


Abstract
The in vitro inhibitory effects of gemfibrozil on cytochrome P450 (CYP) 1A2 (phenacetin O-deethylation), CYP2A6 (coumarin 7-hydroxylation), CYP2C9 (tolbutamide hydroxylation), CYP2C19 (S-mephenytoin 4′-hydroxylation), CYP2D6 (dextromethorphan O-deethylation), CYP2E1 (chlorzoxazone 6-hydroxylation), and CYP3A4 (midazolam 1′-hydroxylation) activities were examined using pooled human liver microsomes. The in vivo drug interactions of gemfibrozil were predicted in vitro using the [I]/([I] + Ki) values. Gemfibrozil strongly and competitively inhibited CYP2C9 activity, with a Ki (IC50) value of 5.8 (9.6) μM. In addition, gemfibrozil exhibited somewhat smaller inhibitory effects on CYP2C19 and CYP1A2 activities, withKi (IC50) values of 24 (47) μM and 82 (136) μM, respectively. With concentrations up to 250 μM, gemfibrozil showed no appreciable effect on CYP2A6, CYP2D6, CYP2E1, and CYP3A4 activities. Based on [I]/([I] +Ki) values calculated using peak total (or unbound) plasma concentration of gemfibrozil, 96% (56%), 86% (24%), and 64% (8%) inhibition of the clearance of CYP2C9, CYP2C19, and CYP1A2 substrates could be expected, respectively. In conclusion, gemfibrozil inhibits the activity of CYP2C9 at clinically relevant concentrations, and this is the likely mechanism by which gemfibrozil interacts with CYP2C9 substrate drugs, such as warfarin and glyburide. Gemfibrozil may also impair clearance of CYP2C19 and CYP1A2 substrates, but inhibition of other CYP isoforms is unlikely.
Gemfibrozil is a fibric acid derivative, which is used in the treatment of patients with lipid disorders (Dollery, 1999). A combined use of gemfibrozil and a statin can result in severe myopathy and rhabdomyolysis (Pierce et al., 1990; Tal et al., 1997). Recently, gemfibrozil has been found to elevate markedly plasma simvastatin acid and lovastatin acid levels, indicating a pharmacokinetic mechanism in the gemfibrozil-statin interactions (Backman et al., 2000; Kyrklund et al., 2001). In addition, gemfibrozil has been reported to enhance the effects of some other drugs, such as warfarin and glyburide (Ahmad, 1990; Ahmad, 1991;Rindone and Keng, 1998). However, the mechanisms of these gemfibrozil-related drug interactions are unclear.
We have investigated the effects of gemfibrozil on the major cytochrome P450 (CYP1) isoform activities in human liver microsomes in vitro, using selective marker reactions for CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 to clarify the mechanisms of the drug-drug interactions of gemfibrozil.
Experimental Procedures
Materials.
Gemfibrozil, dextromethorphan, and dextrorphan were obtained from Orion Pharma (Espoo, Finland). Phenacetin, paracetamol, coumarin, 7-hydroxycoumarin, tolbutamide, chlorzoxazone, and NADPH were purchased from Sigma (St. Louis, MO). Hydroxytolbutamide, 6-hydroxychlorzoxazone,S-mephenytoin, and 4′-hydroxymephenytoin were purchased from Ultrafine Chemicals (Manchester, UK). Midazolam and 1′-hydroxymidazolam were kindly provided by Hoffmann-La Roche (Basel, Switzerland). Pooled human liver microsomes (prepared from five male and five female human liver microsomal samples) containing representative activities of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 were obtained from GENTEST (Woburn, MA). Other chemicals and reagents were obtained from Merck (Darmstadt, Germany).
Inhibition Studies.
The effects of gemfibrozil on seven different CYP isoform-specific marker reactions were studied: phenacetin O-deethylation for CYP1A2, coumarin 7-hydroxylation for CYP2A6, tolbutamide hydroxylation for CYP2C9, S-mephenytoin 4′-hydroxylation for CYP2C19, dextromethorphan O-deethylation for CYP2D6, chlorzoxazone 6-hydroxylation for CYP2E1, and midazolam 1′-hydroxylation for CYP3A4. The incubation conditions used to study the metabolism of the various substrates and the effects of specific inhibitors have been reported elsewhere (Wen et al., 2001). The time of incubation and concentration of microsomal protein (100 μg/ml) used in each assay were determined to be in the linear range for the rate of metabolite formation.
All incubations were performed in duplicate, and the mean values were used. Briefly, gemfibrozil was dissolved in acetonitrile. After evaporation of acetonitrile to dryness, gemfibrozil was reconstituted in an incubation medium (final concentrations, 0–250 μM) containing 0.1 M sodium phosphate buffer, pH 7.4, 5 mM MgCl2, and 1.0 mM NADPH. The reaction was started by the addition of the marker substrate either without or after preincubation of the sample at 37°C for 15 min. After a specific period of time, the reactions were terminated by adding the appropriate chemicals to precipitate the proteins (Wen et al., 2001). After centrifugation at 10,000g for 5 min, an aliquot of the supernatant was subjected to analysis of the metabolites by high-performance liquid chromatography, as described previously (Wen et al., 2001). The intra- and interday coefficients of variation were <7% at relevant concentrations (n = 6).
Data Analysis.
The IC50 values were determined graphically. The apparent inhibitory constant (Ki) values were calculated by nonlinear regression analysis by fitting different models of enzyme inhibition to the kinetic data using SYSTAT for Windows 6.0.1 (SPSS, Inc., Chicago, IL).
Results and Discussion
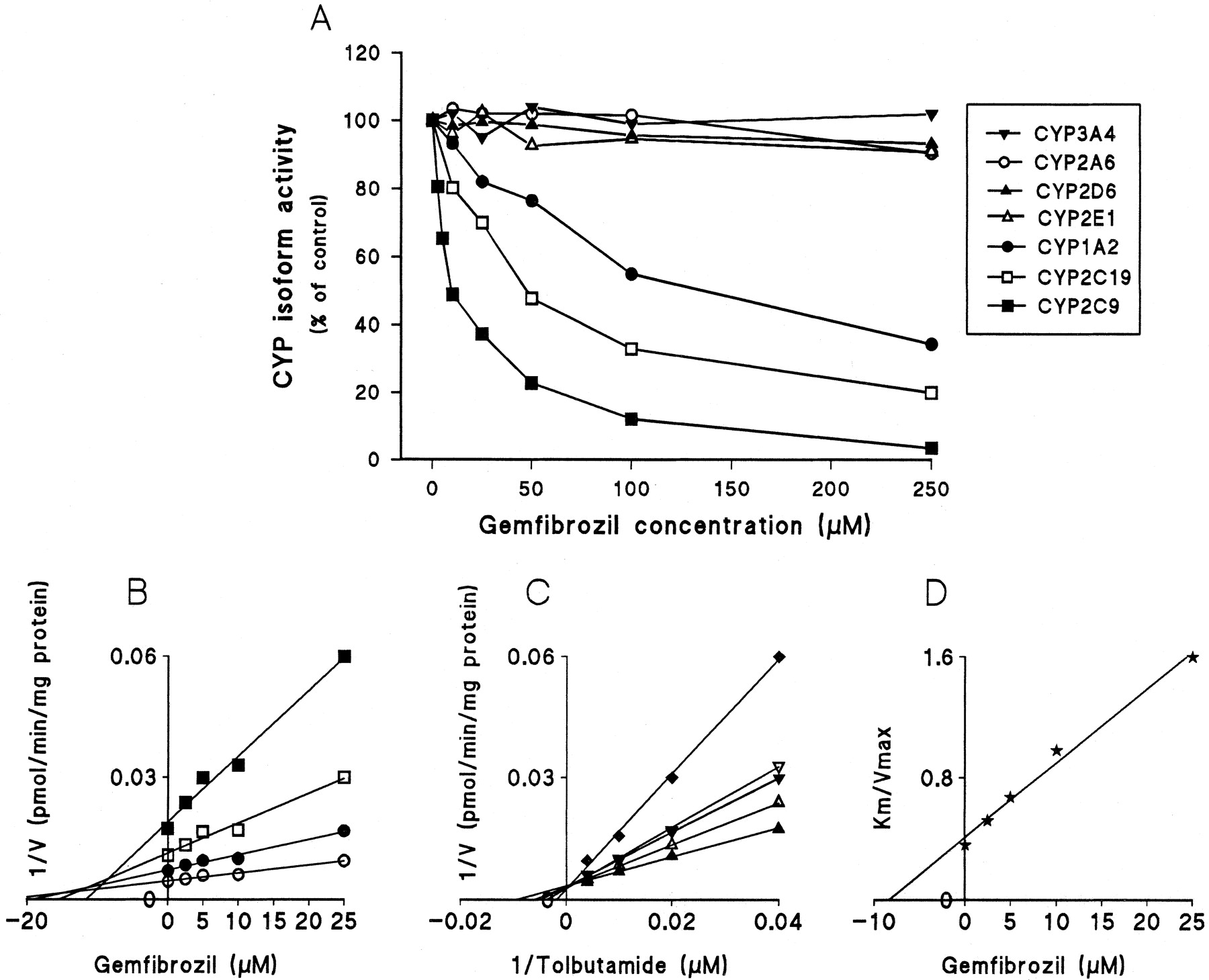
Gemfibrozil strongly inhibited CYP2C9-catalyzed tolbutamide hydroxylation (Fig. 1A). The double reciprocal plots, Dixon plots, and the secondary plot of the slopes of double reciprocal plots versus gemfibrozil concentration indicated that gemfibrozil competitively inhibited CYP2C9 activity, with aKi (IC50) value of 5.8 (9.6) μM (Fig. 1, B–D; Table 1). Gemfibrozil exhibited a modest but significant inhibitory effect on CYP2C19 and CYP1A2 activities, with apparentKi (IC50) values of 24 (47) μM and 82 (136) μM, respectively. The pattern of inhibition of CYP2C19 and CYP1A2 by gemfibrozil was compatible with mixed inhibition. With concentrations ranging from 5 to 250 μM, gemfibrozil showed no remarkable effects on CYP2A6, CYP2D6, CYP2E1, and CYP3A4 activities (Fig. 1A). Preincubation of gemfibrozil with NADPH for 15 min before the addition of the specific substrates did not increase the degree of inhibition (data not shown).

Inhibitory effect of gemfibrozil on CYP-catalyzed reactions in pooled human liver microsomes.
A, effects of gemfibrozil (0–250 μM) on CYP1A2-catalyzed phenacetinO-deethylation (●), CYP2A6-catalyzed coumarin 7-hydroxylation (○), CYP2C9-catalyzed tolbutamide hydroxylation (▪), CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation (■), CYP2D6-catalyzed dextromethorphan O-demethylation (▴), CYP2E1-catalyzed chlorzoxazone 6-hydroxylation (▵), and CYP3A4-catalyzed midazolam 1′-hydroxylation (▾). When 50 μM phenacetin, 1 μM coumarin, 50 μM tolbutamide, 40 μMS-mephenytoin, 1.5 μM dextromethorphan, 25 μM chlorzoxazone, and 2 μM midazolam were used as substrates in the absence of inhibitor; the corresponding metabolic activities were 722, 701, 90, 100, 104, 294, and 1464 pmol/mg/min, respectively. B, a representative Dixon plot obtained from a 60-min incubation with 25 (▪), 50 (■), 100 (●), and 250 μM (○) of tolbutamide (CYP2C9 marker) in the absence or presence of gemfibrozil (2.5–25 μM). C, a double reciprocal plot obtained from a 60-min incubation of human liver microsomes with NADPH and tolbutamide (25–250 μM) in the absence (▴) or presence of 2.5 (▵), 5 (▾), 10 (▿) or 25 μM (♦) gemfibrozil. D, a secondary plot of slopes taken from double reciprocal plots versus gemfibrozil concentration. Each data point represents the average of duplicate determinations.
The constants and types of inhibition of gemfibrozil for human P450 isoform activities and the predicted in vivo inhibition of the metabolism of coadministered CYP2C9, CYP2C19, and CYP1A2 substrates by gemfibrozil from in vitro data
After the usual daily doses of gemfibrozil (600 mg b.i.d.), the peak total plasma concentrations of gemfibrozil range from 30 to 60 mg/liter (approximately 120–240 μM), and the average total plasma gemfibrozil concentrations during the 12-h-dosing interval range from 5 to 23 mg/liter (20–92 μM) (Backman et al., 2000). Thus, theKi values for CYP2C9, CYP2C19, and CYP1A2 fall in the range of the total plasma concentrations of gemfibrozil observed in humans in vivo.
Theoretically, drug interactions based on inhibition of hepatic metabolism (i) can be predicted by theKi value and the concentration of inhibitor ([I]) around the metabolic enzyme in the liver using the following predictive model: i = [I]/([I] +Ki), assuming that the substrate concentration is much lower than its Kmvalue (von Moltke et al., 1998). However, the in vivo concentration of an inhibitor around the enzyme is not known. Based on a hypothesis stating that only the unbound concentration in plasma is available for diffusion to intrahepatic sites of metabolic activity, the unbound plasma concentration of an inhibitor has been used to predict in vivo drug interactions (Ito et al., 1998). However, this hypothesis is not suitable for a highly lipophilic compound since the liver concentrations of many lipophilic compounds greatly exceed those in plasma, despite extensive plasma protein binding (von Moltke et al., 1998). In these cases, successful prediction of in vivo drug interactions has been achieved by using the total plasma concentration of an inhibitor or multiplying total plasma concentration by a liver/plasma partition ratio (von Moltke et al., 1998).
Gemfibrozil is a lipophilic compound, and about 95% of gemfibrozil is bound to serum albumin (Dollery, 1999). In vivo, gemfibrozil accumulates into tissues, such as liver and kidney, during long-term therapy. However, the exact liver/plasma partition ratio is unknown. If the mean peak total plasma concentration of gemfibrozil (150 μM) is used in the scaling model described above, one would expect approximately 96, 86, and 64% inhibition of the clearance of CYP2C9, CYP2C19, and CYP1A2 substrates by gemfibrozil, respectively (Table 1). On the other hand, the mean total gemfibrozil concentration during the 12-h-dosing interval (40 μM) would cause approximately 85, 58, and 30% inhibition, respectively. Finally, even the mean plasma concentrations of unbound gemfibrozil (approximately 2 μM) would cause about 24, 7, and 2% inhibition, respectively.
Gemfibrozil has been reported to enhance the anticoagulant effect of warfarin, resulting in severe hypoprothrombinemia and bleeding (Ahmad, 1990; Rindone and Keng, 1998). The biotransformation of the pharmacologically more active S-enantiomer of warfarin is catalyzed mainly by CYP2C9, whereas the metabolism ofR-warfarin is catalyzed by CYP1A2 and CYP3A4 (Yamazaki and Shimada, 1997). Because the binding of warfarin to human serum albumin is not influenced by gemfibrozil (Hamberger et al., 1986), the gemfibrozil-warfarin interaction can be explained by inhibition of the CYP2C9-mediated metabolism of S-warfarin. Minor inhibition of the CYP1A2-mediated metabolism of R-warfarin by gemfibrozil may also contribute to the interaction. In one case report, gemfibrozil has been reported to interact with glyburide, resulting in hypoglycemia (Ahmad, 1991). Glyburide is extensively metabolized in the liver, with CYP2C9 being the predominant enzyme (Brian, 2000). Our in vitro inhibition studies suggest that inhibition of CYP2C9 activity by gemfibrozil is the likely mechanism of the gemfibrozil-glyburide interaction.
Gemfibrozil can interact with several statins, such as lovastatin (Pierce et al., 1990) and simvastatin (Tal et al., 1997), resulting in an increased incidence of myopathy and rhabdomyolysis. The exact mechanism that underlies these drug interactions is unknown. However, it was found recently that gemfibrozil markedly increases the plasma concentrations of active simvastatin acid and lovastatin acid, whereas the concentrations of parent simvastatin and lovastatin are not altered (Backman et al., 2000; Kyrklund et al., 2001). Parent simvastatin and lovastatin are metabolized mainly by CYP3A4 (Vickers et al., 1990). Because gemfibrozil is not a CYP3A4 inhibitor, the gemfibrozil-simvastatin and gemfibrozil-lovastatin interactions cannot be explained by inhibition of the CYP3A4-mediated simvastatin and lovastatin metabolism, as is the case with itraconazole-lovastatin and itraconazole-simvastatin interactions. The present finding that gemfibrozil strongly inhibits the activity of CYP2C9 and, to a lesser extent, that of CYP2C19 and CYP1A2 raises the possibility that some of these CYP isoforms might be involved in the metabolism of simvastatin and lovastatin acid. However, inhibition of some other pathways that regulate the levels of the statin acids by gemfibrozil cannot be ruled out.
To conclude, the present in vitro study demonstrates that gemfibrozil in clinically relevant concentrations is a potent inhibitor of CYP2C9 and a modest inhibitor of CYP2C19 and CYP1A2. However, the activity of CYP2A6, CYP2D6, CYP2E1, and CYP3A4 is not affected by gemfibrozil. Inhibition of CYP2C9 seems to explain the observed interactions of gemfibrozil with warfarin and glyburide. Also, because other substrates of CYP2C9 with a narrow therapeutic range may be affected by gemfibrozil, care is warranted in the use of such drug combinations.
Acknowledgments
We thank Jouko Laitila for skillful technical assistance.
Footnotes
-
This study was supported by grants from the Helsinki University Central Hospital Research Fund and the National Technology Agency of Finland (Tekes).
- Abbreviation used is::
- CYP
- cytochrome P450
- Received May 1, 2001.
- Accepted July 10, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}