


Abstract
Clevidipine is a new vascular selective calcium channel antagonist of the dihydropyridine type, structurally related to felodipine. Clinical trials have shown that the drug can be used to effectively control the blood pressure in connection with cardiac surgical procedures. The compound is tailored to be a short-acting drug and, due to incorporation of an ester linkage into the drug molecule, clevidipine is rapidly metabolized by ester hydrolysis. The pharmacokinetics of clevidipine and its primary metabolite, H 152/81, were studied in rats, rabbits, and dogs. In addition, the influence of the pharmacokinetics on the effect on mean arterial blood pressure was evaluated in anesthetized dogs. Compartmental nonlinear mixed effect regression analysis was used to calculate the population mean and individual pharmacokinetics of clevidipine, whereas nonlinear regression analysis of individual data was used to determine the pharmacokinetics of the primary metabolite. A linkedEmax model was fitted to the individual pharmacodynamic/pharmacokinetic data in dogs. According to the results, clevidipine is a high-clearance drug with a relatively small volume of distribution, resulting in an extremely short half-life in all species studied. The median initial half-life of the individual value (Bayesian estimates) is 12, 20, and 22 s in the rabbit, rat, and dog, respectively. The primary metabolite is a high-clearance compound in the dog, whereas it is a low-clearance compound in the rat. A significant gender difference in the clearance of the metabolite was observed in the rat. The mean maximum reduction in arterial blood pressure is 38 ± 12% (Emax) and is achieved at 85 ± 46 nM (EC50). The half-life for reaching equilibrium between the central and the effect compartment (T1/2ke0) is 47 ± 49 s.
Systemic hypertension after coronary artery bypass grafting was first described by Estafanous et al. in 1973 as a sustained arterial pressure elevation with no obvious cause. At that time, the authors recommended rapid and effective treatment of postoperative hypertensive episodes in an effort to reduce the risk of increased blood loss and hemorrhage from the vascular suture line. A more recently performed study (Vuylsteke et al., 1996) showed that more than 80% of the patients were treated at least once, during or immediately following the cardiac surgical procedure, to lower arterial blood pressure. The agents used for this indication need to have a rapid onset and recovery from effect to be able to control the blood pressure properly. The most commonly used drugs for control and reduction of blood pressure in connection with cardiac surgical procedures are sodium nitroprusside and glyceryl trinitrate, whose pharmacokinetic profiles allow for rapid titration to the desired effect. However, both of these drugs are associated with known drawbacks (Mason and Braunwald, 1965; Kaplan et al., 1980; Zäll et al., 1993; Friederich and Butterworth, 1995). A calcium channel antagonist would be the ideal drug to used for this indication for several reasons, according to Levy (1993). Among the calcium channel antagonists available in i.v. dosage forms, nicardipine has become a drug of choice for reduction and control of blood pressure in association with cardiac surgery (David et al., 1991). However, the pharmacokinetic profile of nicardipine is far from optimal for rapid control of blood pressure. Particularly, the postinfusion decline of the blood concentration is affected by the dose and the duration of the infusion (Cook et al., 1990; Hughes et al., 1992)
Clevidipine (Fig. 1) is a new vascular selective calcium channel antagonist of the dihydropyridine type (Levy et al., 1997) developed for reduction and control of blood pressure in connection with cardiac surgery. Clevidipine is structurally related to another calcium channel antagonist of the dihydropyridine type, felodipine, but incorporation of an ester group into the drug molecule results in a rapid hydrolysis of the compound by esterases in blood and extravascular tissues to the corresponding acid (Ericsson et al., 1999a,b). Available data in animals and humans suggest that this metabolite is devoid of any effect on the blood pressure (M. Nordlander, unpublished data; Ericsson et al., 1999b). As indicated in this Fig. 1, clevidipine is a racemic mixture of two enantiomers.
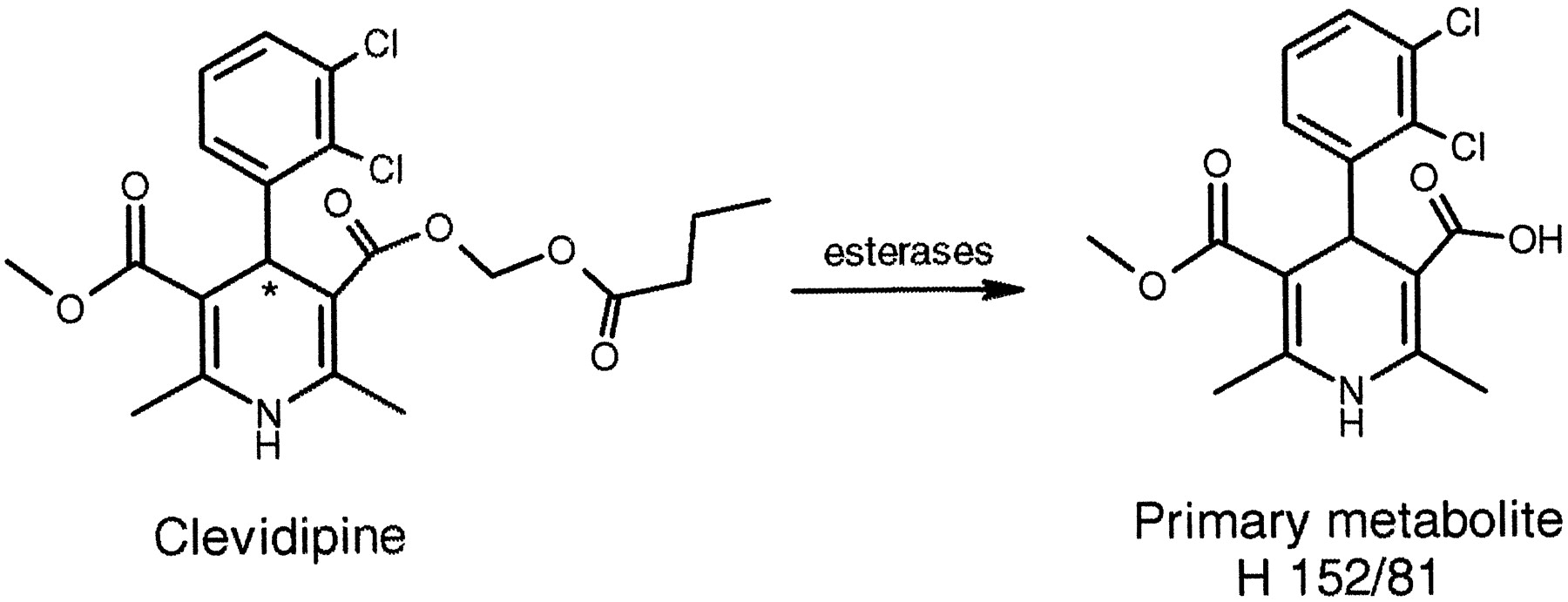
Initial metabolic pathway of clevidipine to primary metabolite and internal standards used in HPLC analysis.
Clevidipine is metabolized by esterases in blood to the carboxylic acid metabolite, H 152/81. Asterisk indicates chiral center of clevidipine.
The rat, rabbit, and dog are the species used in the toxicity studies of clevidipine. This report describes the pharmacokinetics of clevidipine in these species following i.v. infusion of clevidipine. Furthermore, the pharmacokinetics of the primary metabolite, H 152/81, have been evaluated in the rat and the dog in association with the clevidipine dose and after bolus administration of the metabolite. The doses were selected to cover those used in the toxicity studies and to produce blood levels of clevidipine and its primary metabolite relevant to therapeutic blood levels in humans. Finally, the pharmacokinetic/pharmacodynamic (PK/PD)1 relationship in anesthetized dogs is reported.
Materials and Methods
Chemicals.
Clevidipine and the internal standard, felodipine, were manufactured at Astra Pharmaceutical Production AB (Södertälje, Sweden). The purity of clevidipine was 99.6% based on HPLC analysis and the purity of felodipine was 99.4%.The primary metabolite (H 152/81) and H 172/99 (internal standard for H 152/81; Fig. 1) were synthesized at Astra Hässle AB.
Animals.
Rats
Sixteen Sprague-Dawley rats of each gender were studied. The age of the rats varied between 11 and 14 weeks. The body weight of the female rats ranged between 248 g and 284 g and that of male rats between 347 g and 439 g. The animals were acclimatized for at least 1 week before surgery. They were prepared 2 to 3 days before dosing by cannulation of the carotid artery (blood sampling) and the jugular vein (dose administration) during anesthesia. The cannulas were exteriorized at the nape of the neck, filled with heparin (100–200 International Units/ml) and sealed. The rats were housed individually after surgery and they had free access to food and water until the studies were completed.
Dogs.
Six beagle dogs of both sexes (4 males/2 females) were administered clevidipine, whereas seven (4 males/3 females) beagle dogs were studied in the vehicle experiment. The body weight ranged from 11.7 to 17.2 kg. During anesthesia a polyethylene catheter was inserted via arteria grasilis into the abdominal aorta for blood sampling. In addition, a polyethylene catheter was inserted into the right saphenous vein for clevidipine infusion. The body temperature of the animals was maintained at 38°C throughout the experiment by external heating. Heart rate and mean arterial pressure (MAP) were recorded continuously throughout the study.
Rabbits.
Seven female Dutch rabbits were studied. The weight of the rabbits varied between 2.0 and 3.4 kg. The rabbits were acclimatized for 2 weeks before the study. During the study day, a polyethylene catheter connected to a Neofly (21G, 0.8 mm, Boc Ohmeda AB, Helsingborg, Sweden) was inserted into a peripheral ear vein for clevidipine infusion and a Venflon (22G, 0.8 mm, Helsingborg, Sweden) was inserted into the opposite ear artery for blood sampling.
Formulations.
Rats: clevidipine
A formulation based on 20% lipid emulsion, with the same constituents as 20% Intralipid (Pharmacia, Uppsala, Sweden) and containing 3 mg/ml clevidipine, was manufactured. The formulation was further diluted with 20% Intralipid to yield 0.14, 0.46, and 1.37 mg of clevidipine per ml emulsion.
Rats:H 152/81.
The compound was dissolved in 99% ethanol and further diluted with polyethylenglycol (400) and 0.9% saline to yield a final concentration of 0.4 mg/ml. The final composition of the polyethylenglycol/ethanol/H2O-vehicle was 40/10/50 (v/v/v).
Dogs: clevidipine.
A formulation based on 10% lipid emulsion, with the same constituents as 10% Intralipid and containing 1.37 mg/ml clevidipine, was manufactured and used at the two highest infusion rates with clevidipine. The formulation was further diluted with 10% Intralipid to yield 0.46 mg clevidipine per ml emulsion. This dilution was used for the lowest infusion rate with clevidipine. Vehicle, 10% Intralipid, was administered to the control dogs at the same infusion rate as in the clevidipine study.
Rabbits: Clevidipine.
A formulation based on 20% lipid emulsion, with the same constituents as 20% Intralipid and containing 1 mg/ml of clevidipine, was manufactured and used in the study.
Administration and Blood Sampling.
Rats: clevidipine
The drug was administered to three groups of four male and four female rats by infusion into a jugular vein at different constant rates for 120 min with a CMA/100 pump (CMA/Microdialysis AB, Stockholm, Sweden). The target dose rates were 20, 67, and 200 nmol/min/kg, respectively. To obtain the exact time for the formulation to reach the systemic circulation, the time for the emulsion to pass 10 cm of a polyethylenglycol 50 catheter was determined in triplicate. The pump was then connected to the implanted catheter marked 10 cm from the edge. A stopwatch was started when the formulation passed the mark and reset to zero and restarted (T = 0) when the emulsion was estimated to reach the systemic circulation. Blood samples were to be drawn from the carotid artery at 0.5, 1, 1.5, 2, 30, 110, and 115 min during ongoing infusion and at 0.25, 0.5, 0.75, 1, 5, and 20 min following termination of the infusion.
Rats: H 152/81.
The metabolite was administered as an i.v. bolus into a jugular vein over a period of 1 min. The target dose was 2.25 μmol/kg (2 ml/kg). Blood samples were to be drawn from the carotid artery at 5, 10, and 30 min and at 1, 2, 7, 12, 24, and 31 h after the bolus administration.
The rats were given an overdose of pentobarbital sodium after collection of the last blood samples and the localization of the cannula in the jugular vein was confirmed visually.
Dogs: vehicle and clevidipine.
Each experiment started with one 45-min sequence when no drug or vehicle was infused, whereafter clevidipine or vehicle was given by stepwise dose escalation at three different dose rates for 45 min each with a CMA/100 pump. The dose rates of the clevidipine infusions were 6, 18, and 54 nmol/kg/min, respectively. In the control experiment, vehicle was administered at the same rate as for clevidipine; consequently the dogs receiving clevidipine or vehicle were given equal amounts of lipid. Blood samples for determination of clevidipine andH 152/81 were to be drawn at 0, 81, 126, 165, and 171 min after start of the run in period and at 0.25, 0.50, 1, 2, 4, 6, 15, and 30 min following termination of the clevidipine or vehicle infusion.
Because in vitro studies in animal and human whole blood have shown that clevidipine is quantitatively metabolized to its corresponding primary metabolite (Ericsson et al., 1999a), it was anticipated that the same reaction would occur in the in vivo situation. Accordingly, the dose of the metabolite was assumed to be equal to the clevidipine dose.
Rabbits: clevidipine.
Two different doses of clevidipine were infused at a constant rate into a peripheral ear vein for a total infusion time of 60 min with a CMA/100 pump. The target dose rates were 54 and 83 nmol/kg/min, respectively, and the infusion time of each dose was 30 min. Blood samples were to be drawn at 10, 20, 28, 40, 50, and 58 min after the start of the infusion. An additional three to four blood samples were drawn between 0 to 1, 1 to 5, and 5 to 15 min following termination of the infusion. The blood concentration of H 152/81 was not determined.
All blood samples were immediately transferred to preweighed test tubes containing 10% (w/v) SDS-solution and 0.5 M ascorbic acid, which stopped the hydrolysis of clevidipine and prevented the primary metabolite from undergoing oxidation. The samples were then weighed, frozen (within 1 h) and stored at −70°C until analysis.
Analytical Methods.
Blood standards of clevidipine kept at room temperature for 4 h gave 100% recovery and blood samples from animals stored at −70°C for 12 months showed no decrease in concentration (Fakt and Stenhoff, 1999). The blood concentrations of clevidipine in the rat were determined by a method based on gas chromatography-mass spectrometry, whereas the content of the primary metabolite H 152/81 in the blood was analyzed by liquid chromatography (Fakt and Stenhoff, 1999). The limit of quantitation was set to 5 nmol/liter for clevidipine and 50 nmol/liter for the metabolite. The clevidipine concentrations in the rabbit blood were analyzed with the gas chromatography-mass spectrometry method, having a limit of quantitation of 1 nmol/liter (Fakt and Stenhoff, 1999).
Pharmacokinetic Analysis.
Clevidipine
Due to the study design and the extremely rapid postinfusion decline of clevidipine in the blood in all three species, it was not possible to perform model fitting to each animal’s blood concentration versus time data. In some animals the data became too sparse, because the concentration of clevidipine reached the limit of quantitation almost immediately after termination of the infusion. Instead, the pharmacokinetic parameters of clevidipine were determined by nonlinear mixed effect modeling using the software program P-Pharm (ver. 1.4, SIMED, Creteil, Cedex, France). The time course of clevidipine concentrations in the blood of the population data was fitted to mono- or biexponential disposition functions. In all analyses, a heteroscedastic (1/ypred2) distribution of error of the data was assumed. The blood clearance (Clb), initial volume of distribution, and the rate constants for drug transfer between the central and the peripheral compartments (k12,k21,) were obtained directly from the fitting function and were assumed to be normally distributed. The goodness of fit was evaluated by the Akaike Information Criterion (Yamaoka et al., 1976) and visual inspection of residual plots. The potential influence of covariates such as dose rate, weight, and gender on the individual posterior estimates (Bayesian estimates) was examined in the rat by graphical exploration and statistical evaluation using a multiple linear stepwise algorithm (Gomeni et al., 1994).
The Bayesian estimates were used to derive the rest of the reported parameters for clevidipine with standard pharmacokinetic equations (Wagner, 1976). The fractional area under the initial phase of the blood concentration-time curve following an i.v. bolus (AUCλ1) was calculated as:
C1/λ1/(C1/λ1+ C2/λ2), where C1 and C2 represent the fractional zero time intercept following a unit i.v. bolus dose and λ1 and λ2 represent the initial and the terminal rate constants.
H 152/81.
The pharmacokinetics of the primary metabolite H 152/81 in rats and dogs were calculated by fitting one- or two-compartment models to individual blood concentration versus time data by nonlinear regression analysis. The analyses were performed with the computer program WinNonlin (ver. 1.5, Pharsight Corporation, Palo Alto, CA) using the weight 1/(ypred2). The goodness of fit was evaluated by the Akaike Information Criterion and visual inspection of residual plots. The maximum blood concentration of the metabolite (Cmax) and the time to reach this concentration (Tmax) were read directly from the blood concentration versus. time curves.
PK/PD Analysis.
The relationship between the effect on MAP and the blood levels of clevidipine in anesthetized dogs were analyzed using a two-compartment, linked sigmoid Emax PK/PD-model (Sheiner et al., 1979). The individual PK/PD parameters were estimated by nonlinear regression analysis (WinNonlin). The individual Bayesian estimates of the pharmacokinetics of clevidipine, obtained in the dogs by the population approach, were fixed into the pharmacokinetic model and used to drive the dynamics. The pharmacodynamic model consisted of the Hill equation;
Statistical Analysis.
The individual pharmacokinetic parameters of H 152/81 in rats were analyzed by Student’s t test with gender as factor. Statistical significance was assumed for p < .05.
Results
The pharmacokinetic analysis of clevidipine included 282 blood concentrations of clevidipine from the rats, 62 blood concentrations from the rabbits, and 61 blood concentrations from the dogs. A two-compartment model gave the best fit to the population blood concentration versus time data in all species studied. The simulated blood concentration-time curves, based on the mean population values, the individual time course of the blood concentration, and the individual fit of a two-compartment model for a representative rat, dog, and rabbit are shown in Fig. 2. The population mean and the mean values of the individual pharmacokinetics were virtually identical and the mean and the range of the individual pharmacokinetic parameters (Bayesian estimates) are given in Table1.

Simulated clevidipine concentration-time curves, based on mean population values, following a constant rate infusion of clevidipine at 50 nmol/kg/min for 60 min (A) and observed blood concentration of clevidipine versus time following clevidipine infusion for a representative rat (B), rabbit (C), and dog (D).
Solid lines denote posterior individual fit of a two-compartment model to data (B, C, and D).
Mean and range of individual pharmacokinetic parameters (Bayesian estimates) of clevidipine derived from the rat, rabbit, and dog
Clevidipine was a high-clearance drug in all three species studied. The median CLb values were 0.331, 0.165, and 0.291 liters/min/kg in the rat, dog, and rabbit, respectively. The corresponding values for the volume of distribution at steady state (VSS) were, 0.42, 0.21, and 0.14 liters/kg, respectively. The small volume of distribution and the high clearance resulted in an extremely short half-life. The median half-life of the initial phase of the blood concentration versus time curve was 20, 22, and 12 s in the rat, dog and rabbit, respectively.
There was no significant effect of dose rate, gender, or weight on the estimated pharmacokinetic parameters in the rat. Due to the small number of dogs and rabbits studied, no covariate analysis was performed for these species.
A two-compartment model was fitted to the individual blood concentration versus time data of the metabolite following i.v. bolus administration of H 152/81 to male and female rats, whereas a one-compartment model was used to describe the time course of the metabolite in the dog. The results are shown in Table2. A significantly (p < .05) lower Clb and a longer terminal half-life were observed in female rats compared with the male rats. The metabolite clearance was 0.022 liters/h/kg in male rats and 0.013 liters/h/kg in female rats. The corresponding values of the VSS were 0.38 and 0.33 liters/kg. The terminal half-life was 12.8 h in male rats and 18.9 h in female rats. In the dog, the clearance of the metabolite was 3.18 liters/h/kg and the volume of distribution was 0.96 liters/kg. The half-life was 12.6 min and the maximum metabolite concentration was reached approximately 0.6 min following termination of the clevidipine infusion (Fig. 3).
Mean and range of individual pharmacokinetics of H 152/81in male rats (n = 4), female rats (n = 4), and dogs (n = 6, 2 females/4 males) obtained by fitting a two-compartment model to individual concentration of H 152/81versus time data following i.v. bolus administration of metabolite to rats and a one-compartment model following clevidipine infusion in dogs

Blood concentrations of H 152/81versus time following i.v bolus administration of H 152/81(2.25 μmol/kg) to a representative rat (A) and blood concentration of H 152/81 following clevidipine administration at 6, 18, and 54 nmol/kg/min for 45 min at each dose rate to a representative dog (B).
Solid lines denote fit of models to data.
A representative graph of the effect on MAP during the run-in period, the three different dose rates with clevidipine, and postinfusion in one dog is shown in Fig.4. Plotting the reduction in MAP versus the blood concentration during infusion at three different dose rates and the postinfusion concentrations resulted in counterclockwise hysteresis, as shown in Fig. 5. The pharmacodynamics of clevidipine were characterized by a sigmoidEmax model. Fitting a two-compartment, linked PK/PD-model with fixed individual pharmacokinetic parameters resulted in a collapse of the loop, and the reduction in MAP versus the concentration in the theoretical effect compartment is shown for one dog in Fig. 6. The individual PK/PD parameters in the anesthetized dogs are given in Table3. According to the model, the maximum reduction in MAP (Emax) was 38%, the blood concentration producing 50% of the maximal effect (EC50) was 85 nmol/liter, and the slope factor was 1.5. The median half-life for reaching equilibrium between the arterial blood concentration and effect compartment was 30 s (T1/2ke0). The vehicle infusion into control animals did not show any effect on the mean arterial blood pressure at infusion rates and for durations similar to the ones used for clevidipine infusion in the present study.

Mean arterial blood pressure versus time following run in period and clevidipine infusion at three different dose rates (6, 18, and 54 nmol/kg/min) in a representative dog.

Effect (% reduction in MAP) versus observed clevidipine concentrations in a representative dog.
Data are plotted in time order as indicated by arrows, and numbers refer to time (min) after start of infusion.
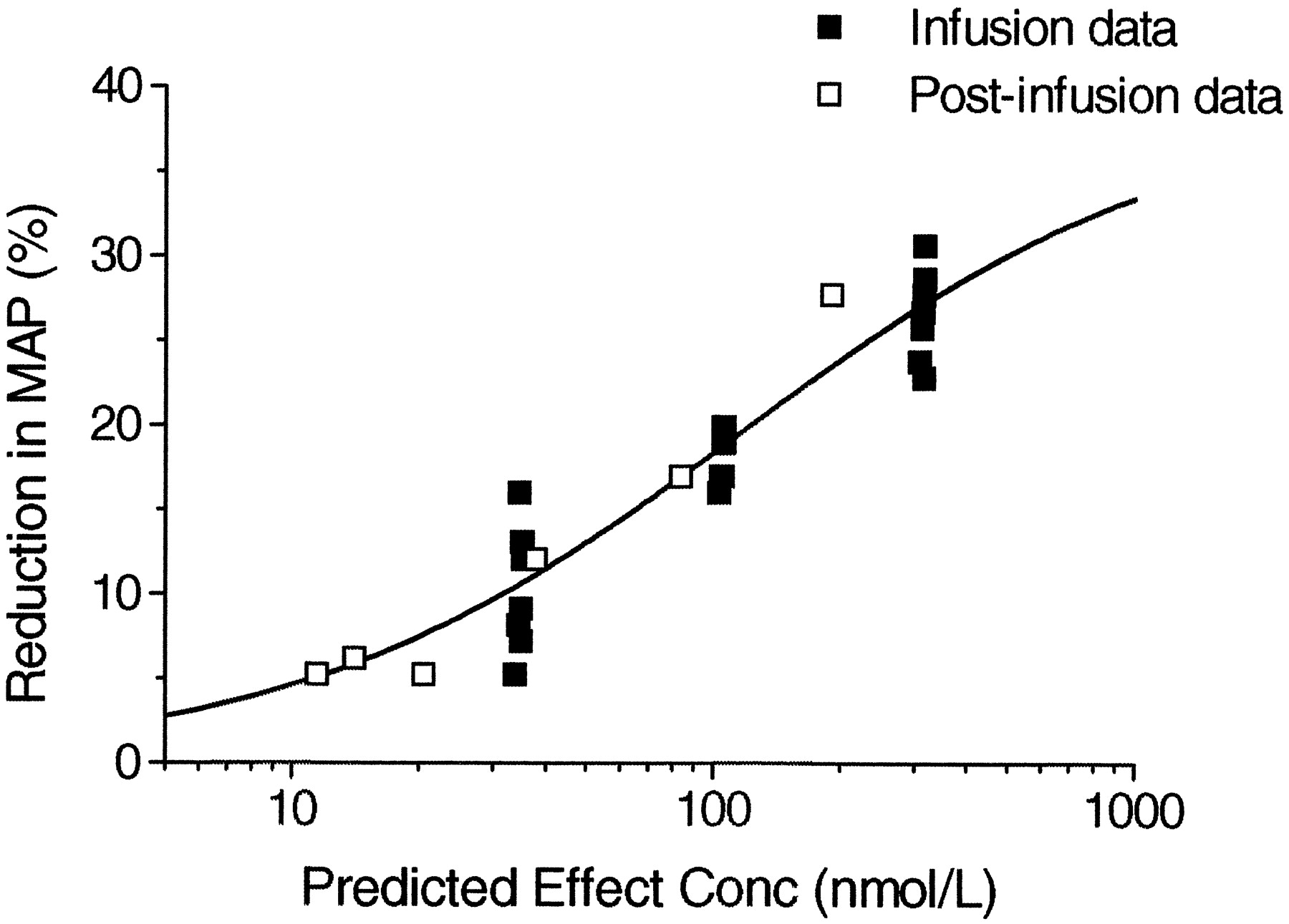
Reduction in MAP versus predicted effect site concentration of clevidipine in a representative dog.
Individual pharmacodynamic parameters of clevidipine obtained by fitting a two-compartment, linked sigmoid Emax PK/PD model following i.v. clevidipine infusion in anesthetized dogs at three different dose rates (n = 6)
Discussion
The aim of the present study was to determine the pharmacokinetics of the ultrashort-acting calcium channel antagonist clevidipine and its primary metabolite in species used in the safety assessment of clevidipine and to evaluate the relationship between the arterial blood concentration of clevidipine and its effect on MAP in the anesthetized dog. The dose rates were selected to cover those used in the toxicity studies and to produce blood levels of clevidipine and its primary metabolite relevant to therapeutic blood levels in humans (10–200 nmol/liter). These levels were covered and exceeded in the present study.
The pharmacokinetics of clevidipine were similar in all three species studied. Thus the Clb of clevidipine was extremely high and was approximately 5-fold higher than the liver blood flow in all three species (Davies and Morris, 1993). Compared with an ultrashort-acting μ-opioid receptor agonist with an ester structure, remifentanil, the clevidipine clearance in anesthetized dogs was approximately four times the clearance of remifentanil in conscious dogs, whereas the Clb of remifentanil in conscious rats was slightly higher than for clevidipine in the rat (Chism and Rickert, 1996; Haidar et al., 1997). The relatively small volume of distribution of clevidipine resulted in extremely short half-lives. The half-life of the initial phase of the blood concentration versus time curve, estimated to cover more than 85% of the total AUC following an unit i.v. bolus, was less than 25 s in all three species.
The extremely rapid in vivo and in vitro hydrolysis rate of clevidipine in rat blood, 0.6 min (Ericsson et al., 1999a), necessitated very short blood collection times in this species to avoid extrapolation of observed concentrations back to the starting time of the blood collection. With a sampling time less than 8 s, the underestimation of the blood concentrations is maximum 7% when mixing with the hydrolysis inhibitor is taken as the actual sampling time. The relatively slow hydrolysis rate of clevidipine in whole blood from dogs, 16 min (Ericsson et al., 1999a), did not motivate the same time correction as in the rat study. At present, there is no information about the hydrolysis rate of clevidipine in whole blood from Dutch rabbits. However, in vitro studies with another ester-containing compound, esmolol, in rabbit and rat blood indicate relatively low in vitro esterase activity in rabbit blood compared with the rat blood (Yang et al., 1995). Therefore, the start of blood withdrawal was used as the sampling time also in the rabbit.
Interestingly, there was a much closer agreement between the in vivo pharmacokinetics, approximately a 2-fold difference in the clearance value in rats and dogs, compared with the in vitro hydrolysis rate in whole blood from the rat and dog, 0.6 and 16 min, respectively (Ericsson et al., 1999a). This difference is probably a consequence of the fact that other tissues than blood and plasma contribute to a large extent to the elimination of clevidipine, especially in the dog. Support for this hypothesis is obtained from a study on remifentanil in anesthetized dogs (Chism and Rickert, 1996). According to these authors, the muscle and intestine had the highest tissue clearance of remifentanil, whereas the liver, kidneys, and blood each accounted for 1% or less of the systemic clearance.
The maximum blood levels of the metabolite were observed approximately 0.5 min following termination of the clevidipine infusion in dogs, showing that the rapid postinfusion decline of clevidipine is due to the elimination rather than the distribution of clevidipine. The pharmacokinetics of the primary metabolite differ markedly from those of the parent compound in both the rat and the dog. Furthermore, there is a substantial interspecies difference in the pharmacokinetics of the metabolite. The mean half-life of the metabolite in the dog, determined over a postinfusion period of 30 min following clevidipine infusion, was 12 min, suggesting that the metabolite will be at steady-state levels in the dog within 1 h of clevidipine infusion. In the rat, the terminal half-life was on average 13 and 19 h, depending on sex, and it will take 1.5 to 2.5 days before the metabolite reaches steady state during continuous clevidipine infusion. In both species, the metabolite is further metabolized before being excreted, mainly by glucuronidation and excretion into the bile (Fryklund L., unpublished data from distribution, metabolism, and excretion studies in rats and dogs). The gender effect on the further metabolism of the primary metabolite is in agreement with previous reports on other compounds metabolized by glucuronidation (Greenblatt et al., 1980;Divoll et al., 1981; Bonate, 1991; Hansen and Stentoft, 1995) and probably the difference in the rate of elimination of the metabolite between male and female rats is mainly to be found in the glucuronidation capacity. Similarly, the species difference in the clearance of the metabolite is probably an effect of a different glucuronidation capacity in the rat and dog, of which the latter species efficiently metabolizes the primary metabolite H 152/81 by glucuronidation. The rat seems to be a better model than the dog for predicting the pharmacokinetics of the metabolite in humans. Thus, the half-life of the metabolite in male rats in the present study is close to the reported pharmacokinetics for the metabolite in healthy male volunteers (Ohtake et al., 1992; Ericsson et al., 1999b).
Plotting the percentage reduction in MAP versus the arterial blood concentration of clevidipine in the anesthetized dog resulted in an anticlockwise hysteresis loop. This type of relationship between effect and concentration is usually explained by a delayed distribution into the site of action or the formation of an active metabolite. However, many drug effects may be considered as indirect responses i.e., the drug acts by stimulation or inhibition of factors controlling the response(s). Different PK/PD models have been suggested in the literature to account for this delay in response in relation to the blood or plasma concentration. Among these models, the effect compartment model and the indirect response models are commonly used (Sheiner et al., 1979; Sharma and Jusko, 1996). The effect compartment model accounts for the time delay by adding a theoretical effect compartment connected to the pharmacokinetic model without affecting the mass transport for the pharmacokinetic model. An effect compartment model was used in the present study to account for the short delay between the effect and blood concentration of clevidipine. The pharmacodynamics of clevidipine were well characterized by a sigmoidEmax model and a two-compartment, linked PK/PD with fixed individual pharmacokinetic parameters resulted in a collapse of the hysteresis loop.
The interindividual variability in the PK/PD parameters in the anaesthetized dogs was rather large and the estimates of these parameters could potentially be influenced by the predetermined dose rate studied and by the fact that the maximal reduction in MAP was not achieved in some dogs. However, the extremely short equilibrium half-life into the effect compartment (T1/2ke0) suggests a rapid onset of effect and a short effect duration following termination of the clevidipine infusion, which is in agreement with the findings in this study.
In conclusion, clevidipine is a high-clearance drug in the rat, the dog, and the rabbit. The high clearance and the small volume of distribution results in extremely short half-lives. The initial half-life accounting for more than 85% of the total AUC following i.v. bolus administration in all three species is less than 25 s. The pharmacokinetics of the metabolite are markedly different in the dog and rat. This compound is cleared rapidly in the dog, presumably due to extensive glucuronidation by the liver, whereas in the rat the metabolite is a typical low-clearance compound.
Acknowledgments
We thank Pia Thalen for help with the pharmacokinetic/pharmacodynamic study in dogs, Liselotte Andersson and coworkers for help and assistance in blood sampling in the rabbit study, and Christina Fakt and coworker for analyzing clevidipine and metabolite samples.
Footnotes
-
Send reprint requests to: Hans Ericsson, Pharmacokinetics and Drug Metabolism, Astra Hässle AB, S-431 83 Mölndal, Sweden. E-mail: Hans.Ericsson{at}hassle.se.astra.com
- Abbreviations used are::
- PK/PD
- pharmacokinetic/pharmacodynamic
- MAP
- mean arterial pressure
- Clb
- blood clearance
- AUCλ1
- area under initial phase of blood concentration-time curve following an i.v. bolus
- Vss
- volume of distribution at steady state
- Received September 28, 1998.
- Accepted January 19, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}