


Abstract
In view of conflicting data in the literature regarding the enzyme(s) responsible for metabolism of selegiline, a drug used in the treatment of Parkinson's disease, investigations were carried out in vitro using the human cytochrome P450 enzymes CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 recombinantly expressed in yeast to elucidate the enzyme specificity in selegiline metabolism. In the yeast microsomes used, desmethylselegiline and levomethamphetamine were formed from selegiline at significant rates. The highest contribution to the hepatic clearance of selegiline was calculated to be exerted by CYP2B6 (124 l/h) CYP2C19 (82 l/h), whereas CYP3A4 (27 l/h) and CYP1A2 (21 l/h) were of less importance. Antibodies against CYP2B6 inhibited metabolism of selegiline in microsomes containing CYP2B6 but not in microsomes without significant amounts of the enzyme. In contrast to previous reports, we could not find any role for CYP2D6 in the metabolism of selegiline. The data strongly indicate that the high extent of interindividual variation seen in vivo for selegiline clearance is caused by the metabolism of the compound by the highly polymorphic CYP2B6 and CYP2C19.
Selegiline [(−)-(R)-N,α-dimethyl-N-2-propynylphenethylamine hydrochloride] is a selective irreversible inhibitor of monoamine oxidase B (MAO-B1) and is used in combination with levodopa in the treatment of Parkinson's disease. Levodopa is converted to dopamine, which is responsible for the therapeutic effect. Dopamine is further metabolized by monoamine oxidases of which MAO-B is the most abundant in brain. By inhibiting MAO-B, selegiline decreases dopamine metabolism at the target location and thereby prolongs the effect of medication (Knoll, 1978). It has also been proposed that selegiline itself increases endogenous dopamine levels by inhibiting reuptake at the synaptic cleft and also causes an increase in the synthesis of dopamine by blocking regulatory presynaptic dopamine autoreceptors (Knoll, 1987; Tetrud and Langston, 1989)
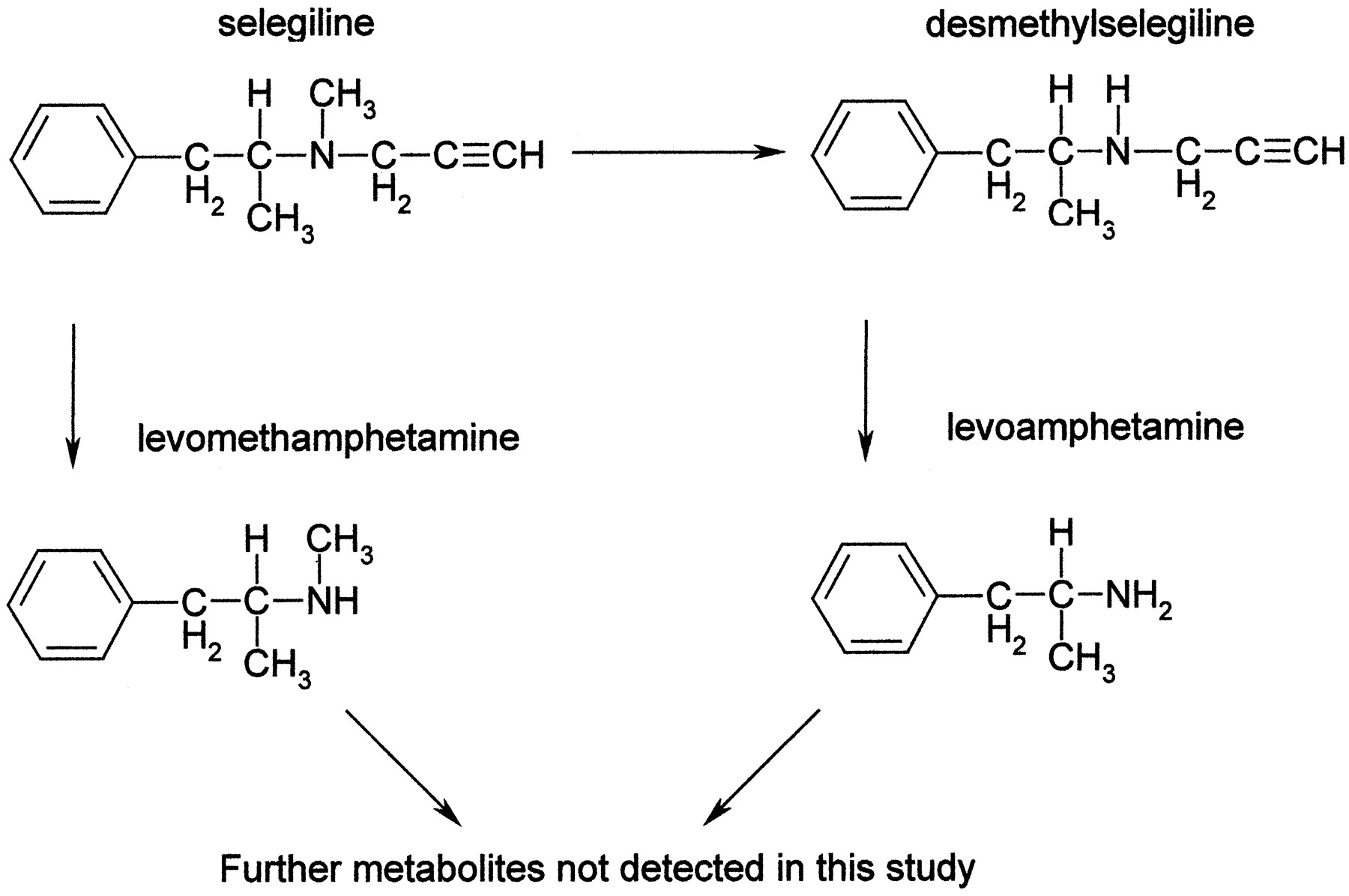
Selegiline is rapidly absorbed after oral administration and metabolized to desmethylselegiline, levomethamphetamine, and levoamphetamine, which in turn are further metabolized (Fig.1) (Shin, 1997). The interindividual variability in the rate of metabolism is very large (Mahmood, 1997). In vitro studies suggest that hepatic cytochromes P450 are involved, and some reports have proposed a role for CYP2D6 in the metabolism of selegiline (Grace et al., 1994; Bach et al., 2000). In contrast, CYP2D6 was not predicted to be important for selegiline elimination in an in vivo study where the metabolism of selegiline was compared between extensive and poor metabolizers of debrisoquine (Scheinin et al., 1998). The same study showed essentially very high IC50 values for selegiline inhibition on CYP2D6 catalyzed activity in vitro. Based on inhibition experiments carried out in microsomes, CYP1A2 and CYP3A4 have also been implicated to participate in the metabolism of selegiline (Taavitsainen et al., 2000). Moreover, analyses of drug-drug interactions with fluvoxamine in vivo have indicated a possible interaction of selegiline with CYP1A2, CYP2D6, 2C19, and 3A4 (Figgitt and McClellan, 2000).

Metabolic pathways of selegiline.
Additional metabolites are found in vivo, as described by Shin (1997)
Because of the open question regarding the principle cytochrome P450 enzyme(s) responsible for the metabolism of selegiline, we considered it of importance to use human cytochrome P450 enzymes heterologously expressed in Saccharomyces cerevisiae to be able to identify the enzymes in question. A prediction of hepatic clearance based on theVmax and Kmvalues obtained with the recombinant enzymes was also done, and this revealed the polymorphic enzymes CYP2B6 and CYP2C19 to be of major importance.
Materials and Methods
Selegiline hydrochloride was from Orion Pharma (Turku, Finland). Desmethylselegiline was from Chinoin Pharmaceutical and Chemical Works (Budapest, Hungary), and l-methamphetamine andl-amphetamine were obtained from Sigma (St. Louis, MO).
cDNAs encoding the human cytochrome P450 enzymes 1A1, 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4 were cloned into the pYeDP60 (V60) (Bellamine et al., 1994; Pompon et al., 1995) and expressed in aS. cerevisiae strain W(R) genetically engineered to overexpress yeast NADPH cytochrome P450 reductase (Truan et al., 1993), essentially as described elsewhere (Truan et al., 1993; Bylund et al., 2000; Andersson et al., 2001). In brief, the yeast were transfected with the expression vector V60 containing a galactose inducible promoter in front of a cDNA encoding one of the above-mentioned P450 enzymes. To achieve higher expression levels, the yeast cells were first grown to high density with glucose as the main energy source; thereafter, galactose was added to induce expression. After expression, the cells were harvested and mechanically disrupted as previously described (Bylund et al., 2000). Microsomes containing the recombinant enzyme were thereafter prepared by differential centrifugation. All P450 enzymes were expressed at levels sufficient for quantification by carbon monoxide binding spectral analyses, as described by (Omura and Sato, 1964). The expression varies between different enzymes, and the concentrations of the specific P450s in this study were for CYP1A1, 66; CYP1A2, 62; CYP2A6, 17; CYP2B6, 33; CYP2C9, 300; CYP2C19, 49; CYP2D6, 30; CYP2E1, 149 and CYP3A4, 291 pmol/mg of microsomal protein. The amount of cytochrome P450 reductase was estimated to be more than 10-fold higher than the amount of P450. Levels of reductase and total protein content were determined according to Yasukochi and Masters (1976) and Lowry et al. (1951), respectively. The enzyme specificity and catalytic properties were characterized with P450-specific probe substrates, and the kinetics for metabolism were in accordance with published values (Andersson et al., 2001). Yeast microsomes expressing CYP2C8 and CYP3A4 with human cytochrome P450 reductase and cytochrome b5 in a 1:1:3 ratio were kindly donated by Astra CYP Products and Dr. Tommy B. Andersson (AstraZeneca, Mölndal, Sweden). The source of human livers and methods for preparation of human liver microsomes has previously been described in (Ekstrom et al., 1989). Monoclonal CYP2B6 antibodies used both for inhibitory studies as well as for immunodetection was purchased from GENTEST (Woburn, MA).
Immunodetection.
Human liver microsomes corresponding to 1 μg were loaded on a 10% SDS-polyacrylamide gel using a mini-PROTEAN II electrophoresis cell (Bio-Rad, Hercules, CA) (Laemmli, 1970). After a 40-min separation at 200 V, the proteins were transferred to nitrocellulose membranes (Hybond-C extra; Amersham Pharmacia Biotech, Piscataway, NJ), as described (Towbin et al., 1979). Unspecific binding was blocked by incubating the membranes in 5% (w/v) nonfat dry milk in Tris-buffered saline-Tween. Thereafter, the membranes were incubated with primary monoclonal mouse CYP2B6 antibodies followed by horseradish peroxidase conjugated goat-antimouse antibodies, both diluted 1:2000 in 1% (w/v) nonfat dry milk containing Tris-buffered saline-Tween. The immunoblots were detected by using enhanced chemiluminescence reagent from Amersham Pharmacia Biotech according to manufacture's instruction and visualized by a Fuji Las-1000 luminescent image analyzer (Fujifilm, Stockholm, Sweden).
Incubation Conditions.
All incubations were carried out in 100 mM potassium phosphate buffer, pH 7.4, at a temperature of 37°. The final incubation volume was in all cases 200 μl. After 2 min of preincubation at 37°, the reactions were initiated by adding NADPH to a concentration of 1.2 mM and terminated by addition of 20 μl 2 M NaOH. Screening of P450 enzymes involved in selegiline metabolism was done with microsomes corresponding to 200 μg of total protein and 1 mM the parent compound using an incubation time of 20 min. As negative controls, we performed incubations with microsomes prepared from yeast transfected with a vector lacking insert as well as incubations without NADPH. For subsequent determination of kinetic properties, selegiline concentrations ranging from 0 to 640 μM were incubated for 5 min with 200 μg of total microsomal protein. Linearity with time and total protein content was assured in all cases.
An inhibitory study on human liver microsomes using antibodies (3.3 mg of antibodies/100 μg of microsomes) was carried out according to the manufacture's recommendation. In brief, 300 μg of microsomes with or without inhibitory antibodies were incubated in a total volume of 250 μl at a selegiline concentration of either 25 or 100 μM. Incubations without NADPH were used as negative controls. The reaction was terminated after 40 min by the addition of 25 μl of 2 M NaOH.
Analysis of Selegiline Metabolites.
Concentrations of selegiline, desmethylselegiline, levoamphetamine, and levomethamphetamine were either determined by gas chromatography using nitrogen selective detection (Scheinin et al., 1998) or a reverse phase high-pressure liquid chromatography method previously described (Taavitsainen et al., 2000).
Data Analysis.
The kinetic results were analyzed by nonlinear regression analysis (Sigma Plot for Windows 4.00; SPSS Science, Inc., Chicago, IL) using a one enzyme Michaelis-Menten model. The Kmand Vmax values obtained were used to estimate the intrinsic clearance (CLint) according to CLint =Vo/C =Vmax/(Km +C), where V0 is the initial metabolic rate and C the unbound drug concentration (Ito et al., 1998). At substrate concentrations considerably lower thanKm, CLint can be approximated byVmax/Km. When estimating CLint in vivo from CLint in vitro the following constants were used.
The content of each P450 in human liver microsomes used was the average enzyme content in microsomes from 12 different human livers, as published by GENTEST (see Table 1). A value of 45 mg of microsomal protein/g of liver (Houston, 1994) was used, and the liver weight was approximated to 1500 g. To calculate the hepatic clearance, the well stirred model was applied, and since selegiline has basic properties, we used the formula excluding protein binding. It has been shown by (Obach, 1999) that this concept gives very good predictability for various basic compounds: CLH = QH ∗ CLint/(QH + CLint), where the hepatic blood flow (QH) was approximated to 1450 ml/min (Davies and Morris, 1993).
Vmax and Km values for P450-mediated selegiline metabolism to desmethylselegiline and levomethamphetamine
Results
An initial screening of selegiline metabolism was carried out with yeast microsomes containing CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2E1, or CYP3A4, as well as with human liver microsomes. Three metabolites, levomethamphetamine, desmethylselegiline, and levoamphetamine, which are detected in vivo, were formed in human liver microsomes, whereas only levomethamphetamine and desmethylselegiline were formed with the recombinant enzymes. The major metabolite formed in the human liver microsomes was levomethamphetamine, a major product also seen for all recombinant enzymes except for CYP2B6, which produced equal amounts of levomethamphetamine and desmethylselegiline, and CYP2C8, which produced slightly higher amounts of desmethylselegiline. Only minor amounts of levoamphetamine were detected in the control human liver microsomal incubations compared with the other two metabolites.
Recombinant enzymes active in selegiline metabolism were chosen for further kinetic characterization, and in addition, CYP3A4 and CYP2C8 in yeast microsomes containing human cytochromeb5 and cytochrome P450 reductase were included. The yeast-expressed CYP3A4 was inactive without these additional human forms of reductase and b5. Incubation conditions were established such that the formation of metabolites was linear with respect to time and microsomal protein content. Selegiline at concentrations ranging from 0 to 640 μM was thereafter incubated with a fixed amount of microsomal protein. The rate of formation at each concentration was calculated as picomole per picomole per minute on the basis of spectrally determined P450 holoenzyme levels. Representative plots on the kinetic evaluation of selegiline-metabolizing enzymes are shown in Fig.2. Vmax andKm were estimated by nonlinear regression analysis, and the values obtained were used to calculate the intrinsic clearance (Table 2). CYP2B6 displayed the highest intrinsic clearance for both of the metabolites formed, and CYP2C19 was the second most efficient enzyme, especially in levomethamphetamine formation. Taken together, the data indicated that CYP2B6 and CYP2C19 accounted for 81% of desmethylselegiline formation and 76% of levomethamphetamine formation. In these calculations, the contribution of CYP1A2 was 6 and 9% for desmethylselegiline and levomethamphetamine, respectively, and for CYP3A4 the corresponding values were 7 and 12%, respectively (Table 2). Calculations of the hepatic clearance of selegiline based on the formation of both metabolites, as outlined under Materials and Methods, revealed the highest values exerted by CYP2B6 (124 l/h) and CYP2C19 (82 l/h), whereas CYP3A4 (27 l/h) and CYP1A2 (21 l/h) were of less importance. Thus, the total hepatic clearance was calculated, based on the relative expression of the various P450s in liver as detailed in Table 2, to be 265 l/h, indicating that selegiline is a high clearance drug.
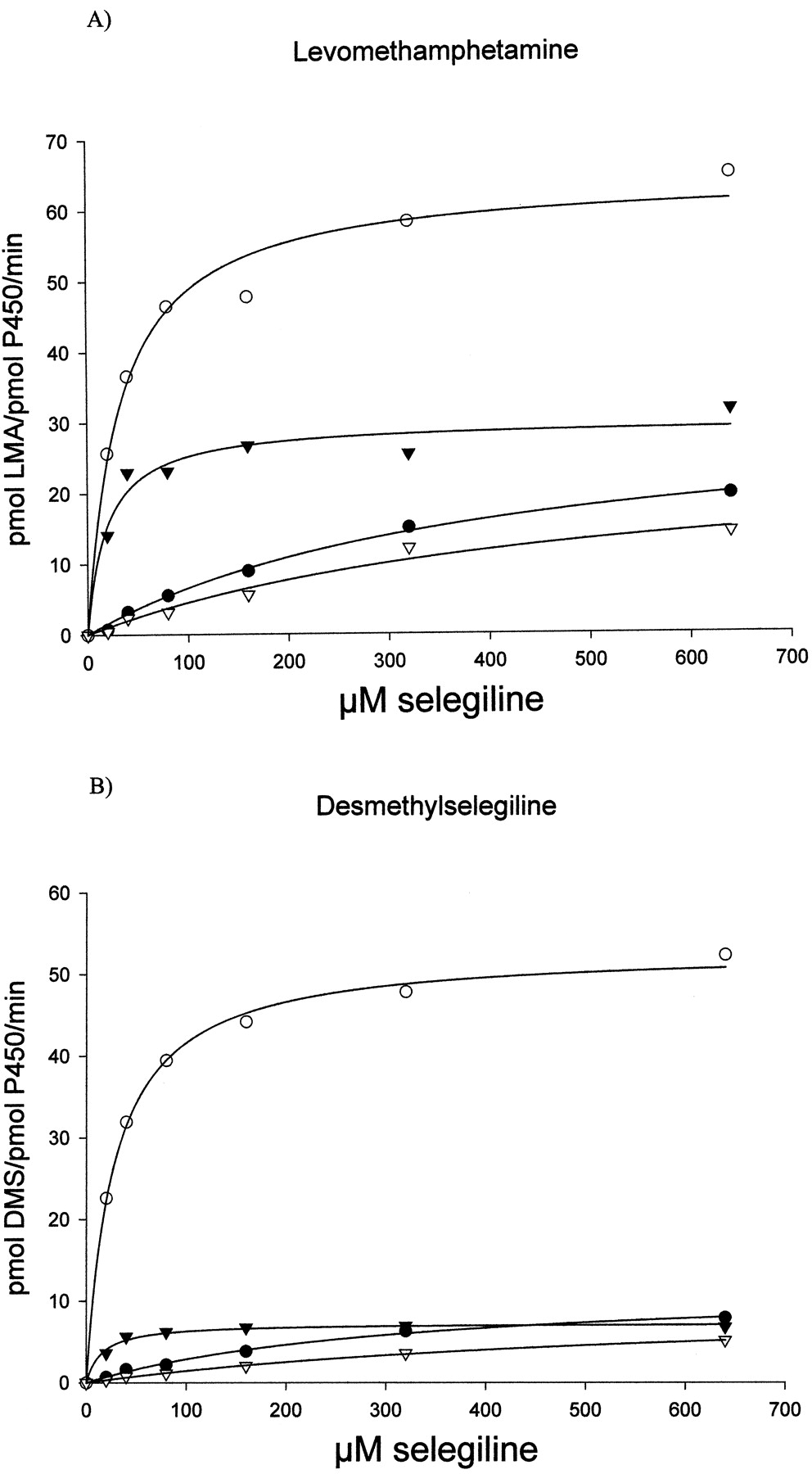
Kinetics of levomethamphetamine (LMA) (A) and desmethylselegiline (DMS) (B) formation in yeast microsomes with recombinantly expressed CYP1A2 (●), CYP2B6 (○), CYP2C19 (▾), and CYP3A4 (▿).
Inhibition of selegiline metabolite formation in human liver microsomes using mouse monoclonal inhibitory antibodies against human CYP2B6
Because of the high capacity of CYP2B6 to metabolize selegiline, we decided to do additional experiments with human liver microsomes and inhibitory antibodies. For these studies, we screened seven human liver microsomal preparations for their CYP2B6 content by Western blot (Fig.3). Two of these preparations with high-CYP2B6 content (HL 33 and HL 44) and one with low-CYP2B6 content (HL 41) were chosen for inhibitory studies at one concentration well above the Km value observed for CYP2B6 in yeast microsomes and one around theKm value. A 20% inhibition was observed in HL 33 for both desmethylselegiline and levomethamphetamine at the high concentration (100 μM), whereas at 25 μM a 40% inhibition was achieved. HL 44 showed a 20% reduction at both concentrations for levomethamphetamine and for desmethylselegiline at the high concentration, and a 7% decrease was observed at the 25 μM concentration. In HL 41 in which no CYP2B6 could be detected, there was only a 3% inhibition of levomethamphetamine, and in contrast, the antibodies against CYP2B6 caused an increase in the formation of desmethylselegiline.
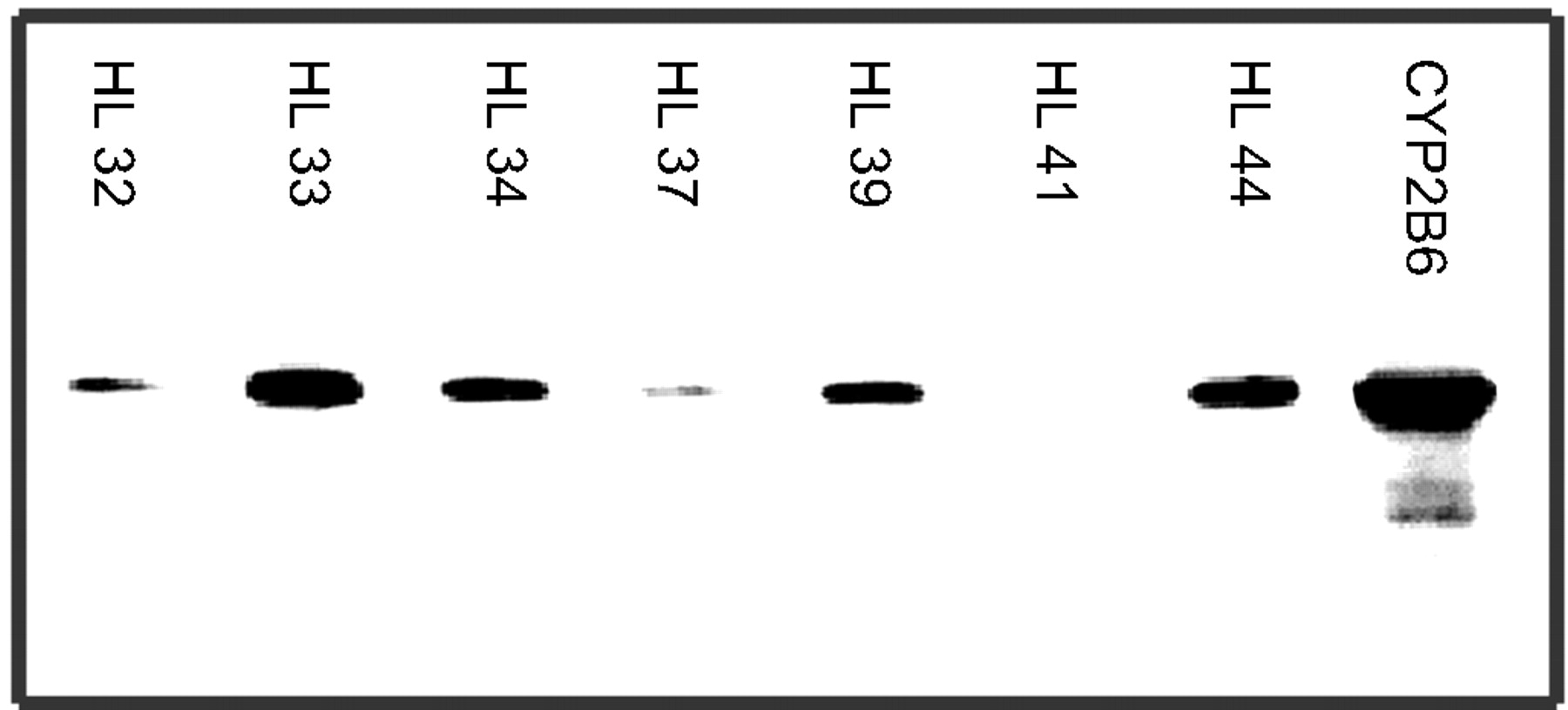
Detection of CYP2B6 in human liver microsomes and yeast-expressing recombinant CYP2B6 by Western blot analysis.
Discussion
In this investigation, we have used microsomes from S. cerevisiae expressing different human P450s to evaluate the metabolism of selegiline, a drug used in the treatment of Parkinson's disease. We found that CYP2B6 and CYP2C19 were the enzymes almost entirely responsible for the hepatic clearance of selegiline. Two of the three major metabolites found in vivo, desmethylselegiline and levomethamphetamine, were detected in this in vitro system. The third metabolite, levoamphetamine, is a secondary metabolite (Shin, 1997). Accordingly, only small amounts of levoamphetamine were formed in the control liver microsomes, and the formation in the recombinant systems, if any, was lower than the detection limit. In humans, most of the metabolites are found in urine, and only approximately 15% are discharged via feces. No unchanged selegiline is found in urine. Almost 60% of the excreted metabolites are levomethamphetamine, and around 25% are the secondary product levoamphetamine. Desmethylselegiline accounts for 3% of the recovered metabolites (Heinonen et al., 1994).
CYP2B6 displayed the highestVmax/Km for selegiline in this study. It is interesting that another drug with a related structure and a similar capacity to increase dopamine levels, bupropion, was recently also described to be a CYP2B6 substrate (Faucette et al., 2000; Hesse et al., 2000). Since few substrates have been characterized with respect to their affinity for CYP2B6, selegiline in combination with other substrates could be used to elucidate the active site of CYP2B6 and thereby increase the possibility to predict whether a compound is a substrate for CYP2B6. In comparison to other substrates found in the literature, selegiline has a rather high affinity for the CYP2B6 active site (Ekins et al., 1999).
The calculation of hepatic clearance of selegiline based on the relative content of the active enzymes in human liver and theVmax and Kmvalues revealed a slightly higher value (265 l/h) than presented in two in vivo studies in which the oral clearances was estimated to be 236 l/h and 56 l/h (Heinonen et al., 1994; Mahmood, 1997). The differences, however, are not significant. This reveals that our in vitro system could correctly predict the in vivo clearance value and thus be able to classify the compound as a high clearance drug.
Previous results regarding the P450 enzyme specificity for selegiline metabolism have been contradictory. A study by Grace et al. (1994)using recombinant CYP2D6 showed that quinidine and quinine inhibited selegiline metabolism, and another study using AHH-1 TK± cells also supported the involvement of this enzyme (Bach et al., 2000). In contrast, in vivo data showed that selegiline metabolism did not correlate to that of other CYP2D6 substrates as seen in poor, extensive, or ultrarapid metabolizers of debrisoquine (Scheinin et al., 1998). Furthermore, in vitro inhibition studies using human liver microsomes (Scheinin et al., 1998; Taavitsainen et al., 2000) did not show any evidence for the involvement of CYP2D6 in selegiline metabolism. Based on our present findings, it might be concluded that CYP2D6 does not play an important role in selegiline metabolism. A recent paper by Taavitsainen et al. (2000) describes selegiline as a potent inhibitor of the CYP2C19-catalyzed mephenytoin 4′-hydroxylation in human liver microsomes, implicating the importance of this enzyme, a finding supported by our studies. The involvement of CYP2B6, however, has not been suggested before, probably due to lack of inhibitory agents for this enzyme.
The participation of the CYP2B6 enzyme in selegiline metabolism is further strengthened by the fact that the recently available inhibitory CYP2B6 antibodies decreased the activity when incubated with human liver microsomes containing CYP2B6 but not when incubated with microsomes lacking or containing very small amounts of this enzyme. The inhibition was more pronounced at low-substrate concentrations where, according to the Km values obtained in yeast microsomes, one would expect the specific inhibition of CYP2B6 to be greater.
The interindividual variability in the capacity for selegiline metabolism has been shown to be considerable (Mahmood, 1997). This could partly be explained by the fact that it is being metabolized by several P450 enzymes with variable activity and expression levels. In our current investigation, we find the polymorphic enzymes CYP2B6 and CYP2C19 to be the major catalysts of the metabolism. The expression level of CYP2B6 and CYP2C19 in human liver is considerably variable (Goldstein and de Morais, 1994; Code et al., 1997), and alleles that cause enzymes with altered activity have been described for both CYP2B6 and CYP2C19. Interestingly, a recently found polymorphism in theCYP2B6 gene was reported to increase this enzyme's 7-ethoxycoumarin-O-deethylation activity (Ariyoshi et al., 2001). Additional alleles of both CYP2B6 andCYP2C19 that explain the variability have been described (see http://www.imm.ki.se/CYPalleles/). At present however, there are no data available on the involvement of these enzymes in selegiline metabolism in vivo, and, thus, there is a need to carry out such studies using enzyme-specific inhibitors.
In conclusion, we have investigated the in vitro metabolism of selegiline and found that CYP2B6 is the major P450 enzyme responsible for its metabolism. Significant contribution is also seen by CYP2C19 and to a small extent by CYP3A4 and CYP1A2. We have shown that use of in vitro systems based on heterologously expressed enzymes could predict the in vivo clearance with a reasonable high accuracy.
Acknowledgments
We thank Dr. Charlotta Otter (AstraZeneca, Mölndal, Sweden) for the generous gift of CYP2C8 and CYP2C19 cDNAs.
Footnotes
-
This work was supported by grants from the Swedish Medical Research Council, AstraZeneca, and by the European Union Framework 4 Program (EUROCYP Project BMH4-CT96-0254).
- Abbreviations used are::
- MAO-B
- monoamine oxidase B
- P450
- cytochrome P450
- CLint
- intrinsic clearance
- Received March 28, 2001.
- Accepted August 6, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}