


Abstract
The COMT inhibitors entacapone and tolcapone are rapidly metabolized in vivo, mainly by glucuronidation. In this work, the main UGT isoforms responsible for their glucuronidation in vitro were characterized by using a subset of representative cloned and expressed human UGT isoforms. Entacapone in particular was seen to be an exceptionally good substrate for UGT1A9 with an even higher reaction velocity value at 500 μM substrate concentration compared with that of the commonly used substrate, propofol (1.3 and 0.78 nmol min−1 mg−1, respectively). Neither entacapone nor tolcapone was glucuronidated by UGT1A6. Tolcapone was not detectably glucuronidated by UGT1A1, and the rate of glucuronidation of entacapone was also low by this isoform. However, UGT1A1 was the only UGT capable of catalyzing the formation of two glucuronides of the catecholic entacapone. Both COMT inhibitors were glucuronidated at low rates by the representative members of the UGT2B family, UGT2B7 and UGT2B15. Michaelis-Menten parameters were determined for entacapone and tolcapone using recombinant human UGT isoforms and human liver microsomes to compare the kinetic properties of the two COMT inhibitors. The kinetic data illustrates that UGT1A9 exhibited a much greater rate of glucuronidation and a far lowerKm value for both entacapone and tolcapone than UGT2B15 and UGT2B7 whose contribution is minor by comparison. Entacapone showed a 3 to 4 times higher Vmaxvalue and a 4 to 6 times lower Km value compared with those of tolcapone both in UGT1A9 cell lysates and in human liver microsomes.
Entacapone and tolcapone are novel drugs that are used to improve the bioavailability of l-dopa in the treatment of Parkinson's disease. When l-dopa is administered with a peripheral dopadecarboxylase inhibitor, 3-O-methylation by COMT1 (EC 2.1.1.6) becomes an important consideration as 3-O-methyldopa is potentially harmful and may compete with l-dopa for transport into the brain (Männistö et al., 1992). Entacapone and tolcapone are both designed to inhibit COMT with high affinity and thus reduce the formation of 3-O-methyldopa when coadministered under this treatment regime. Both compounds have a nitro-group in position 5 and another strongly electron-withdrawing hydrophobic substituent in position 1 (Fig. 1), which are essential for their pharmacological action as COMT inhibitors (Lotta et al., 1992). Recently, tolcapone has been withdrawn from the market in the European Union countries being a possible causative agent of liver damage by elevation of expression of alanine and aspartate aminotransferases in certain susceptible individuals (Assal et al., 1998).

Structural formulae of entacapone and tolcapone.
The elimination half-life of entacapone is very short (0.3 h) (Keränen et al., 1994), while that of tolcapone, despite the structural similarities with entacapone, is much longer (2.3 h) (Dingemanse et al., 1995). Both COMT inhibitors are almost completely metabolized before excretion in the urine and feces (Wikberg et al., 1993; Jorga et al., 1999). A small amount of tolcapone is methylated by COMT to form 3-O-methyltolcapone, but the most important metabolic pathway of both compounds is glucuronidation. Glucuronides of entacapone and its main phase I metabolite, entacapone(Z)-isomer, have been found to represent even over 95% of all urinary metabolites in humans (Wikberg et al., 1993).
Characterization of the metabolism of new drugs has been successfully achieved by the use of cloned and expressed isoforms of human drug-metabolizing enzymes, and the use of expressed cytochrome P450s in this process is now routine. The application of a similar approach with other drug-metabolizing enzymes, particularly phase II isoforms, has not been as forthcoming, as the commercial availability of expressed human enzymes has been limited until recently. The cloning and expression of human UGTs (EC 24.1.17) have demonstrated that many isoforms of this enzyme have the capability to metabolize xenobiotics and drugs including 17α-ethinylestradiol (UGT1A1) (Ebner et al., 1993), paracetamol (UGT1A6) (Bock et al., 1993), propofol (UGT1A9) (Ebner and Burchell, 1993), and morphine (UGT2B7) (Coffman et al., 1997). Furthermore the existence of polymorphic variations in UGTs present a new level of complexity in the elucidation of the metabolic role of individual isoforms (Ciotti et al., 1997; Levesque et al., 1997, 1999; Coffman et al., 1998). Identification of isoforms contributing to the metabolism of drugs biotransformed primarily by glucuronidation, such as entacapone and tolcapone, would be of great help in assessing the risk of metabolic interactions with endogenous compounds, other drugs, and dietary chemicals.
In this study, UGT isoforms involved in the metabolism of entacapone and tolcapone were identified and characterized using recombinant human enzymes. Where a UGT isoform was found to be able to metabolize either of these compounds, kinetic parameters were determined and compared to those determined for human liver microsomes in an attempt to better explain the variation in clearance of these drugs.
Materials and Methods
Chemicals.
Entacapone and tolcapone were kindly donated by Orion Pharma (Espoo, Finland). Uridine 5′-diphosphate-glucuronic acid (sodium salt) was purchased from Boehringer-Mannheim (Mannheim, Germany) and uridine diphospho[U-14C]glucuronic acid from NEN DuPont (Stevenage, Hertfordshire, UK). 3-O-glucuronides of entacapone and tolcapone were synthesized and identified in the Department of Pharmaceutical Chemistry, University of Helsinki, Finland, as described by Luukkanen et al. (1999). β-Glucuronidase (Escherichia coli) was supplied by Boehringer-Mannheim. Other reagents used in the assays were of the highest grade commercially available.
Tissue Culture and Human Liver Microsomes.
V79 and recombinant cell lines were grown up in Dulbecco's Modified Eagle's Medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin (Gibco Life Technologies, Paisley, Scotland) maintained under constant selection by optimized concentrations of geneticin. Pooled human liver microsomes from several donors were purchased from Human Biologics Inc. (Scottsdale, AZ).
UGT Assays.
Cells were disrupted by a standardized sonication method: four 5-s bursts (MSE soniprep, Sanyo Gallenkamp, Leicester, UK) allowing at least 1 min on ice for cooling between bursts. Assays were performed according to the HPLC gradient radiochemical method of Ethell et al. (1998). The composition of a screening assay was 100 mM Tris/maleate, pH 7.4, 5 mM MgCl2, 2 mM UDPGA (0.1 μCi/assay [14C]UDPGA), 250 to 500 μg of cellular sonicate, and 500 μM entacapone or tolcapone in a volume of 100 μl. Screening assays were also incubated at UDPGA concentrations of 50 μM to increase the levels of radiolabel incorporation at subKm concentrations of the cosubstrate. Assays were incubated at 37°C for 60 min before termination by addition of an equal volume of methanol that had been prechilled to −20°C. Protein was removed by centrifugation at 1000g for 10 min. The supernatant was injected onto HPLC comprising a binary gradient of 0 to 100% acetonitrile in 0.05 M ammonium acetate developed over 13 min on a 0.46- × 25-cm Techsphere ODS2 column (HPLC Technology, Macclesfield, UK) allowing a 2-min re-equilibration step between sample injections. 14C-Labeled UDPGA and glucuronide were detected using a Reeve 9701 radioactivity flow monitor (Reeve Analytical, Glasgow, Scotland, UK) fitted with a 200-μl flowcell packed with cerium-activated lithium glass as scintillant.
Enzyme Kinetic Measurements.
Apparent enzyme kinetic parameters (Km andVmax) were determined for entacapone and tolcapone using recombinant human UGT isoforms and human liver microsomes by varying the substrate concentration (usually 10–500 μM) at a fixed concentration of UDPGA (5 mM). The samples containing 5 to 300 μg of cellular homogenate were incubated for 40 min at 37°C before terminating the reaction by 10 μl of 4 M perchloric acid and removing the precipitated proteins by centrifugation (1000g, 10 min). Control samples were incubated without UDPGA. The linearity of the reaction catalyzed by different UGT sources was tested with respect to incubation time and protein concentration. The samples were analyzed by HPLC (HP1100, Hewlett Packard, Waldbronn, Germany) using an RP-18 column (Hypersil, 0.4- × 25-cm, 5 μm, Hewlett Packard) and a mixture of phosphate/citrate buffer (25 mM NaH2PO4, 10 mM citric acid, with pH adjusted to 2.2 with o-phosphoric acid), and methanol (52:48 and 42:58 v/v for entacapone and tolcapone, respectively). The mobile phase flow rate was 1.0 ml/min and the injection volume was 80 μl. The formed glucuronides of entacapone and tolcapone were identified and quantitated by UV detection with the aid of authentic reference standards at 305 and 278 nm, respectively. The initial velocities obtained were fitted to the Michaelis-Menten equation using the Leonora Steady State Enzyme Kinetic Program (version 1.0 by Cornish-Bowden, 1994).
Hydrolysis by β-Glucuronidase.
After centrifugation of the reaction mixture, the supernatant was evaporated to dryness and redissolved in 100 μl of 50 mM phosphate buffer (pH 6.1). β-Glucuronidase (10 U/ml) was added and the sample was incubated at 37°C for 30 min before the reaction was terminated by 10 μl of 4 M perchloric acid.
Results
The ability of different recombinant human UGT isoforms as well as human liver and kidney microsomes to catalyze the glucuronidation of the COMT inhibitors entacapone and tolcapone was tested. Each screening assay was performed in tandem using a control assay with a marker substrate for each UGT isoform. Results of the screening, shown in Table 1, clearly suggest that UGT1A9 is the most important isoform catalyzing the glucuronidation of entacapone and tolcapone. Entacapone was observed to be an excellent substrate of this isoform showing even higher reaction velocity value than propofol, the UGT1A9 probe substrate used. Both of the nitrocatechols were glucuronidated at a high rate by human liver and kidney microsomes. Entacapone was shown to be a substrate of all UGT isoforms studied except UGT1A6, whereas tolcapone was not detectably glucuronidated by either UGT1A1 or UGT1A6. However, glucuronidation of these compounds by isoforms other than UGT1A9 seemed to be of minor importance. Entacapone was glucuronidated at a 2 to 3 times higher rate compared with tolcapone by most of the UGT sources at the relatively high substrate concentration that was used in the screening experiments.
Glucuronidation of entacapone and tolcapone (500 μM) by different UGT sources at 50 μM and 2 mM UDP-glucuronic acid
Although catechols contain two hydroxyl groups, which in theory are both available for conjugation, the chromatograms from assays with both compounds usually showed only a single peak, corresponding to the retention times of 3-O-glucuronides of entacapone and tolcapone. Interestingly, the only UGT that catalyzed the formation of two glucuronides of entacapone was UGT1A1 (Fig.2C). The second peak in the UGT1A1 assay has the same elution time as the synthetic 3-O-glucuronide of entacapone as illustrated in Fig. 2B. The two peaks in Fig. 2C were both absent in assays that were treated with β-glucuronidase (Fig.2D) verifying that they were both glucuronide conjugates, most probably 3-O- and 4-O-glucuronides of entacapone.

Glucuronidation of entacapone by human UGT1A1 and hydrolysis of the respective glucuronides by β-glucuronidase.
A, blank incubation of 500 μM entacapone (retention time 28 min, not shown in the chromatogram) with V79 cells expressing UGT1A1 without UDPGA. B, synthesized entacapone 3-O-glucuronide; C, incubation of 500 μM entacapone and 5 mM UDP-glucuronic acid with V79 cells expressing UGT1A1 (0.3 mg of protein, 60 min at 37°C) and 30 min without β-glucuronidase; D, the same as sample C but incubated with β-glucuronidase (10 U/ml) for 30 min at 37°C. Chromatographic conditions: 33% methanol in 25 mM NaH2PO4/10 mM citric acid, pH 2.2, flow rate 1.0 ml/min, RP-18 column (Hypersil, 0.4- × 25-cm, 5 μm, Hewlett Packard), UV detection at 305 nm.
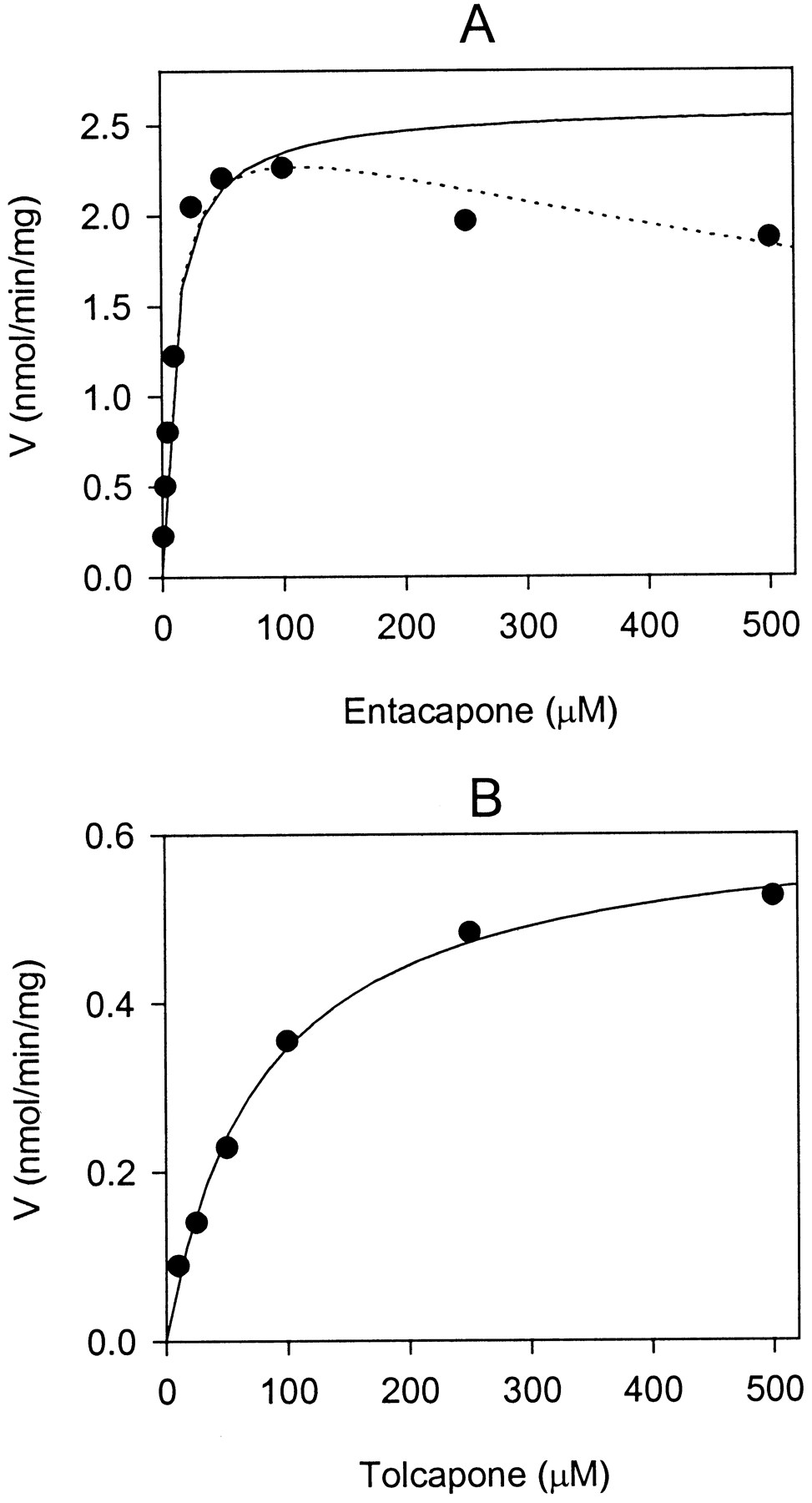
To compare properties of entacapone and tolcapone as substrates of UGTs, apparent enzyme kinetic parameters (Km, Vmax,Vmax/Km) were determined for these compounds using human liver microsomes and recombinant UGT isoforms. The apparent enzyme kinetic parameters were estimated by fitting the initial reaction velocity values, measured as a function of nitrocatechol concentration, to the Michaelis-Menten equation. Glucuronidation of entacapone and tolcapone by human liver microsomes was shown to be linear for at least 60 min and up to 0.2 mg of protein/ml. The reaction catalyzed by UGT1A9 was also linear for at least 60 min and up to 0.5 and 5 mg of protein/ml for entacapone and tolcapone, respectively. The reactions followed Michaelis-Menten kinetics reasonably well, although when UGT1A9 was used the glucuronidation velocity of entacapone started to decrease at concentrations exceeding 100 μM instead of approaching asymptoticallyVmax (Fig.3). A possible explanation for this phenomenon is substrate inhibition although no inhibition was apparent in the kinetic determination with human liver microsomes. Fitting the data on entacapone glucuronidation catalyzed by UGT1A9 to substrate inhibition equation gave Vmax = 2.2 ± 0.65 nmol min−1 mg−1,Km = 14.4 ± 2.8 μM,Vmax/Km = 156 ± 50 × 10−3 ml mg−1 min−1, which are directly comparable to the results derived using the Michaelis-Menten equation (Table 2).

Glucuronidation of entacapone (A) and tolcapone (B) by recombinant human UGT1A9.
The solid lines representing the fitting of the data to the Michaelis-Menten equation give Vmax = 2.60 ± 0.10, Km = 10.7 ± 0.7 and Vmax = 0.62 ± 0.02,Km = 78.3 ± 11.9 for entacapone and tolcapone, respectively. Fitting of the entacapone data to the substrate inhibition equation (dotted line) givesVmax = 2.74 ± 0.14,Km = 11.4 ± 0.9.
Apparent kinetic parameters for the glucuronidation of entacapone and tolcapone by human recombinant UGT isoforms and human liver microsomes (HLM). The values represent mean ± S.D. of two to five determinations (standard deviation shown when n = 3–5).
The apparent kinetic parameters further demonstrate that entacapone is an excellent substrate of UGT1A9 with both high turnover rate and good affinity. When entacapone was used as a substrate for both UGT1A9 cell lysates and human liver microsomes the Vmaxwas 3 to 4 times higher and the Km was 4 to 6 times lower compared with the kinetic parameters of tolcapone. Determination of the kinetic parameters for tolcapone and entacapone glucuronidation by UGT2B7 indicated a considerably higherKm compared with any of the other isoforms used in this study (data not shown). The enzyme failed to saturate at the same substrate concentrations of the COMT inhibitors as used for UGT1A1, UGT1A9 and UGT2B15; this fact combined with the low reaction velocities observed for UGT2B7 in vitro greatly affected the reliability of the determination. However, these experiments did indicate that in comparison to UGT1A9, UGT2B7 is unlikely to have a significant role in the turnover of either entacapone or tolcapone in vivo. UGT2B15 did show normal Michaelis-Menten kinetics under the conditions used and did not discriminate between the two substrates tested presenting very similar Km andVmax values for both.
Discussion
The in vitro glucuronidation of two extensively glucuronidated drugs in vivo, entacapone and tolcapone, was studied by characterizing the UGT isoforms that were shown to be most important in their conjugation. The properties of the nitrocatechols as UGT substrates were compared by determining apparent kinetic parameters for them using human recombinant UGT isoforms and liver microsomes.
Although the contribution of UGT1A1 to the glucuronidation of entacapone was small, it is noteworthy that it was the only UGT capable of glucuronidating this substrate at both of the catecholic hydroxyl groups. UGT1A1 is known to catalyze the formation of both bilirubin IXaC8 and IXaC12 monoglucuronides as well as bilirubin IXaC8C12 diglucuronide (Senafi et al., 1994). However, the formation of a diglucuronide from entacapone is unlikely as conjugation of one catecholic hydroxyl group with UDP-glucuronic acid may cause steric hindrance for the conjugation of the other. In addition, the very similar chromatographic behavior of the glucuronides supports the interpretation that two regioisomeric monoglucuronides were formed. The fact that UGT1A1 showed a lower Km value for the formation of 3-O-glucuronide compared with the kinetics of the second entacapone glucuronide suggests that the binding mode resulting in that regioisomer is also favored with UGT1A1. The low activity exhibited by UGT1A1 in conjunction with the large capacity of other isoforms to turn over these compounds indicate that competition with bilirubin glucuronidation is unlikely. Consistently, only one glucuronide of entacapone has been detected in human urine (Wikberg et al., 1993).
UGT1A6 has been shown to catalyze the glucuronidation of phenolic substrates with the steric requirements of small size and planarity (Ebner and Burchell, 1993). Lack of activity toward entacapone and tolcapone may be due to the relatively big size and bulkiness of their R1 substituents, which probably exceed the limitations of the active site of this enzyme. Of the representative members in the UGT2B family included in the study, UGT2B7 has been reported to metabolize some carboxylic acid drugs and S-naproxen (Jin et al., 1993). UGT2B15 has been shown to accept phenolic compounds, certain drugs, and their hydroxylated metabolites [5-(m-hydroxyphenyl)-5-phenylhydantoin, 5-(p-hydroxyphenyl)-5-phenylhydantoin, and dienestrol] (Green et al., 1994). Both entacapone and tolcapone were glucuronidated by both isoforms, although at about 10 times lower rates compared with the control substrates used to verify the activity of the expressed UGTs. Based on these results, the contribution of the UGT2B family to the metabolism of the nitrocatecholic drugs seems likely to be of lesser importance compared with the activity of UGT1A9, but to verify this, other members of the family have to be investigated.
Previous studies (Ebner and Burchell, 1993) have shown that UGT1A9 is capable of glucuronidating a wide diversity of drug and xenobiotic substrates including β-adrenoceptor agonists, nonsteroidal anti-inflammatory analgesics, diuretics, and propofol. It has been considered to be an important isoform in the metabolism of xenobiotics including a range of therapeutic agents (Burchell et al., 1995). Entacapone and tolcapone were glucuronidated at a high rate by this isoform, which is consistent with previous observations on substrate acceptance. In fact, entacapone proved to be an exceptionally good substrate; it exhibited an almost 2 times higher glucuronidation rate in vitro compared with the anesthetic propofol, which is widely used clinically and is also cleared extensively by glucuronidation in vivo. It is of interest to note that the half-life of propofol using an open two-compartmental model was calculated at 1.5 to 1.8 h (Langley and Heel, 1988). This, relative to the in vivo clearances for entacapone and tolcapone stated earlier (0.3 and 2.3 h, respectively) compares very well to the in vitro rates of glucuronidation with UGT1A9 (r2 = 0.98, the mean value for propofol clearance used). Although the correlation between the rapid clearance in vivo and the high UGT1A9 glucuronidation rate in vitro for all three substrates is not statistically significant, it may be suggestive of a role for this UGT isoform in the clearance of entacapone and tolcapone in vivo.
Entacapone was a clearly better substrate of UGT1A9 with higherVmax and lowerKm value than tolcapone. In the absence of any detailed study on the structure-activity relationships of this isoform, it remains unclear which structural features make the two very similar molecules two such different substrates of this enzyme.
Entacapone and tolcapone were glucuronidated at a high rate by both human liver and human kidney microsomes. This again is substantiating evidence of the role of UGT1A9 in the glucuronidation of these compounds as it has previously been shown to be expressed to a high degree in the kidney (McGurk et al., 1998). This result also demonstrates the importance of renal metabolism in the clearances of both COMT inhibitors and emphasizes that the contribution of the kidney to the metabolic fate of any drug that is glucuronidated should not be overlooked although the restrictions on tissue availability meant that full kinetic analysis was not possible. Comparable differences in the glucuronidation of entacapone and tolcapone by liver microsomes and UGT1A9 (3–4-fold Vmax and 4–6 times lowerKm for entacapone) further suggest that this particular isoform plays a significant role the in overall glucuronidation of these compounds.
The absence of substrate inhibition in human liver microsomes, which was observed with UGT1A9, was thought to be due to the effect of other UGT isoforms that can also bind entacapone. The results from kinetic analysis showed that at least three other UGT isoforms can bind entacapone and these are also represented in the heterogenous population of UGTs in human liver microsomes. TheKm values for two of these isoforms, UGT2B7 and UGT2B15, were shown to be considerably higher than for UGT1A9 but the Vmax values were much lower. As the substrate concentration increases, these lower affinity isoforms will be able to bind substrate, effectively reducing the actual concentration that UGT1A9 would be exposed to. This, in conjunction with the relatively small degree of inhibition of UGT1A9, could explain the apparent discrepancy between human liver microsomes and UGT1A9.
Experiments with human liver microsomes have shown that there is a 14-fold greaterVmax/Km value for entacapone compared with tolcapone, which differs greatly from the situation seen in rat liver microsomes where, conversely, tolcapone exhibits twice theVmax/Km value of entacapone (Lautala et al., 1997). Consequently, it appears that the rat is a poor model species for predicting human glucuronidation for these compounds. Similar findings have also been reported in studies on the in vitro glucuronidation of propofol and the antithrombotic drug LF 4.0212 (Le Guellec et al., 1995; Pless et al., 1999). The understanding of the in vitro glucuronidation of entacapone and tolcapone by human liver and kidney microsomes and human UGT1A9 is a significant step forward in understanding the difference in the clearances of these two important drugs in man in contrast to the data obtained in the rat model.
Footnotes
-
Send reprint requests to: Pia Lautala, Orion Pharma, P.O. Box 65, FIN-02101 Espoo, Finland. E-mail pia.lautala{at}orion.fi
-
This work was supported by the Biotechnology and Biological Sciences Research Council, the Commission of the European Communities (BMH4-CT97-2621), and by Wellcome Trust.
- Abbreviations used are::
- COMT
- catecholO-methyltransferase
- HPLC
- high-performance liquid chromatography
- UDPGA
- UDP-glucuronic acid
- UGT
- UDP-glucuronosyltransferase
- Received February 11, 2000.
- Accepted August 17, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}