


Abstract
The O-demethylation of dextromethorphan to dextrorphan in humans is catalyzed primarily by cytochrome P450 2D6 (CYP2D6). However, contrary to conventional wisdom, preparations of recombinant cytochrome P450 (P450) expressed fromCYP2D6*1 cDNA also appear to produce significant amounts of 3-methoxymorphinan, the N-demethylated metabolite of dextromethorphan, when assayed in vitro. We hypothesized that both pathways were intrinsic to 2D6 and here further examine the kinetics of formation using a highly purified preparation of CYP2D6 in a reconstituted lipid system. Purified CYP2D6 protein with a measured molecular weight of 55772.0 (55769.6 Da predicted) was reconstituted into an active, lipid-vesicle environment with purified rat cytochrome P450 reductase before the addition of substrate and NADPH. Reaction kinetics were followed, and apparent Michaelis-Menten constants were determined for the appearance of each metabolite by high-pressure liquid chromatography, using both UV and fluorescence detection. In a 2-min assay, purified 2D6 catalyzed the formation of dextrorphan with an apparent Km value of 1.9 ± 0.2 μM and a Vmax value of 8.5 ± 0.2 nmol/nmol of P450/min and measured simultaneously the formation of 3-methoxymorphinan with an apparentKm value of 5000 ± 700 μM andVmax value of 176 ± 12 nmol (nmol of P450)−1 min−1. These results indicate that at least two distinct binding orientations exist for dextromethorphan within the active site of CYP2D6.
Dextromethorphan (DXM1), a notable cough-suppressing synthetic analog of codeine, undergoes parallel O-demethylation to dextrorphan (DXO), N-demethylation to 3-methoxymorphinan (MEM), and secondary N- and O-didemethylation to 3-hydroxymorphinan (HYM) in humans and human liver microsomes. TheO-demethylation of dextromethorphan is an established and widely used phenotypic index reaction both in vivo (Dayer et al., 1988;Guttendorf et al., 1988; Baumann et al., 1992; Jacqz-Aigrain et al., 1993; Wu et al., 1993) and in vitro (Jacqz-Aigrain et al., 1993; Kerry et al., 1993) for the metabolic capacity of cytochrome P450 2D6 (CYP2D6) due to its relative specificity, safety, and over-the-counter availability. Historically, the N-demethylation pathway has been attributed solely to cytochrome P450 3A4 (Smith & Jones, 1992;Jacqz-Aigrain et al., 1993; Gorski et al., 1994; Schmider et al., 1997); hence, dextromethorphan has been used to simultaneously estimate both CYP2D6 and CYP3A4 activities by measuring the relative amounts ofO- and N-demethylated metabolites produced (Ducharme et al., 1996; Jones et al., 1996; Krecic-Shepard et al., 1999). More recently, significant contributions from 2C9, 2C19, 2B6, and 2D6 itself have been predicted to contribute toward theN-demethylation reaction, and the use of MEM as a 3A4 probe has been questioned (von Moltke et al., 1998; Kashuba et al., 1999;Wang & Unadkat, 1999; McGinnity et al., 2000).
Rationale for the role of 2D6 in DXM O-demethylation was provided with the characterization of aspartic acid 301 (D301) mutants (Ellis et al., 1995; Mackman et al., 1996). This amino acid appears in the active site of 2D6 by homology modeling (Mackman et al., 1996; Modi et al., 1996), making it a likely candidate for affecting substrate preference and orientation. Additional pharmacophore modeling finds that typical 2D6 substrates often contain a cationic nitrogen 5 or 7Å from their major site of oxidation (de Groot et al., 1997), ideally situated to form ion-pair interactions with D301. Clinically, dextromethorphan O-demethylation by CYP2D6 is estimated to constitute 90% or greater toward the overall clearance of this drug in human liver. Given the strong apparent preference for a particular dextromethorphan binding orientation, it is not surprising that CYP2D6-catalyzed N-dealkylation, initially considered as an atypical and rare pathway, is only gradually being recognized as an accepted metabolic pathway as more and more basic drugs are found to beN-dealkylated by CYP2D6 (Coutts et al., 1994). Recently, a combined protein and pharmacophore model for CYP2D6 was reported to explain CYP2D6-catalyzed N-dealkylation reactions (de Groot et al., 1997).
We desired to investigate the N-demethylation of dextromethorphan phenomenon further without the uncertainty of unknown and/or unwanted enzyme activities found in multienzyme systems of variable composition, such as liver microsomes or heterologous whole-cell/membrane homogenates. We hypothesized that theN-demethylation is intrinsic to 2D6 and not a contaminant P450 such that, in this highly purified preparation, CYP2D6 would be capable of catalyzing the N-demethylation of dextromethorphan. In addition, we reason that if indeed 2D6 is intrinsically capable of catalyzing both demethylation reactions of DXM, it may be possible to use theN-/O-demethylation ratio as an indicator of active site alterations for other natural variants or site-directed mutants of CYP2D6. In the present study, we adapted a baculovirus-mediated insect cell system for high-level protein expression and heme incorporation to achieve the purification of unmodified CYP2D6.1 holoenzyme.
Materials and Methods
Reagents.
Dextromethorphan HBr, dextrorphan d-tartrate, 3-methoxymorphinan HCl, and 3-hydroxymorphinan HBr were purchased from Sigma/RBI (Natick, MA). Glycerol, sodium cholate, NADPH,l-α-dilauroylphosphatidylcholine (DLPC), dithiothreitol (DTT), phenylmethylsulfonyl fluoride (PMSF), and octyl Sepharose CL-4B were purchased from Sigma (St. Louis, MO). DEAE Sepharose Fast-Flow was obtained from Amersham Pharmacia Biotech (Piscataway, NJ), and ceramic hydroxyapatite was purchased from Bio-Rad (Hercules, CA). Emulgen 911 was a gift from Kao-Atlas (Tokyo, Japan). HPLC solvents and other chemicals were of the highest grade commercially available and were used as received.
Baculovirus-Mediated CYP2D6 Expression and Enzyme Purification.
The CYP2D6 cDNA clone used to make recombinant baculoviral stocks was sequenced in its entirety before use and contained noN-terminal alterations or histidine tags. Expression of 2D6 in Trichoplusia ni suspension cultures (Invitrogen, Carlsbad, CA) was carried out as described for CYP2C9 allelic variants (Haining, 1996). All purification steps were carried out at 4°C, and all buffers were at pH 7.4. The crude insect cell pellet was homogenized in 1 ml of solubilization buffer [20% (v/v) glycerol, 1 mM EDTA, 0.1 mM DTT, 0.2 mM PMSF, and 1% (w/v) cholate in 100 mM potassium phosphate buffer]/nanomole of P450 by making 5 to 10 passes with a Teflon homogenizer. The P450 was solubilized by stirring the homogenized insect cell pellet for 30 min, followed by removal of insoluble material by centrifugation at 100,000g for 40 min. Supernatant was loaded onto an octyl Sepharose CL-4B column (1 ml/nmol of P450) pre-equilibrated with buffer A [20% (v/v) glycerol, 1 mM EDTA, 0.1 mM DTT, 0.2 mM PMSF, and 0.5% (w/v) cholate in 10 mM potassium phosphate buffer] at 30 ml/h. After loading, the column was washed with four columns of buffer A and eluted with buffer A plus 0.4% (v/v) Emulgen 911 (buffer B) at 40 ml/h.
The P450-containing fractions were combined and loaded onto a DEAE Sepharose Fast-Flow column pre-equilibrated with buffer B without cholate (buffer C) at 30 ml/h. The column was washed with four columns of buffer C and eluted with buffer C containing 100 mM potassium phosphate. The fractions were dialyzed overnight against 200 volumes of buffer D [20% (v/v) glycerol, 0.1 mM EDTA in 3 mM potassium phosphate buffer]. The dialyzed enzyme was adsorbed onto a ceramic hydroxyapatite column (1 ml/10 nmol) pre-equilibrated with buffer D. The column was washed with buffer D until A280 < 0.018. The P450 was eluted with buffer E [20% (v/v) glycerol, 0.1 mM EDTA, and 0.5% cholate in 350 mM potassium phosphate buffer]. The fractions were dialyzed overnight against buffer F [20% (v/v) glycerol and 0.1 mM EDTA in 100 mM potassium phosphate buffer]. Purified P450 was aliquoted and stored at −80°C until further use.
Incubation Conditions.
Incubation reactions were performed in 100 mM potassium phosphate, pH 7.4, containing 0.1 μM CYP2D6, 0.2 μM P450 reductase, 10 μg of DLPC, 1 mM NADPH, and DXM in a final volume of 200 μl. 2D6 and reductase were added together first and left to incubate at room temperature for 15 min. DLPC was then added for an another 15-min incubation period before the addition of buffer and substrate. Reactions were initiated by the addition of NADPH and terminated after a 2-min incubation at 37°C by the addition of 10 μl of 60% perchloric acid. Alternatively, reactions were terminated using 200 μl of acetonitrile to avoid DXM precipitation at higher concentrations. The mixtures were centrifuged for 10 min, and the supernatant was used for HPLC and LC-MS analyses to determine the demethylated metabolites.
LC-MS and HPLC Conditions.
Liquid chromatography/electrospray ionization-mass spectrometry analyses of whole proteins were performed essentially as described (Koenigs et al., 1999). Mass spectral analysis of metabolites was carried out using standard procedures on the same instrument as that used for whole proteins. HPLC analyses were carried out on a Waters Alliance System (Milford, MA) consisting of the 2690 separation module, the 2487 dual λ absorbance detector, and the 474 scanning fluorescence detector. The Alliance HPLC System was controlled with Millennium32 software (NuGenesis Technologies, Westborough, MA). A 250- × 4.6-mm i.d., hi-chrome phenyl column (Regis Technologies, Inc., Morton Grove, IL) was used to separate the metabolites. The composition of the mobile phase was 50% acetonitrile and methanol mixture (250:200, v/v) and 50% 10 mM potassium phosphate buffer, adjusted to pH 3.5 with orthophosphoric acid. The flow rate through the column at ambient temperature was 1 ml/min. The excitation and emission wavelengths of the fluorescence detector were 280 and 310 nm, respectively. The UV detector was set at a wavelength of 280 nm.
Results and Discussion
Final CYP2D6.1 protein product was estimated at greater than 95% purity by SDS-polyacrylamide gel electrophoresis with a total yield of 52.3%. The carbon monoxide difference spectrum exhibited a Soret maximum at 450 nm with no evidence of cytochrome P420 formation. Additionally, protein was analyzed by Western blot using an anti-peptide anti-2D6 antibody raised in rabbit. As expected, CYP2D6.1 protein products were detected during each of the purification stages. The isolated CYP2D6 was subjected to liquid chromatography/electrospray ionization-mass spectrometry analyses as a quality control for the expression and purification procedure to reveal the presence of any natural post-translational modifications or unwanted alterations. The experimentally determined molecular weight (mol. wt.) was calculated using the observed charge state distribution or ion envelope. The experimental mol. wt. was 55772.0 for CYP2D6.1 compared with a mol. wt. of 55769.6 predicted based on the primary sequence. Therefore, given the error of 43 ppm, it appears that 2D6 is expressed in active form inT. ni cell cultures with no necessary or applied post-translational modifications (not shown).
Kinetic studies of dextromethorphan O-demethylation were carried out with this highly purified CYP2D6.1 following reconstitution with requisite cofactors in a lipid environment (see Material and Methods). Although the addition of cytochromeb5 has been shown to enhance the activity of, or indeed be requisite for, the metabolic turnover of many cytochromes P450, we chose to eliminate this variable in the present experiments since this cofactor is not required for significant turnover of DXM by 2D6 (not shown). Following incubation, reaction mixtures were subjected to centrifugation and then injected directly onto the column for HPLC peak separation and identification. Under the conditions used, the MEM peak was clearly separated from substrate and DXO peaks, as well as doubly demethylated HYM peaks. HYM, DXO, MEM, and DXM eluted at 7.7, 8.9, 11.7, and 13.8 min, respectively, were verified by fluorescence and UV detection, and separately confirmed by LC-MS. As expected, the omission of CYP2D6, NADPH, DXM, or P450 reductase resulted in a complete loss of metabolite production (not shown).
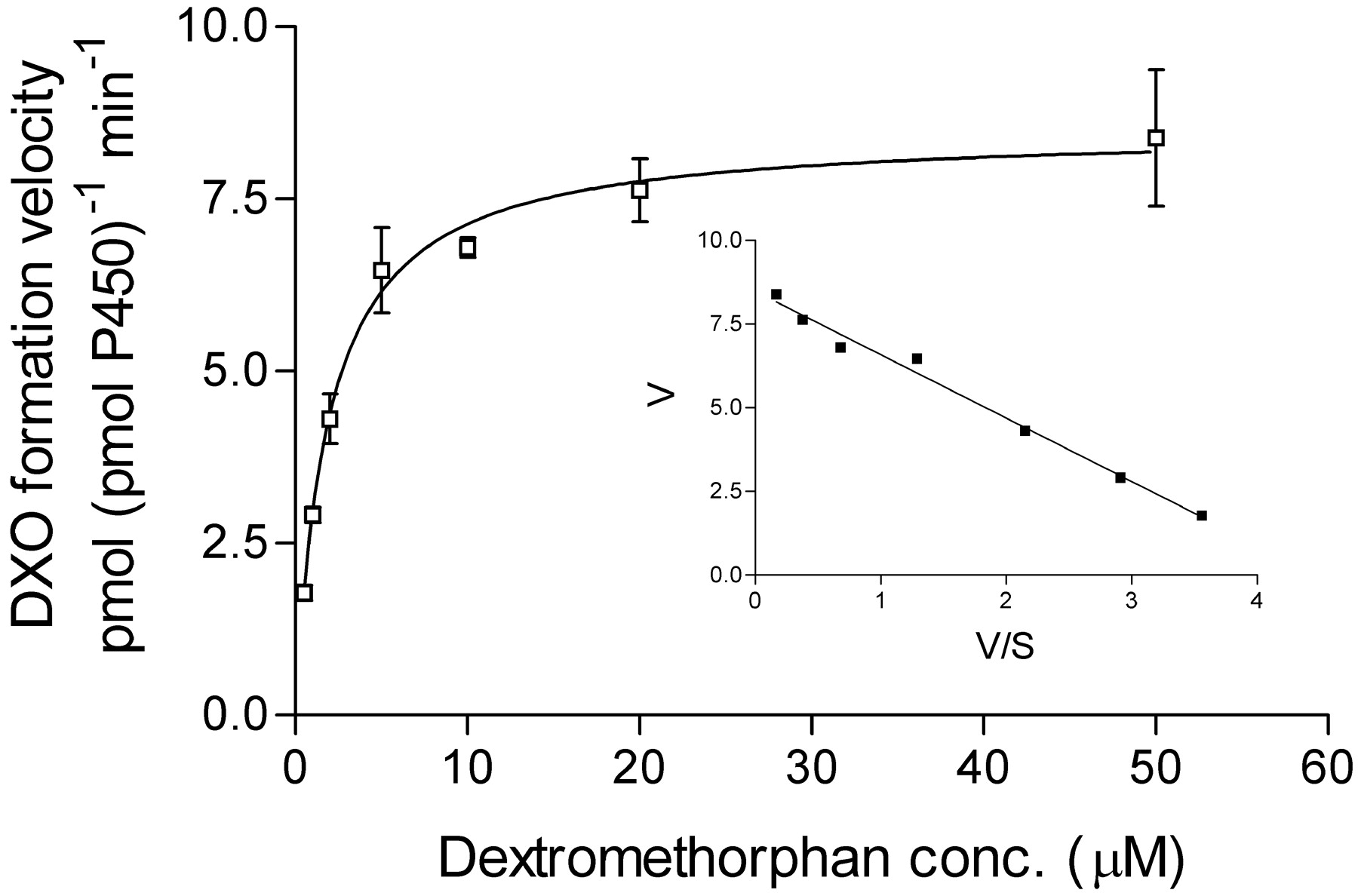
Figure 1 shows the kinetics of dextrorphan formation (O-demethylation) catalyzed by purified CYP2D6 at substrate concentrations of 0 to 50 μM. At these low concentrations, chosen to be nearer a physiological range, the incubation of highly purified CYP2D6 with dextromethorphan produces mainly DXO, as expected, and appears to follow simple saturation kinetics. The Eadie-Hofstee plot for DXM O-demethylation is approximately linear (Fig. 1, inset); thus, we were able to estimate kinetic parameters by fitting them to a simple Michaelis-Menten equation. Data were analyzed using GraphPad Prism v3.02 (GraphPad Software, Inc., San Diego, Ca). As stated in the abstract, purified 2D6 catalyzed the O-demethylation of dextromethorphan with an apparent Km value of 1.9 ± 0.2 μM and Vmax value of 8.5 ± 0.2 nmol/nmol of P450/min, as determined from a 2-min assay (Fig. 1). However, a truer measure of Km requires that initial reaction velocities be determined. The low-turnover rate of many P450s has required the use of longer assay times in many instances, precluding shorter reaction times. However, due to the use of purified enzyme, we were able to shorten our assay to 30 s and still obtain reliable data for DXO formation. In one such 30-s assay, the kinetics of dextrorphan formation proceeded with an apparentKm value of 0.7 ± 0.1 μM, whereas a 5-min assay yielded a Km value of 3.8 ± 1.4. By extrapolation to time 0, perhaps a “true”Km is nearer 0.26 μM (not shown).

NADPH-dependent formation of DXO from DXM as catalyzed by highly purified recombinant CYP2D6.1 during a 2-min assay in vitro at low-DXM concentrations (0.5–50 μM).
P450 was reconstituted into an active lipid (DLPC) environment with rat cytochrome P450 reductase before the addition of DXM and NADPH. Reactions were terminated after 2 min with 10 μl of 60% perchloric acid. Metabolites were quantified by HPLC using fluorescent detection. Points shown are the mean of triplicates ± S.D. Data were fit using a simple Michaelis-Menten equation to yield aKm value of 1.9 ± 0.2 μM andVmax value of 8.5 ± 0.2 nmol (nmol of P450)−1 min−1. Inset, Eadie-Hofstee transformation and plot of the same data set.
Figure 2 shows a representative experiment (one of seven) for the velocity of appearance of DXO and MEM over a wide range of dextromethorphan concentrations, from 0.5 μM to 10 mM (shown on a log scale). Data points shown are the mean of triplicates ± S.D. A detectable contribution from contaminant MEM in the DXM stocks was subtracted out by running reaction blanks (minus NADPH) and deducting the residual peak area. At the highest DXM concentrations used (above 5 mM), contaminant MEM accounted for less than 10% of the total observed peak area. Interestingly, the appearance of 3-methoxymorphinan is not evident until initial DXM concentrations reach the saturation point (20 μM, or approximately 10 × Km) for dextromethorphan binding to the CYP2D6 active site, as determined from the data shown in Fig. 1. Also, the N-demethylase activity does not result in a significant inhibition of dextrorphan turnover even at the highest concentrations, suggesting that binding in the “N-demethylation orientation” is noncompetitive with binding in the “preferred” orientation. Notably, MEM production was not subject to delayed onset, as evidenced from experiments ranging in length from 30 s to 15 min (not shown). Nevertheless, the data for MEM formation could also be fit to a simple kinetic equation; thus, we estimate a second Km value of 5000 ± 700 μM and Vmax value of 176 ± 12 nmol (nmol of P450)−1min−1 for the data shown in Fig. 2 during a 2-min assay. In other experiments of varying times, estimates of theKm value ranged from 1 to 5 mM.

NADPH-dependent formation of MEM (dotted line) and DXO (solid line) from dextromethorphan as catalyzed by highly purified CYP2D6.1 at higher (up to 10 mM) initial substrate concentrations (shown here on a log scale).
Reactions were terminated after 2 min with 200 μl of acetonitrile. Error estimates shown are mean ± S.D. for samples run in triplicate. Data for the production of MEM could be fit to a simple Michaelis-Menten equation to yield an apparentKm value of 5000 ± 700 μM andVmax value of 176 ± 12 nmol (nmol P450)−1 min. Despite the fact that formation of dextrorphan clearly saturates at lower concentrations (Fig. 1), this additional activity (N-demethylation) appears to “kick-in” at higher concentrations and have its own substrate saturation point.
Given the high Km for DXMN-demethylation by CYP2D6, this reaction is unlikely to proceed to any significant extent at DXM concentrations encountered in vivo. Indeed, in a separate work (submitted for publication), we argue that despite potential contributions from CYP2D6 and CYP2B6, DXMN-demethylation is still a valid indicator of CYP3A4/5 activity in vivo except in individuals with unusually high CYB2B6 expression. However, we believe there is a strong theoretical interest in our result. Given the clear correlation of CYP2D6 with DXMO-demethylation, N-demethylation of DXM by 2D6 seems to be a paradox because it suggests DXM has bound in the completely opposite or “upside-down” orientation in a single site model. Although variable regioselectivity of oxidation of substrates by P450 is well accepted, the simultaneous N- andO-demethylation for this substrate is more difficult to visualize because it would also not allow the noted and most probable ionic interaction of the cationic nitrogen found in 2D6 substrates with aspartic acid 301 in the active site (Ellis et al., 1995). This unusual characteristic of DXM metabolism by CYP2D6 may lead to a further understanding of important protein-substrate interactions and better pharmacophore models for CYP2D6.
In recent years, the exhibition of non Michaelis-Menten (atypical) kinetics has been noted for P450s, such as 3A4 and 2C9, in which it is postulated that two substrates can occupy a single active site simultaneously (Korzekwa et al., 1998; Schrag and Wienkers, 2001a,b). We have shown that in the case of the metabolism of dextromethorphan by CYP2D6, a single P450 isoform may exhibit two distinct apparentKm values for the appearance of two metabolites from the same parent compound. These results indicate that the substrate must have available at least one other binding mode to the active site of CYP2D6. How this is possible is not know at this time. It may be that high-substrate concentrations effect an allosteric change, which allows an equilibrium to form with a second binding mode, resulting in N-demethylation. However, given the saturability of this reaction while the rate ofO-demethylation does not change and the strong evidence for a preferred substrate orientation (by interaction with D301), it is tempting to suggest that 2D6 can bind two DXM molecules simultaneously. Indeed, the observed situation is very much like the theoretical situation described by Korzekwa et al. (1998) for a two-site model, with one site being low Km/lowVmax and the other being a highKm/high Vmaxsite where both substrates are identical. We are continuing our investigations to further elucidate these possibilities.
Acknowledgments
We thank Frank Gonzalez (National Institutes of Health) and Ulrich M. Zanger for CYP2D6 cDNA clones. We also thank Steve Leeder for providing the rabbit anti-2D6-peptide antibody.
Footnotes
-
This work was supported by National Institute of Environmental Health Sciences Grant ES09894. Abstracts of this work have been presented at the 13th International Symposium on Microsomes and Drug Oxidations (July 2000, Stresa, Italy) and the Sixth International Symposium on Biological Reactive Intermediates (July 2000, Paris, France).
- Abbreviations used are::
- DXM
- dextromethorphan
- DXO
- dextrorphan
- MEM
- 3-methoxymorphinan
- HYM
- 3-hydroxymorphinan
- P450
- cytochrome P450
- DLPC
- l-α-dilauroylphosphatidylcholine
- DTT
- dithiothreitol
- PMSF
- phenylmethylsulfonyl fluoride
- HPLC
- high-pressure liquid chromatography
- LC-MS
- liquid chromatography-mass spectrometry
- Received April 24, 2001.
- Accepted August 6, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}