S-mephenytoinN-demethylation (Heyn et al., 1996; Ekins et al., 1998) as in vitro markers of CYP2B6 activity. In these studies, high activity HLMs that are outliers as a result of induced catalytic activity could significantly expand the range of CYP2B6 activity determined in vitro, such that intrinsic in vivo differences in catalytic activity observed in the absence of induction are much smaller. In fact, at least three high activity HLMs (2, 17, and 25) used in this study were known to be isolated from donors treated with phenobarbital (Chang et al., 1997) or phenytoin (E. L. Lecluyse, unpublished data), which have been shown to increase CYP2B6 protein levels in human hepatocyte cultures. When four HLMs with the highest catalytic activity at 500 μM BUP were omitted from data analysis, the range of BUP hydroxylase activity among the HLM bank decreased from 80- to 10-fold. A decrease in the range ofS-mephenytoin N-demethylase activity from 26- (Ekins et al., 1998) to 5-fold was also noted when 5 of 19 HLMs, representing the highest quartile of catalytic activity, were omitted from data analysis. The 10-fold range in rates of HBUP hydroxylation in the absence of the four highest activity HLMs is similar to the 13-fold difference in rates of BUP hydroxylation among six HLMs assayed at 10 μM BUP, a concentration near the range of BUP concentrations achieved clinically. This observation strengthens support for the potential use of BUP as an in vivo probe of CYP2B6 catalytic activity.
In conclusion, this study validates BUP hydroxylation as an in vitro diagnostic marker for CYP2B6 catalytic activity when assayed at 500 μM BUP. The availability of BUP, an in vitro selective CYP2B6 substrate probe, should facilitate detection of phenotypic differences in CYP2B6 activity and further exploration of the contribution of CYP2B6 to xenobiotic metabolism. Further study is required to determine whether the selectivity of BUP hydroxylation for CYP2B6 in vitro is also observed in vivo.
Acknowledgments
We thank Samantha Desmarias, B.A., and Summer Jolley for technical assistance; Michael Morlan, B.S., and Scott Smith, Ph.D., for assistance with HPLC development and analysis; and Morris Clarke, Ph.D., and Gary Pollack, Ph.D., for helpful discussions.
Footnotes
-
Send reprint requests to: Celeste M. Lindley, Pharm.D., M.S., FCCP, FASHP, Division of Pharmacotherapy, School of Pharmacy, CB #7360, Beard Hall, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7360. E-mail: celeste_lindley{at}unc.edu
-
This work was supported in part by a Hollingsworth Faculty Scholarship, awarded to C.M.L. by the School of Pharmacy, University of North Carolina at Chapel Hill, and by a Junior Faculty Development Award, awarded to E.L.L. by the University Research Council, University of North Carolina at Chapel Hill.
- Abbreviations used are::
- CYP or P450
- cytochrome P450
- 7-EFC
- 7-ethoxy-4-trifluoromethylcoumarin
- HLM
- human liver microsome
- MAB
- monoclonal antibody
- MAB-2B6
- monoclonal antibody specific to CYP2B6
- MAB-2E1
- monoclonal antibody specific to CYP2E1
- BUP
- bupropion
- Received June 30, 2000.
- Accepted July 6, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}